
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA “waiting” carbon nitride radical anion: a charge storage material and key intermediate in direct C–H thiolation of methylarenes using elemental sulfur as the “S”-source†
Aleksandr
Savateev
 *a,
Bogdan
Kurpil
a,
Artem
Mishchenko
b,
Guigang
Zhang
a and
Markus
Antonietti
a
*a,
Bogdan
Kurpil
a,
Artem
Mishchenko
b,
Guigang
Zhang
a and
Markus
Antonietti
a
aDepartment of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: oleksandr.savatieiev@mpikg.mpg.de
bDepartment for Heterophase Synthesis of Inorganic Compounds and Materials, V.I. Vernadsky Institute of General and Inorganic Chemistry, Palladina Ave., 32/34, Kiev, 03142, Ukraine
First published on 14th March 2018
Abstract
Potassium poly(heptazine imide), a carbon nitride based semiconductor with high structural order and a valence band potential of +2.2 V vs. NHE, in the presence of hole scavengers and under visible light irradiation gives the corresponding polymeric radical anion, in which the specific density of unpaired electrons reaches 112 μmol g−1. The obtained polymeric radical anion is stable under anaerobic conditions for several hours. It was characterized using UV-vis absorption, time resolved and steady state photoluminescence spectra. The electronic structure of the polymeric radical anion was confirmed by DFT cluster modelling. The unique properties of potassium poly(heptazine imide) for storing charges were employed in visible light photocatalysis. A series of substituted dibenzyldisulfanes was synthesized in 41–67% yield from the corresponding methylarenes via cleavage of the methyl C–H bond under visible light irradiation and metal-free conditions.
Introduction
C–H functionalization of hydrocarbons mediated by heterogeneous visible light photocatalysts is highly attractive for sustainable chemistry.1 This strategy can afford high value-added chemicals from inexpensive reagents, minimize the number of steps and hence auxiliary chemicals, reduce the number of by-products and could potentially use only sunlight as the energy source, while the photocatalyst can be easily separated from the solution and used again.2 Numerous examples of heterogeneous photocatalysts and photocatalytic reactions have been reported in the literature – the hydrogen evolution reaction3,4 and overall water splitting,5 CO2 reduction6,7 and styrene production, just to name a few.8 Due to their chemical inertness, arising from very high C–H bond energies and ionization potentials, selective functionalization of hydrocarbons is quite challenging.9The literature, describing C–H functionalization of hydrocarbons mediated by heterogeneous photocatalysts, is mostly represented by synthesis of aldehydes, ketones and carboxylic acids, since O2 in all these cases is used as an electron scavenger and hydrogen oxidation as the thermodynamic driving force.10–13
In order to transcend the borders of heterogeneous photocatalysis applied to organic synthesis and to broaden the spectrum of products, semiconductors with high oxidation power of photogenerated holes may be envisioned for use. According to this strategy, a potent thermodynamic driving force for substrate oxidation is created, eliminating the need for O2, while mild electron acceptors can then be used to remove the electron from the conduction band (CB) and close the photocatalytic cycle.
In this regard, potassium poly(heptazine imide), K-PHI hereafter (Fig. 1a),14 a carbon nitride based material, is a promising candidate to assay this strategy.15–17 The band structure of K-PHI is schematically shown in Fig. 1d. Due to the highly positive valence band (VB) potential, +2.22 V vs. NHE, photogenerated holes are effective oxidants for application in organic synthesis. For example, K-PHI was successfully used in the oxidation of alcohols to aldehydes and 1,4-dihydropyridines to pyridines,18 in the synthesis of 1,3,4-oxadizoles by oxidative cyclization of N-acylhydrazones,19 in thioamide synthesis by photocatalytic Kindler reaction,20 and in oxygen and hydrogen evolution reactions.21
 | ||
| Fig. 1 (a) Idealized structure of K-PHI. (b) PXRD pattern of K-PHI. (c) HR-TEM image of K-PHI. (d) Band structure of K-PHI. The CB position of K-PHI was determined by Mott–Schottky analysis15 and the VB position calculated as EVB = ECB + BG, where BG is the optical band gap of K-PHI determined from the Tauc plot (Fig. S1e†). CB and VB potentials of g-CN were taken from the literature.14 (e) Suspension of K-PHI (10.39 mg) and 4-methylanisole (0.6 mL) in acetonitrile (6.05 mL). (f) The same suspension after stirring at +50 °C for 24 h under light irradiation (461 nm, 0.0517 ± 3 × 10−5 W cm−2). (g) The same suspension immediately after methylviologen dichloride dihydrate (6.9 mg, 24 μmol) addition. The blue rectangle shows the solution of MV˙+ in acetonitrile, and the orange rectangle shows the precipitated K-PHI. (h) UV-vis absorption spectra of the K-PHI suspension in a 4-methylanisole/MeCN mixture: prior to irradiation (orange, the suspension appearance is also shown in (e)) and after irradiation for 2 h with a 461 nm LED (green, the suspension appearance is also shown in (f)), both recorded under anaerobic conditions. (i) Time resolved PL spectra of the K-PHI suspension in a 4-methylanisole/MeCN mixture: prior to irradiation (orange, the suspension appearance is also shown in (e)) and after irradiation for 2 h with a 461 nm LED (green, the suspension appearance is also shown in (f)), both recorded under anaerobic conditions. The steady state PL spectra of the suspended materials, recorded using a 360 nm excitation wavelength, are given as the inset. Experiments aimed at checking the ability of K-PHI to store electrons without any external energy input under anaerobic conditions are enclosed with a dashed line. (j) Absorption spectrum of MV˙+ solution in acetonitrile. | ||
On the other hand, the CB in K-PHI is located at −0.5 V vs. NHE, which is significantly lower compared, for example, to that of graphitic carbon nitride (g-CN), −1.29 V vs. NHE. Therefore, it is reasonable to assume that the K-PHI radical anion, hereafter denoted as K-PHI˙−, formed from K-PHI upon its single electron reduction, would be stable and have a lifetime above the μs–s timescale (Fig. 2).22 The viability of this idea was recently proved by Lotsch et al. who have reported radical carbon nitride species with a lifetime exceeding hours, while the material was applied in the hydrogen evolution reaction and a sunlight harvesting device.23,24
 | ||
| Fig. 2 A schematic representation of the photocatalytic reaction proceeding via reductive quenching of the photocatalyst. K-PHI* represents the excited state of the photocatalyst, K-PHI˙− stands for the radical anion, HC stands for “hydrocarbon”, and ES stands for electron scavenger. | ||
In view of hydrocarbon C–H bond activation, the long living K–PHI˙− might be a key intermediate permitting employment of reagents different from the abovementioned O2. The mechanism describing this approach is summarized in Fig. 2.
