
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad†
Xueyan
Wu
a,
Jixi
Guo
 *a,
Yali
Cao
a,
Jianzhang
Zhao
ab,
Wei
Jia
a,
Yi
Chen
a and
Dianzeng
Jia
*a
*a,
Yali
Cao
a,
Jianzhang
Zhao
ab,
Wei
Jia
a,
Yi
Chen
a and
Dianzeng
Jia
*a
aKey Laboratory of Energy Materials Chemistry, Ministry of Education, Key Laboratory of Advanced Functional Materials, Autonomous Region, Institute of Applied Chemistry, Xinjiang University, Urumqi, 830046, P. R. China. E-mail: jxguo1012@163.com; jdz@xju.edu.cn
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, Dalian 116024, P. R. China
First published on 18th May 2018
Abstract
A novel single organic molecule–carborane conjugate, CAN, was synthesized in a high yield via a modified nickel-catalyzed cross-coupling reaction incorporating an anthracene unit and an o-carborane moiety. CAN exhibits multiple functions of tricolored mechanochromism and mechanically triggered thermochromism. The fluorescence could be switched from blue to bright yellow then to pink by grinding. The robust and reversible thermochromic process was triggered by the mechanical force. The locally excited (LE) state emission, intermolecular excimer formation and twisted intermolecular charge transfer (TICT) are the primary origins of this tricolor switching property. High temperature sensitivity of the heavily ground CAN powders contribute to the mechanical force induced TICT emission enhancement and color switching.
Introduction
Piezochromic luminescent materials are a series of stimuli-responsive materials that exhibit significant emission color changes with external stimuli such as shearing, grinding, tension or hydrostatic pressure. On the other hand, the emission colors of piezochromic luminescent materials can be returned to their original emission colors by solvent fuming, heating or other external stimuli.1–3 Piezochromic luminescent materials were first discovered by Francis Bacon in 1605. Since then, a series of luminescent materials with such features have been reported on owing to their promising potential as sensors, photoluminescence (PL) switches and optical recording devices.4–13 Interestingly, most emission color changing processes of piezochromic luminescent materials are accompanied by a crystal-to-amorphous phase transition, aggregation state change, excimer formation and pressure-induced chemical structure changes.14–18 Although a few piezochromic luminescent materials display tricolor switching and pressure triggered temperature dependent ICT emission, the mechanism is attributed to the mechanical force breaking the donor–acceptor (D–A) interactions which induces the ICT process.16,19 Design strategies for piezochromic luminescent materials are rarely reported and an understanding of the essential piezochromic mechanism is still missing. To date few examples of external-force-induced multicolored change and pressure triggered temperature dependent organic molecules have been described in the literature.6,20Herein, a structurally well-defined donor–acceptor (D–A) molecule, an anthracene-o-carborane (CAN) dyad connecting a planar anthracene unit as an electron donor and a three-dimensional o-carborane unit as an electron acceptor, has been readily synthesized with high yield via a modified nickel-catalyzed cross-coupling reaction21,22 and the detailed synthetic process is depicted in Scheme 1. The structure and purity of CAN were confirmed using 1H NMR and 13C NMR spectroscopy, as well as HRMS analysis.
 | ||
| Scheme 1 Synthetic routes to CAN. | ||
The design method described herein may afford the product CAN novel properties: (1) the electron-donating capability of the anthracene moiety can enhance the ICT process, which is from the anthracene to the strongly electron-deficient o-carborane.23 (2) the dihedral angle (φ) between the C–C bond in the o-carborane unit and the plane of the anthracene moiety should play a significant role in the emission properties. When the φ = 0°, a LE emission from the anthracene unit is observed.23,29 In contrast, once the φ is changed and the twisted conformation formed, TICT emission can be detected.23,29,31 (3) The good planarity of the anthracene moieties can facilitate the CAN molecule arranging into ordered structures. Unlike traditional piezochromic luminescent materials with two color luminescence switching, the molecule CAN shows an interesting tricolor turn-on luminescence in response to external stimuli. This multicolor-tuned piezochromic material with high contrast luminescence and pressure triggered temperature dependent luminescence change can be applied as a pressure recording medium and a temperature sensor. In addition, CAN represents an intriguing example for exploring the structure–property relationship of solid-state piezochromic luminescent materials under external mechanical stimuli. To gain a deep insight into the tricolor tuning and pressure triggered thermochromic properties of the CAN molecule, we have systematically investigated the optical properties, especially the mechano- and thermochromic luminescence performance in the solid state.