In the present work, we have shown that K-PHI in the presence of hole scavengers, even those as weak as toluene, when triggered by visible light, acts as a capacious electron buffer and forms a stable radical anion under air-free conditions. This feature was applied in visible light photocatalytic activation of the C–H bond of hydrocarbons – we have developed a simple and convenient method to synthesize dibenzyldisulfanes from inexpensive methylarenes and S8 under visible light irradiation using K-PHI as a heterogeneous photocatalyst under metal-free conditions.
Results and discussion
K-PHI was prepared according to a procedure reported by us earlier.18 The powder X-ray (PXRD) pattern shows diffraction peaks typical of this material.25 Among them, the intense peak at ∼27° is due to stacking of layers similar to g-CN, and the peaks at 8.1°, 14.2°, 21.8°, 36.0° and 43.9° are related to an ordered in-layer arrangement (Fig. 1b).25 The high resolution TEM image reveals the crystalline structure of the synthesized K-PHI with interlayer distances of 0.33 nm and an in-plane periodicity of 1.1 nm (Fig. 1c). The X-ray photoelectron, ultraviolet photoelectron, diffuse reflectance UV-vis absorption, photoluminescence, and Fourier-transform infrared (FT-IR) spectra, N2 absorption isotherm and SEM image of K-PHI are given in the ESI (Fig. S1†) and they are consistent with those reported previously.14,26,27In order to check the possibility of using K-PHI as an electron buffer and subsequently applying it in photocatalytic hydrocarbon functionalization, we performed the following experiment. A mixture of K-PHI and p-methylanisole (holes scavenger) in acetonitrile was mixed, degassed and kept under a N2 atmosphere throughout the course of the experiment (Fig. 1e).‡ The suspension was irradiated with a 461 nm LED. After irradiation was stopped, we noticed that the suspended particles of K-PHI had changed colour from orange to “avocado green” (Fig. 1f). The colour change was taken as the first piece of evidence of K-PHI radical anion formation, hereafter denoted as K-PHI˙−.23 Upon addition of excess dimethylviologen dichloride dihydrate (MV2+), yellow K-PHI immediately precipitated, while the solution changed its colour to intense blue, which is due to MV2+ single electron reduction to its radical cation (MV˙+) (Fig. 1g). Measuring the absorbance of the solution at λ = 606 nm (Fig. 1j) and taking into account the extinction coefficient of MV˙+ in acetonitrile of ε = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900,28 the specific number of electrons stored by the polymeric K-PHI˙− was calculated to be 112 μmol per gram of the material. In the control experiment, when MV2+ and K-PHI were mixed together prior to irradiation, no MV˙+ was formed. However, MV2+ was converted into MV˙+ upon subsequent light irradiation. This implies that energy input is essential to enabling electron flux. Note that the capacity of the covalent g-CN in the same experiment was orders of magnitude lower.
900,28 the specific number of electrons stored by the polymeric K-PHI˙− was calculated to be 112 μmol per gram of the material. In the control experiment, when MV2+ and K-PHI were mixed together prior to irradiation, no MV˙+ was formed. However, MV2+ was converted into MV˙+ upon subsequent light irradiation. This implies that energy input is essential to enabling electron flux. Note that the capacity of the covalent g-CN in the same experiment was orders of magnitude lower.
The optical properties of the suspended K-PHI˙− (Fig. 1f) were investigated. The UV-vis absorption spectrum (Fig. 1h) shows a broad absorption band in the 600–750 nm range, which is in agreement with the observed green colour. The photoluminescence (PL) lifetime of K-PHI˙− is 0.35 ns versus 0.66 ns for K-PHI (Fig. 1i). The room temperature steady-state PL spectrum of the K-PHI˙− suspension is significantly lower compared to that of K-PHI in the ground state, while the maximum is shifted to a shorter wavelength (555 nm). This observation supports that p-methylanisole indeed acts as a hole scavenger – it depopulates the number of holes and hence reduces PL.