Results and discussion
First, we investigated the photophysical properties of CAN in solution. UV/vis absorption measurements were performed to examine the optical properties of CAN. As depicted in Fig. S4,† absorption bands were observed at 332, 346, 365 and 385 nm in THF, which was attributed to the π–π* transition in the anthracene moiety. Absorption spectra were also measured in other solvents, such as n-hexane, toluene, DCM, acetonitrile, and DMF (Fig. S5†). Changes in the position of the absorption bands in different solvents were hardly observed, indicating that there is no significant charge transfer at the ground state. PL spectra were measured in various solvents, such as n-hexane, toluene, DCM, THF, acetonitrile and DMF (Fig. S6†) and the data is summarized in Table S1.† The vibrational emission peaks are at around 400 nm and their intensity is very weak, which corresponds to LE state emission. We proved our deduction using the low temperature luminescence spectra; from the PL spectrum recorded at 77 K, only the LE emission was obtained (Fig. S7†). This phenomenon indicates that o-carborane acts as a strong electron-withdrawing unit in the excited state of conjugated systems originating from the electron deficient nature of the σ*–π* conjugation on the Ccage–Ccage bond of the o-carborane unit. Therefore, through combination with anthracene rings, the ICT state could be readily developed in the excited state. The vibration of the Ccage–Ccage bond in o-carborane should be responsible for quenching the LE state emission and the ICT state emission originating from electronic interactions between the o-carborane and anthracene groups.24On the other hand, the broad emission peak around 520 nm only observed in n-hexane was assigned as the AIE. The AIE of compound CAN was not observed in other solutions as shown in Fig. S6,† especially at the dilute condition of ca. 10−5 M. This is probably because of the poor solubility of compound CAN in n-hexane, while it is very soluble in other solvents. As shown in Fig. S8,† upon adding a polar solvent such as THF (up to 80%) into n-hexane, the AIE shifted from 520 nm to 580 nm, accompanied by a decrease in its intensity. The AIE intensity decreases rapidly with increasing THF fraction, indicating that the aggregates of CAN are easily dissolved in polar THF. In the mixture solution of THF and water, with an increase of fw, the intensities of the LE state emission bands keep the same spectra profile and they become negligible with increasing solvent polarity, and a pale pink emission band emerged (Fig. S9†). Manifestly, the emission was induced by aggregate formation, that is to say, the CAN is AIE-active. These results were different from those of previously reported aggregation- and crystallization-induced emission enhancement (AIEE and CIEE) for carborane–chromophore conjugates.23
CAN displays multicolored piezochromic luminescence, as shown in Fig. 1b. To observe the three emission colors more clearly, we distributed the CAN powders smoothly on an agate mortar. The pristine CAN powder gives bright blue emission, but after being gently grinded, the CAN powder shows extremely bright yellow emission. To our surprise, when we increased the grinding force, the CAN powder changes its emission color from extremely bright yellow to pink. The difference between the emission colors of the pristine, slightly ground and heavily ground powders is distinct, which is visible to the naked eye (for the emission color change process see ESI Fig. S10†).
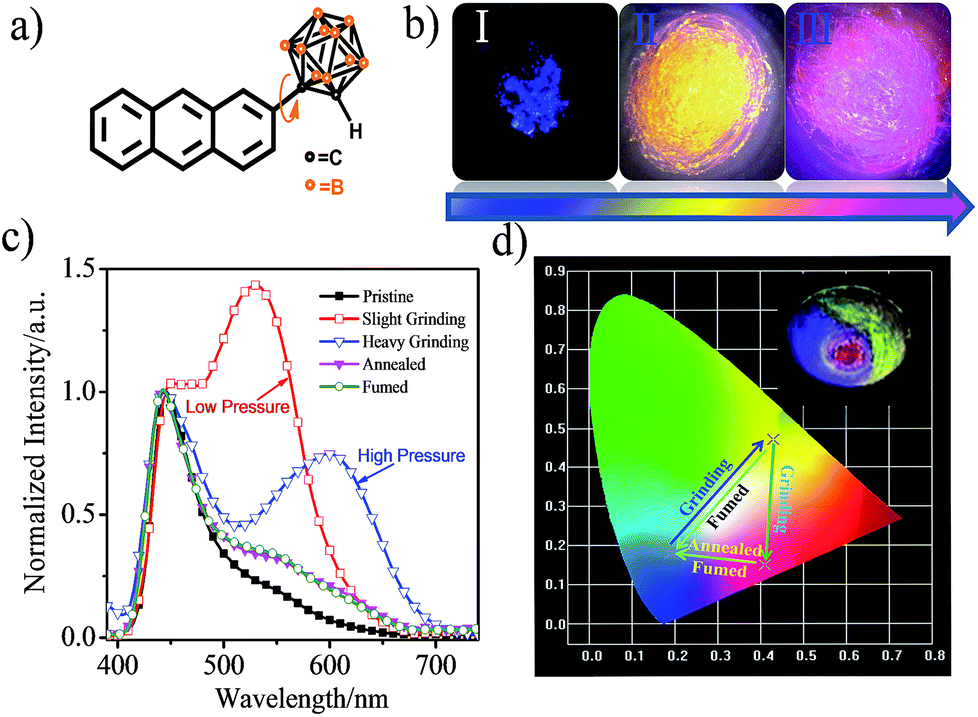 | ||
| Fig. 1 (a) Structural formula of CAN. (b) Fluorescence images under 365 nm irradiation. (I) the original deep-blue powder; (II) bright-yellow powder after slight grinding; and (III) pink powder after continuous heavy grinding. (c) Corresponding fluorescence spectra of CAN with different treatments. (d) CIE diagram of the emission of CAN with different treatments. Inset: different emission colors of CAN on an agate mortar with different grinding force: pristine blue, bright yellow and pink emission upon irradiation with 365 nm UV light. | ||
Fig. 1c shows the fluorescence emission spectra of the CAN powder before and after the application of the changeable external force and the data are summarized in Table S1.† The PL spectra of CAN powder under external pressure underwent a larger red shift of approximately 185 nm, i.e. from 440 nm in the pristine state to 625 nm after heavy grinding. The emission peak centered at 440 nm [Φ = 8.8%] for the initial powder was red-shifted to 530 nm [Φ = 14.4%] after gentle grinding. After further disturbance, a new peak at 625 nm was observed for the pink powder [Φ = 4.3%]. According to previous studies,19,25–32 the emission at 440, 530 and 625 nm mainly originates from the LE state, anthracene excimers and TICT state emission, respectively. After slight grinding, the sandwich packing excimer shows bright yellow emission with a long lifetime (τ = 40.35 ns) and high quantum yield (Φ = 14.4%).7,11 It was indicated that the steric hindrance of o-carborane can suppress aggregation-caused quenching without inhibiting excimer formation.28 Then after heavy grinding the longer emission peak is attributable to TICT emission which shows a short lifetime (τ = 18.35 ns) and low quantum yield (Φ = 4.3%). The emission of ground powders can recover to the pristine state after heating at 150 °C for 5 min or fuming with the organic solvent n-hexane. The change in the emission colors of CAN powder under different external mechanical stimuli were quantified with the CIE coordinates as shown in Fig. 1d. The CIE coordinates change from (0.19, 0.18) to (0.43, 0.47) then to (0.41, 0.15) after applying mechanical force.