We observed a colour change of the suspension from green back to yellow when the suspension was exposed to S8 or O2 implying that these molecules can be used in combination with K-PHI in the subsequent functionalization of hydrocarbons (shown below).
In order to characterize theoretically the electronic structure of K-PHI, we conducted a density functional theory (DFT) study on a model K-PHI cluster containing 6 heptazine units and 2 potassium ions. Its geometry was optimized using several pure (PBE and BLYP) and hybrid (PBE0, B3LYP, and BHHLYP) generalized gradient approximation (GGA) functionals (see the Experimental section and ESI† for details).
To a first approximation the energies of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals of the model cluster can be compared with the edge energies of K-PHI valence and conduction bands respectively. These orbitals are of π-type and are located mainly on the nitrogen atoms of the poly(heptazine imide) matrix (Fig. 3). Among the functionals used, B3LYP gave the best agreement with the optical band gap determined experimentally, 3.02 eV vs. 2.72 eV (Table 1). On the other hand, PBE0 revealed that the HOMO of the K-PHI cluster is located at +2.08 eV, which is comparable with the valence band position, +2.22 eV, obtained by adding the optical band gap value, 2.72 eV (Fig. S1e†), to the conduction band value, −0.5 eV. Moreover, this valence band position agrees well with the VBM, +2.40 eV, determined directly using ultraviolet photoelectron spectroscopy (UPS) (Fig. S1d†). Since the HOMO position is slightly more positive than the oxidation potential of toluene (or even more positive than that of p-methylanisole, due to the M+ effect of the OMe group), electron transfer from toluene to the valence band of K-PHI is thermodynamically allowed (Fig. 3). On the other hand, the predicted LUMO position is −1.32 eV and is significantly more negative than the CB potential, −0.5 eV, measured using Mott–Schottky analysis.15 Nevertheless, the reduction potential of MV2+ to MV˙+, −0.445 V, is less negative than the CB potential; therefore flux of electrons from the CB of K-PHI to MV2+ is thermodynamically allowed.
 | ||
| Fig. 3 CB position determined by Mott–Schottky analysis15 and VB position calculated as EVB = ECB + BG, where BG is the optical band gap, 2.72 eV, of K-PHI determined from the Tauc plot (Fig. S1e†). HOMO and LUMO positions in the K-PHI cluster predicted by the PBE0 functional are given for comparison in parentheses. HOMO and LUMO positions from Table 1 were recalculated taking into account an absolute value of VNHE = 4.18 V for the normal hydrogen electrode. The standard oxidation potential of toluene, MV2+ and sulfur was adapted from the literature.29–31 | ||
| Value | PBE | PBE0 | BLYP | B3LYP | BHHLYP |
|---|---|---|---|---|---|
| E HOMO | −5.33 | −6.26 | −5.17 | −5.99 | −7.13 |
| E LUMO | −3.62 | −2.86 | −3.48 | −2.97 | −1.90 |
| ΔEHOMO–LUMO | 1.71 | 3.40 | 1.69 | 3.02 | 5.22 |
The model reaction of K–PHI˙− quenching by adding MV2+, O2 or S8 allows us to call K-PHI a “waiting” photocatalyst, because in the presence of a hole scavenger, even a weak one such as p-methylaniosle, and light energy input, it is rapidly converted into stable K-PHI˙− and “waits” until an oxidant comes to recover the ground state of K-PHI.
Having evidence that K-PHI is a capacious electron buffer and hence can decouple oxidation and reduction of the substrates, we applied this material in photocatalytic thiolation of toluene. Instead of using sulfur-transferring reagents, we took aim at elemental sulfur (S8) as a cheap raw source. Its redox chemistry is well known and the problem of its utilization exists – the annual excess of sulfur production is about seven millions ton.32 Instead of simply storing it, chemists are searching for possibilities to utilize this huge excess of sulfur as an alternative feedstock for polymeric materials,33 as an electron sacrificial agent in photocatalysis18 and so forth.34 Toluene, having an oxidation potential of +2.019 ± 0.02 V (vs. NHE) and a relatively reactive methyl group,29 has been chosen as a model substrate to screen the reaction conditions. The main results of these tests are summarized in Tables S1 and S2.† The formation of dibenzyldisulfide was unambiguously proved by the fact that its 13C NMR, FT-IR and mass spectra were identical to those of the reference (Fig. S2†).
The developed photocatalytic system is indeed versatile and tolerant to different functional groups. A series of substituted methylarenes was converted into the corresponding disulfanes (Fig. 4). Dibenzyldisulfane 1a was obtained in 56% isolated yield, while p-methoxysubstituted disulfane 1b was obtained with even higher, 67%, yield. Interestingly, that steric effect is negligible and 1,2-bis(2-methoxybenzyl)disulfane 1c was isolated with 63% yield. Ethylbenzene was exclusively oxidized at the α-position of the alkyl chain furnishing disulfane 1d. This result again supports the idea that upon oxidation of alkylarene by K-PHI, first reactive species of benzyl-type is formed, which subsequently reacts with sulfur. Disulfane 1d was isolated as a 1:1 mixture of its diastereomers as proven by 13C NMR because K-PHI does not possess chirality. p-fluorotoluene was oxidized to disulfane 1e with 41% isolated yield. tert-Butyl p-tolylcarbamate gave the corresponding disulfane 1f with 51% isolated yield implying that K-PHI offers quite mild conditions, under which the protection group remains intact. In the case of p-toluonitrile the yield of disulfane 1g was 9%, because the reaction terminated at the stage of polysulfane formation (see below). Finally, p-iodotoluene gave disulfane 1h, though with ca. 3% yield, apparently due to lability of the C–I bond under photoredox conditions.
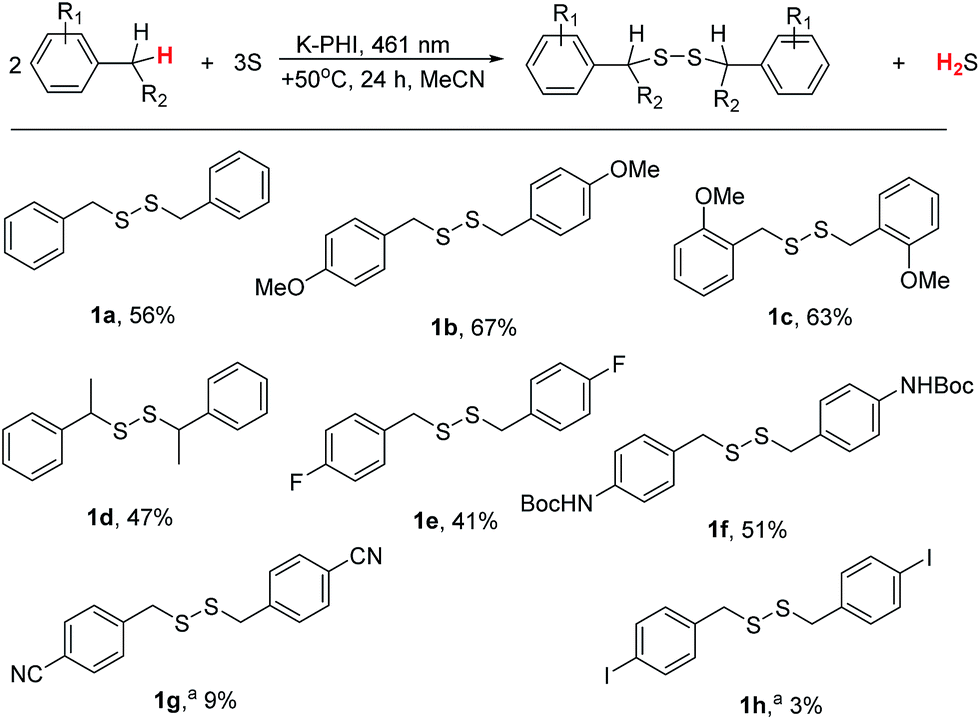 | ||
| Fig. 4 Diaryldisulfane synthesis. Isolated yields are given unless otherwise specified. Conditions: K-PHI 10 mg, methylarene 0.3 mL, S8 0.96 mg, MeCN 2.7 mL, +50 °C, 24 h, 461 nm (0.0517 ± 3 × 10−5 mW cm−2). a Determined by 1H NMR using dibenzylether as an internal standard. | ||
The highest yield of 1,2-dibenzyldisulfane was obtained when toluene oxidation was conducted under blue (461 nm) light (Fig. 5a), as this wavelength matches the K-PHI optical band gap, 2.72 eV. UV light (350 nm) gave disulfane with a slightly lower yield, which could be rationalized by consecutive S–S bond cleavage under the highly energetic UV radiation. On the other hand, under green light (522 nm) almost no disulfane was generated. The apparent quantum yield (AQY) of p-methylanisole photooxidation at 461 nm was calculated to be 0.23 ± 0.02% (Fig. 5a). Our calculations are based on the photon flux measured using an optical power meter equipped with an integrating sphere, taking into account that every polysulfane molecule requires oxidation of two p-methylanisole molecules. It indicates that C–H bond oxidation at close to room temperature is a difficult process especially in the absence of any metal co-catalysts, but still possible due to K-PHI.