Powder X-ray diffraction (PXRD) measurements were performed to understand the microstructure change of the pristine CAN powder treated with different external mechanical stimuli (Fig. 2a). The pristine CAN powder showed intense and sharp reflections in the PXRD patterns, indicating a well-ordered microcrystalline structure. In contrast, some diffraction peaks disappeared and decreased, or changed breadth after slight and heavy grinding, suggesting that the ordered structure of the pristine sample was destroyed. This result indicated that the transformation of molecular packing modes from a crystalline to an amorphous state was triggered by external stimuli, which results in changes in emission color from blue to bright yellow, and then to pink. The broad and red-shifted emission was ascribed to the combined effect of various emission component parts, probably because the anisotropic external grinding force was not strong enough for total phase transition. The applied grinding force may mainly act on the surface of the powder crystal, while the inner parts remain unchanged. Therefore, the microcrystalline and amorphous phase coexist in the ground powder.4 The intensity or the number of diffraction peaks of the ground sample was recovered by heating the sample at 150 °C for 5 min or fuming with the organic solvent n-hexane, indicating the recovery of the microcrystalline structure.15,16,33
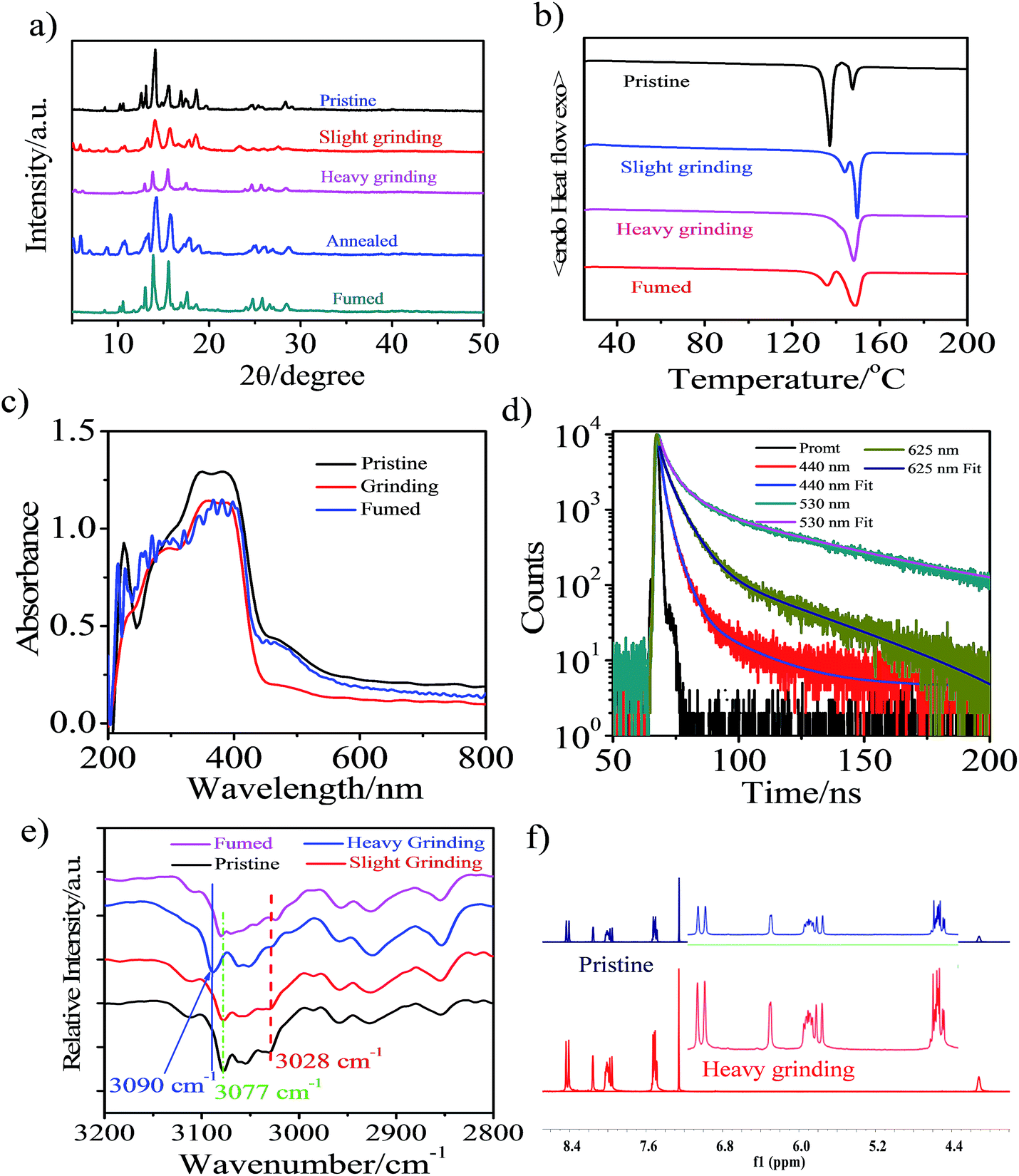 | ||
| Fig. 2 (a) PXRD patterns of CAN powder: pristine, ground, annealed (150 °C for 5 min) and fumed samples (fumed with n-hexane). (b) DSC curves of pristine, ground and fumed powders of CAN. (c) UV-vis absorption spectra of the pristine, ground and fumed powders of CAN performed in diffuse reflectance mode. (d) Lifetimes of the CAN powder: pristine (440 nm), slightly ground (530 nm) and heavily ground (625 nm). (e) Diffuse Reflectance Fourier-Transform Infrared Spectroscopy (DRIFTS) spectra of CAN powder: pristine, ground and fumed samples (fumed with n-hexane). (f) 1H NMR spectra of pristine (up) and ground (down) CAN powders in CDCl3. | ||
Additionally, differential scanning calorimetry (DSC) of pristine sample, samples treated with different external mechanical stimuli and fumed CAN powders was conducted to gain more insight into the thermochromic behavior of CAN powder (Fig. 2b). The DSC curves exhibit an endothermic peak at about 148.5 °C for all measured samples, which corresponds to their melting point. Meanwhile, another endothermic peak at temperatures ranging from 135 to 145 °C was also observed in the DSC curves of the CAN powder under different treatment conditions. In addition, the first endothermic peak gradually reduces from the pristine to the slightly ground to the heavily ground powder, indicating that the pristine powder is present in a metastable microcrystalline phase and converts to a stable amorphous phase via an exothermal recrystallization process. These results are in agreement with the PXRD results as mentioned above. The first endothermic peak reappeared after the heavily ground powder was fumed with the organic solvent n-hexane. All the above-mentioned observations illustrate that uneven grinding induced the transition of molecular assemblies from a metastable crystalline phase to a stable amorphous state, the slight grinding force changed the packing mode to form anthracene excimers emitting bright yellow light, and heavy grinding leads to pink TICT state emission. Importantly, the heavily ground powders are stable, and the pink emission is persistent over five months in an ambient atmosphere.4,12,19
For the CAN powder treated with different external mechanical stimuli the absorption band between 440 and 600 nm decreased and a blue-shift of 8 nm in the absorption spectra was observed, implying H-type aggregation or a decrease of the conjugation system and co-planarity, possibly induced during the grinding process.34 This transformation causes the compact π–π packing of anthracene, which results in the bright yellow emission.35 After fuming with the organic solvent n-hexane, the absorption spectra switched back to the pristine state, although some of the initial aggregation state remained after fuming.
The lifetime of the emission centered at 440 nm [τ1 = 3.15 ns (A1 = 0.57), τ2 = 1.40 ns (A2 = 0.43), χ2 = 0.98] for the initial powder was red-shifted to 530 nm [τ1 = 5.48 ns (A1 = 0.215), τ2 = 49.9 ns (A2 = 78.5), χ2 = 1.05] after slight grinding. After further heavy grinding, a new peak at 625 nm was detected for the pink powder [τ1 = 6.09 ns (A1 = 0.636), τ2 = 38.8 ns (A2 = 0.373), χ2 = 1.06]. The lifetime decay curves fitted well with a double-exponential function, probably due to heterogeneous microenvironments in the solid state. Structural analysis suggests that the newly emerged broad and structureless curves of the yellow emission band after slight grinding are attributed to the anthracene excimer.4,36 This is consistent with the longer fluorescence lifetime of the yellow emission after slight grinding (40.35 ns) compared to those of the pink emission after heavy grinding (18.34 ns) and the blue emission in pristine state (2.40 ns).7 The pristine powder after slight grinding shows a bright yellow emission and also has long-lived fluorescence, which is a characteristic of excimer emission.4 After heavy grinding the more condensed packing results in less conformational freedom and a longer fluorescence lifetime will occur,37 however, the fluorescence lifetime was decreased, revealing that the excited state in the ground solid was altered. The pink emission source in CAN is mainly from TICT emission and this speculation was confirmed by the temperature-dependent luminescence spectra.23
Non-covalent interactions, such as electrostatic, van der Waals and hydrophobic effects and the π–π stacking of phenyl groups may play key roles in the aggregated structure of the pristine powder. We performed diffuse reflectance Fourier-transform infrared spectroscopy (DRIFTS) to further confirm the structure changes after different external mechanical stimuli were applied at the molecular level (Fig. S11†). For the pristine powder, the broad characteristic and fine stretching absorption bands of the B–H and C–H in o-carborane were observed at 2586 and 3057 cm−1,38,39 respectively. After external mechanical stimuli, heating and fuming treating, the relative