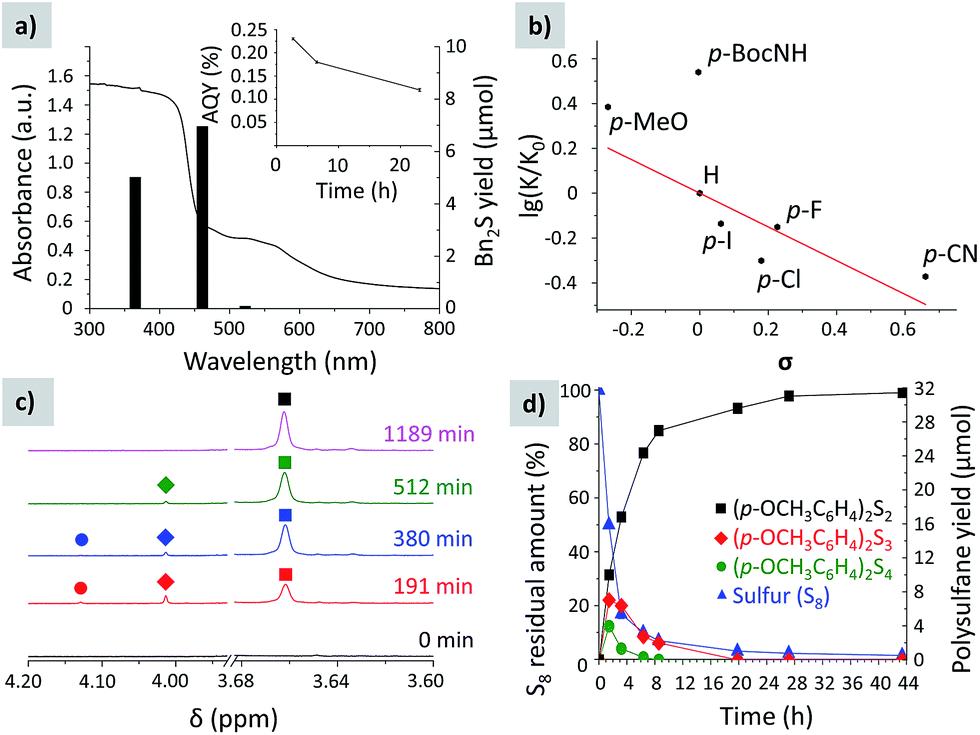 | ||
| Fig. 5 (a) Diffuse reflectance UV-vis spectrum of K-PHI with the yield of 1,2-dibenzyldisulfane depending on the irradiation wavelength. (b) Hammett plot. (c) Time dependent evolution of the methylene group signal of (p-OCH3C6H4)2S2 (squares), (p-OCH3C6H4)2S3 (diamonds) and (p-OCH3C6H4)2S4 (circles) in selected 1H NMR spectra of the reaction mixture. (d) Time dependent yield of (p-OCH3C6H4)2S2 (squares), (p-OCH3C6H4)2S3 (diamonds) and (p-OCH3C6H4)2S4 (circles), and residual sulfur (triangles). | ||
All this behavior indicates that, in contrast to photochemical water splitting with g-CN, this time the reduction process is the slow, rate determining step, as there is obviously no straight chemical reaction channel for the sulfur to quickly take up the photogenerated electron to restore the ground state of the photoredox catalyst.
The Hammett plot (Fig. 5b) supports that the reaction is sensitive to electronic effects. The ρ constant is −0.75, suggesting that the rate limiting step involves positively charged species – the radical cation of benzyl type.
In the reaction mixture we detected intermediate polysulfanes, namely 1,4-bis(4-methoxybenzyl)tetrasulfane and 1,3-bis(4-methoxybenzyl)trisulfane, when oxidation of p-methylanisole was studied. Given that the chemical shift of the methylene groups depends on the number of sulfur atoms between the benzylic fragments, the peak at 4.13 ppm was assigned to the 1,4-bis(4-methoxybenzyl)tetrasulfane CH2 group,35 while the one at 4.01 ppm to the CH2 group of 1,3-bis(4-methoxybenzyl)trisulfane36 (Fig. 5c). The designed photocatalytic system can also produce polysulfides with up to 5 sulfur atoms when excess sulfur was used (Fig. S3†).
The time dependent composition of the reaction mixture is shown in Fig. 5d. Already after 86 min, 50% of sulfur was consumed. However, the yield of disulfane was only 30% of the theoretical value, suggesting that at the beginning of the reaction sulfur is rapidly converted into polysulfides and stored in the form of bis(4-methoxybenzyl)polysulfanes. Nevertheless, the intermediate tetra- and trisulfides were completely converted into 1,2-bis(4-methoxybenzyl)disulfane after ca. 27 h of irradiation, implying that disulfane is the most stable under the given photocatalytic conditions. The amount of target disulfane asymptotically approaches the theoretical value, expected for the given amount of sulfur, and after 44 h 93% of the maximum possible quantity was obtained, proving that the stoichiometric ratios between reagents and products are indeed as described in Fig. 4.