intensity and wavenumber of the absorption band for free B–H in o-carborane have not changed. However, the transformation of the absorption of C–H bonds in o-carborane cannot be negligible. As depicted in Fig. 2e, the absorption band of C–H in o-carborane grows weaker and the wavenumber shows a red-shift of about 13 cm−1 with increasing external grinding force. A possible explanation is that the external force induces the molecular planarity and the π-conjugation is much decreased.23,24,28 The face-to-face sandwich stacking structure is likely to convert to a non-ordered structure. Meanwhile the dihedral angle between the anthryl and Ccage–Ccage bond in the o-carborane also changes.7,40 As a consequence, the bright yellow-emissive excimer no longer exists, resulting in a broad fluorescence band with an emission color converting from blue to bright yellow then to pink.
To further confirm that the piezochromic property is induced by the π–π interaction and transformation of the molecular conformation rather than the disruption of the molecular structure, we performed 1H NMR measurement (Fig. 2f and S12†). The 1H NMR spectra of the heavily ground powder and the annealed samples matched well with that of the pristine powder. This indicates that no chemical structural change occurs during the external stimuli and annealing process, confirming the good thermal stability of CAN. The results are consistent with the DRIFTS measurements.
In order to demonstrate that the altering of the excited state in the ground solid indeed takes place, we measured the temperature dependent luminescence spectra of CAN in the solid state (Fig. S13†). The CT emission should arise from the TICT state which can revert to the stable LE state through a conformational change once the temperature is decreased. In the pristine state (Fig. S13a†), with a temperature decrease from 333 to 77 K, the luminescence spectra of CAN at 440 nm increased gradually and the emission band at about 625 nm was not detected. We can draw the conclusion that only LE state emission is observed in the pristine state of CAN. In contrast, for the heavily ground CAN powder (Fig. S13b†), with the decrease in temperature, the intensity of the emission band at about 625 nm increased at first and then decreased. The luminescence band of CAN at 440 nm and a shoulder emission band at about 530 nm intensified gradually and we speculate that the LE state and excimer formed due to the partially overlapped packing that occurs in the pristine state of CAN. From the above results, heavy grinding destroys the assembled structure and the extreme force induces the change of dihedral angle between the anthryl and Ccage–Ccage bond in the o-carborane, which is a favorable conversion for the formation of the TICT state.27–30,41–44 The reversible piezochromic switching of CAN powder between the blue and pink fluorescence colors was measured (Fig. S14†). After fuming with an organic solvent, such as n-hexane, the original blue emission was regained. After five grinding and fuming cycles of the CAN powder, an excellent reversibility of the switching processes was observed, although some fatigue was observed in the first cycle.
To elucidate the origin of the electronic transitions that are responsible for the distinct photophysical properties of CAN, DFT/TD-DFT computational studies at the B3LYP/6-31g(d, p) level of theory were performed using Gaussian 09,45 and the following findings were obtained. From the electronic density distribution of the molecular orbitals of CAN (Fig. S15†), the largest electron coefficients in the HOMO are located along the anthracene moiety, whereas the LUMO is distributed across the full molecule, suggesting that the molecular orbitals are separate between the anthracene and o-carborane units. According to TD-DFT calculations in the gas phase (Table S2, Fig. S16 and S17†), the lowest energy transition (HOMO → LUMO) of CAN is symmetry-forbidden, while the higher energy transition (HOMO−1 → LUMO) is symmetry-allowed. The forbidden nature of the S0 → S1 transition is consistent with the weak fluorescent character of CAN. Consequently, the transition is predicted to occur at a high energy level, regarded as the LE transition. In addition, the emission spectrum of the CAN monomer in solution is analogous to that of the anthracene monomer in solution (Fig. S18†), further demonstrating that the emission of CAN should originate from the LE state.28
As mentioned above, the emission properties of CAN show a dependence on the change of torsion angle under external mechanical stimuli. To explain the relationship between the dihedral angle, which is defined as the directional difference between the Ccage–Ccage bond in the o-carborane moiety and the anthracene group, and optical properties, quantum calculations were performed using density functional theory (DFT) and time-dependent DFT (TD-DFT) calculated at the B3LYP/6-31G(d, p) level.8,23,28,29 The results are shown in Fig. 3. They reveal the HOMO → LUMO transition switching from a symmetry forbidden to a symmetry allowed transition with the changing torsion angle, while the HOMO−1 → LUMO and HOMO → LUMO+1 transitions in higher energy levels are always allowed. Furthermore, the changing of the torsion angle promotes the delocalization of the electronic cloud density in the anthryl part to the π system of the o-carborane