Summarizing all the abovementioned observations, the tentative mechanism of methylarene photocatalytic oxidative thiolation is shown in Fig. 6. Visible light excites K-PHI giving rise to K-PHI* species with a bound electron–hole exciton pair. At this point two pathways, oxidative and reductive quenching of K-PHI*, are in principle possible.37 However, the reaction proceeds predominantly via the oxidation of methylarene (ArMe) to the corresponding radical cation (ArMe˙+) which occurs relatively fast. The leftover K–PHI˙− is oxidized by S8 back to the initial K-PHI, closing the photocatalytic cycle, while sulfur is then converted into S8˙− (structure a on Fig. 6). We are aware that in solution sulfur can form polysulfides of different lengths as well as cyclic molecules,38,39 but for clarity of presentation we assume that linear S8˙− is the only product of sulfur reduction. Due to the positive charge, ArMe˙+ is a much stronger acid (pKa = −13, PhCH3˙+ in MeCN)40 compared to the neutral ArMe (pKa = 54, PhCH3, in MeCN).41
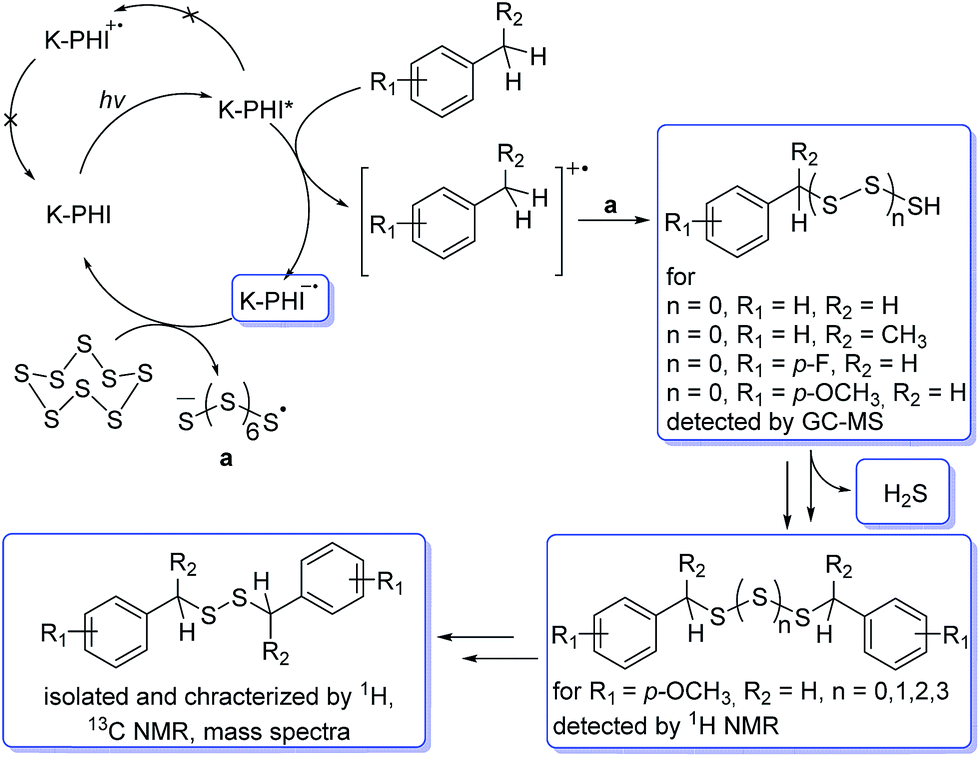 | ||
| Fig. 6 A proposed mechanism of photocatalytic thiolation of toluene with elemental sulfur. Organic molecules or intermediate reactive species that were detected or isolated during the course of reaction are highlighted with rectangles. | ||
On the other hand, S8˙−, similar to the well-studied trisulfur radical anion S3˙−,42 is basic and therefore abstracts a proton from ArMe˙+. This transfer produces HS8˙ and ArCH2˙ respectively. The formation of ArCH2˙ was proven indirectly – we observed 1,2-diphenylethane as a major product when toluene was used both as a reagent and a solvent (Table S1,† entries 2 and 9). In this case, the concentration of ArCH2˙ radicals becomes high enough to favour their recombination. In acetonitrile, in the next step a C–S bond is created by recombination between the HS8˙ and ArCH2˙ radicals. A transition of hydropolysulfides to disulfides requires evolution of H2S and extrusion of extra sulfur atoms, which in turn participate in subsequent reactions with ArMe. Apparently, K-PHI is also involved in all these steps, i.e. the polysulfides obviously react similarly to the original sulfur reactant. H2S was detected as Ag2S, when the reaction mixture headspace was allowed to react with AgNO3 (Fig. S4†). Also traces of intermediate RCH2SH were detected by GC-MS of the reaction mixture additionally supporting the mechanism (mass spectra, Fig. S5†). Finally, the structure of K-PHI remains unaffected as proved by the identical nature of its PXRD patterns and FT-IR spectra before and after the photocatalytic experiment (Fig. S6†).
Conclusions
Potassium poly(heptazine imide) enables the photoredox thiolation of methylarenes with elemental sulfur. This reaction takes place under very mild conditions and blue light irradiation. The reaction proceeds via formation of polysulfides as intermediates, which potentially could also be isolated, if reaction is terminated at the appropriate time. The discovered photocatalytic conversion of methylarenes into disulfides clearly reflects the high oxidation power of K-PHI that activates the methyl C–H bond toward subsequent reaction with sulfur species. Mechanistic studies suggest that the reaction proceeds first via methylarene oxidation by the photogenerated hole of K-PHI*, leaving a K-PHI˙− radical anion. We isolated this long living K–PHI˙− radical anion and characterized it spectroscopically. The exceptionally long lifetime of the K-PHI˙− radical anion arises from its high stability under air-free conditions, as plausibilized by model calculations revealing a narrow band of delocalized states for the extra electron. All of this makes K-PHI a promising photo-responsive material for energy storage applications. The newly disclosed reaction is convenient for the generation of a number of technically very useful disulfanes, as they, for instance, are used for rubber processing, surface modifications, or click chemical modifications of (bio)polymers. We expect the reaction to be more general and applicable to the oxidative modification of all C–H positions where the cation radical is resonance stabilized.Experimental section
1H and 13C NMR spectra were recorded on an Agilent 400 MHz spectrometer using the signal of TMS (0 ppm) as a reference. 13C APT experiments were conducted to distinguish between C, CH2 and CH, CH3 carbon atoms. An Agilent 6890 Network GC System coupled with an Agilent 5975 Inert Mass Selective detector (electron ionization) was used for reaction mixture composition analysis and to obtain mass spectra of the products. Irradiance of the LED module was measured using a PM400 Optical Power and Energy Meter equipped with integrating sphere S142C.Synthetic procedures
tert-Butyl p-tolylcarbamate was prepared according to a literature procedure.43 Mesoporous graphitic carbon nitride (mpg-CN) was prepared according to a literature procedure.44K-PHI was prepared according to a literature procedure.18 A mixture of 5-aminotetrazole (0.99 g) and LiCl/KCl eutectics (4.97 g) was introduced into a steel ball mill cup. The steel ball was placed and the cup was closed. The mixture of precursors was ground for 5 min at a shaking rate of 25 s−1. The resultant flour-like white powder was transferred into a porcelain crucible, covered with a porcelain lid and placed into an oven. The temperature inside the oven was increased from 20 °C to 600 °C within 4 hours under a flow of nitrogen (15 L min−1) after which it was maintained at 600 °C for another 4 hours. The oven was allowed to cool to room temperature. The melt from the crucible was transferred into a beaker; deionized water (50 mL) and a stir bar were placed into the beaker. The suspension was kept stirring at room temperature for 4 hours until the suspension became fully homogeneous and no agglomerated particles were seen. The solid was separated by centrifugation (6500 min−1, 12 min), washed with water (3 × 2 mL) using a centrifuge to separate particles of the material (13500 min−1, 1 min) and dried in a vacuum giving 256 mg of the dark-yellow material.
000 min−1) and washed with acetonitrile (3 × 1.5 mL). The washings were combined and acetonitrile was evaporated under reduced pressure (+50 °C, 80 mbar). The residue was washed with chloroform (3 × 2 mL) and undissolved particles were separated by centrifugation (13000 min−1). The evaporation of chloroform under reduced pressure furnished the corresponding disulfane.
DFT calculations
The calculations were carried out using the GAMESS (US) program package;45 their results were visualized with the MacMolPlt graphical interface.46 The poly(heptazine imide) matrix was modelled by a cluster comprising 6 heptazine units linked by 4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH groups and 2 N atoms. Two K+ cations were added to neutralize the negative charge of the matrix, which originated from the deprotonated imide nitrogen atoms. To reduce computational cost, the symmetry of the model cluster was set to the C2v point group. The basis was a Dunning–Hay double zeta set with one p-type polarization function on hydrogen atoms and one d-type function on other atoms. For the nitrogen atoms the basis set was additionally extended by a diffuse sp-shell. The geometry of K-PHI was optimized using 2 pure GGA functionals (PBE and BLYP) and 3 hybrids with different amounts of Hartree–Fock exchange (25% for PBE0, 20% for B3LYP and 50% for BHHLYP). The spatial distribution of the frontier orbitals in the cluster was found to be independent of the functional. On the other hand, the increase of the Hartree–Fock exchange percentage in the GGA < B3LYP < PBE0 < BHHLYP series increases the HOMO–LUMO gap by lowering the HOMO and raising the LUMO energies (see Table 1).
NH groups and 2 N atoms. Two K+ cations were added to neutralize the negative charge of the matrix, which originated from the deprotonated imide nitrogen atoms. To reduce computational cost, the symmetry of the model cluster was set to the C2v point group. The basis was a Dunning–Hay double zeta set with one p-type polarization function on hydrogen atoms and one d-type function on other atoms. For the nitrogen atoms the basis set was additionally extended by a diffuse sp-shell. The geometry of K-PHI was optimized using 2 pure GGA functionals (PBE and BLYP) and 3 hybrids with different amounts of Hartree–Fock exchange (25% for PBE0, 20% for B3LYP and 50% for BHHLYP). The spatial distribution of the frontier orbitals in the cluster was found to be independent of the functional. On the other hand, the increase of the Hartree–Fock exchange percentage in the GGA < B3LYP < PBE0 < BHHLYP series increases the HOMO–LUMO gap by lowering the HOMO and raising the LUMO energies (see Table 1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Deutsche Forschungsgemeinschaft for the financial support (DFG-An 156 13-1) and thank Christian Wolff for measuring time-resolved PL spectra. A. M. acknowledges the financial support from the scholarship of the President of Ukraine for young scientists. Computing resources for DFT calculations were provided by the SCIT supercomputer (V. M. Glushkov Institute of Cybernetics of the NAS of Ukraine).47Notes and references
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- J. Zhang, M. Zhang, R.-Q. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- L. Lin, C. Wang, W. Ren, H. Ou, Y. Zhang and X. Wang, Chem. Sci., 2017, 8, 5506–5511 RSC.
- Y. Zheng, L. Lin, X. Ye, F. Guo and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 11926–11930 CrossRef CAS PubMed.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CAS.
- F. Guo, P. Yang, Z. Pan, X.-N. Cao, Z. Xie and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 8231–8235 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, in Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, Kluwer Academic Publishers, 2000, ch. 1 Search PubMed.
- Y. Wang, H. Li, J. Yao, X. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4, 2333–2336 CrossRef CAS.
- A. Henríquez, H. D. Mansilla, A. M. M.-d. l. Cruz, J. Freer and D. Contreras, Appl. Catal., B, 2017, 206, 252–262 CrossRef.
- M. A. Gonzalez, S. G. Howell and S. K. Sikdar, J. Catal., 1999, 183, 159–162 CrossRef CAS.
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Chem. Mater., 2015, 27, 5170–5179 CrossRef CAS.
- A. Savateev, S. Pronkin, J. D. Epping, M. Willinger, C. Wolff, D. Neher, M. Antonietti and D. Dontsova, ChemCatChem, 2017, 9, 167–174 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- A. Savateev, S. Pronkin, J. D. Epping, M. G. Willinger, M. Antonietti and D. Dontsova, J. Mater. Chem. A, 2017, 5, 8394–8401 CAS.
- A. Savateev, D. Dontsova, B. Kurpil and M. Antonietti, J. Catal., 2017, 350, 203–211 CrossRef CAS.
- B. Kurpil, K. Otte, M. Antonietti and A. Savateev, Appl. Catal., B, 2018, 228, 97–102 CrossRef CAS.
- B. Kurpil, B. Kumru, T. Heil, M. Antonietti and A. Savateev, Green Chem., 2018, 20, 838–842 RSC.
- B. Kurpil, A. Savateev, V. Papaefthimiou, S. Zafeiratos, T. Heil, S. Özenler, D. Dontsova and M. Antonietti, Appl. Catal., B, 2017, 217, 622–628 CrossRef CAS.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- V. W.-h. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., 2016, 56, 510–514 CrossRef PubMed.
- F. Podjaski, J. Kröger and B. V. Lotsch, Adv. Mater., 2018, 30, 1705477 CrossRef PubMed.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- A. Savateev, S. Pronkin, M. Willinger, M. Antonietti and D. Dontsova, Chem.–Asian J., 2017, 12, 1517–1522 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z.-a. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- T. Watanabe and K. Honda, J. Phys. Chem., 1982, 86, 2617–2619 CrossRef CAS.
- P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, J. Org. Chem., 2009, 74, 5163–5173 CrossRef CAS PubMed.
- A. M. Roy, G. C. De, N. Sasmal and S. S. Bhattacharyya, Int. J. Hydrogen Energy, 1995, 20, 627–630 CrossRef CAS.
- M. Bouroushian, in Monographs in Electrochemistry, Springer-Verlag, Berlin Heidelberg, 2010, vol. 1, ch. 1, pp. 57–75 Search PubMed.
- G. Kutney, Sulfur. History, Technology, Applications and Industry, ChemTec Publishing, 2007 Search PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- J. Lim, J. Pyun and K. Char, Angew. Chem., 2015, 54, 3249–3258 CrossRef CAS PubMed.
- R. Ballini, Synthesis, 1982, 1982, 834–836 CrossRef.
- K. L. Stensaas, A. S. Brownell, S. Ahuja, J. K. Harriss and S. R. Herman, J. Sulfur Chem., 2008, 29, 433–443 CrossRef CAS.
- A. U. Meyer, V. W.-h. Lau, B. König and B. V. Lotsch, Eur. J. Org. Chem., 2017, 2017, 2179–2185 CrossRef CAS.
- A. J. Kamyshny, A. Goifman, J. Gun, D. Rizkov and O. Lev, Environ. Sci. Technol., 2004, 38, 6633–6644 CrossRef CAS PubMed.
- B. Eckert and R. Steudel, in Elemental Sulfur und Sulfur-Rich Compounds II, ed. R. Steudel, Springer Berlin Heidelberg, Berlin, Heidelberg, 2003, pp. 31–98, DOI:10.1007/b13181.
- A. M. d. P. Nicholas and D. R. Arnold, Can. J. Chem., 1982, 60, 2165–2179 CrossRef CAS.
- R. Breslow and J. L. Grant, J. Am. Chem. Soc., 1977, 99, 7745–7746 CrossRef CAS.
- T. Chivers and I. Drummond, Inorg. Chem., 1972, 11, 2525–2527 CrossRef CAS.
- T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848–851 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- B. M. Bode and M. S. Gordon, J. Mol. Graphics Modell., 1998, 16, 133–138 CrossRef CAS PubMed.
- A. L. Holovynsky, A. L. Malenko and I. V. Sergienko, Visn. Nac. Akad. Nauk Ukr., 2013, 50–59 CrossRef.
- J. G. Carey, J. F. Cairns and J. E. Coxhewer, J. Chem. Soc. D, 1969, 1280–1281 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, UPS and FT-IR spectra of K-PHI, N2 absorption isotherm, DFT calculations, toluene oxidative thiolation optimization results, material characterization after photocatalytic tests, and 1H and 13C NMR spectra of disulfanes. See DOI: 10.1039/c8sc00745d |
| ‡ Due to the M+ effect of the methoxy group, the oxidation potential of p-methylanisole is expected to be lower than that of toluene, 2.26 ± 0.02 V vs. SCE.29 Alcohols, commonly used holes scavengers, cannot be applied in this case as they were reported to react with MV2+.48 |
| This journal is © The Royal Society of Chemistry 2018 |