acceptor, leading to an overlap of the frontier orbitals and enhanced electron interaction between the anthracene and o-carborane units. Owing to the increased overlap of the frontier orbitals, the electron density of the HOMO is mainly located in the anthracene moiety, and that of the LUMO is distributed across the full molecular skeleton, indicating the formation of the TICT state.
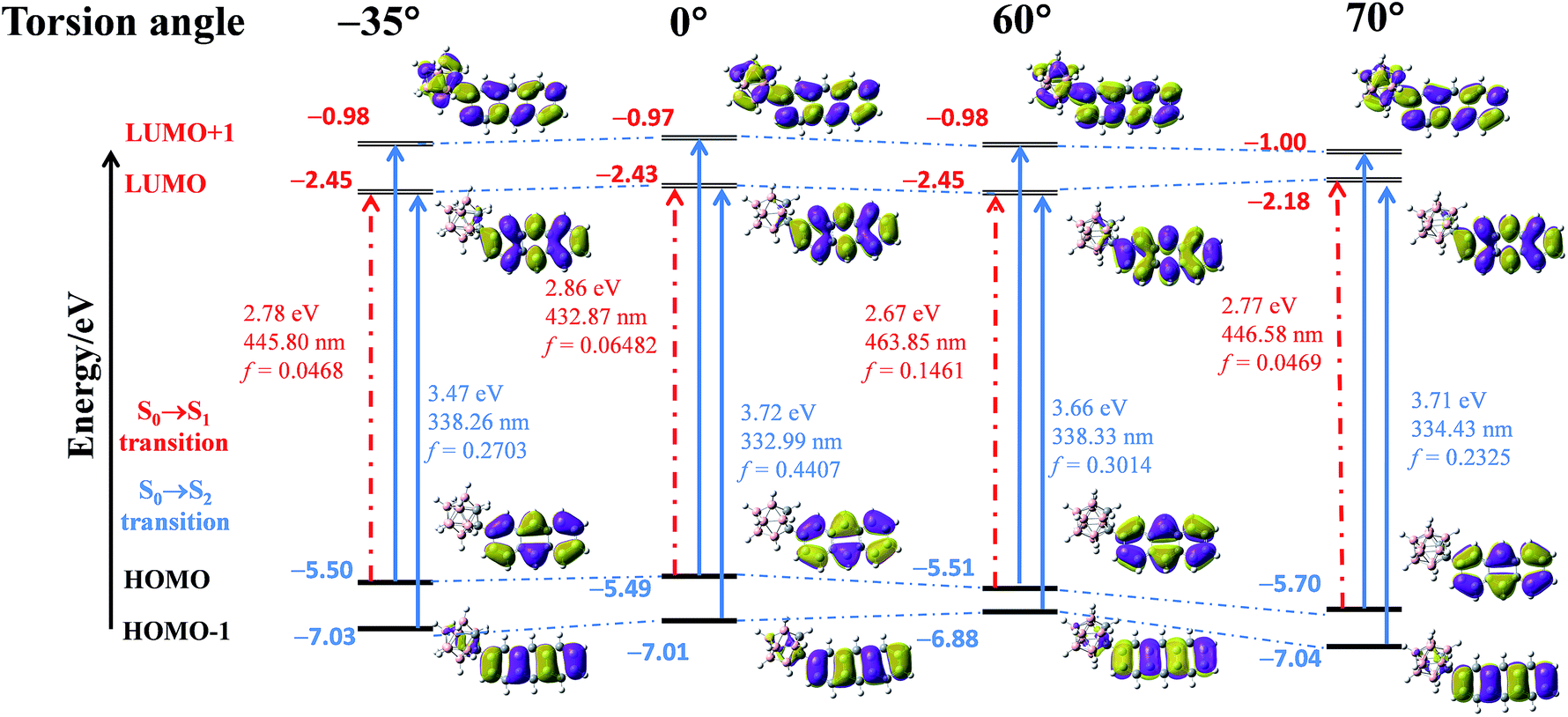 | ||
| Fig. 3 Energy diagrams and the frontier orbital contribution of CAN in fixed conformation based on the gradually decreasing torsion angle between the Ccage–Ccage bond in the o-carborane moiety and the aryl group, and their lowest- and second-lowest-energy transitions estimated by TD-DFT calculations at the B3LYP/6-31G(d, p) level. | ||
On the basis of the above results and analysis, the mechanically induced tricolor switching of CAN powder can be summarized as follows (Fig. 4). Pristine CAN emits blue luminescence (440 nm) because of the LE state emission, although partially overlapped packing of anthracene moieties exists.36 Upon slight grinding, the assembled partially overlapped packing structure is destroyed, which leads to a sandwich stacking of the anthracene units and results in a change in fluorescence color from blue to bright yellow. Then extreme force induces a change in dihedral angle between the anthryl and Ccage–Ccage bond in the o-carborane, which is a favorable conversion for forming the TICT state and the powder emits pink color emission.
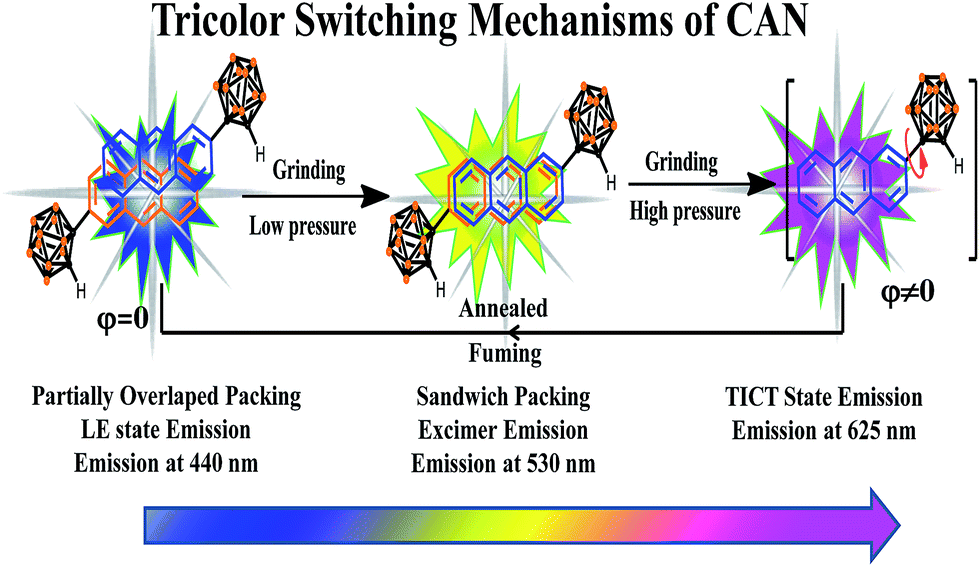 | ||
| Fig. 4 The tricolor switching mechanisms of CAN. | ||
Interestingly, the heavily ground CAN displays a reversible distinct fluorescence color change with variation of temperature. In the pristine state, the emission band was located at ca. 440 nm and the emission intensity becomes stronger with decreasing temperature (Fig. 5a), which is attributed to LE emission. In other words, in the pristine state, LE emission results from the planar conformation, which is the most thermodynamically stable structure.28–32 After heavy grinding, TICT emission was observed from CAN with a maximum at 625 nm at room temperature, the LE emission and another broad emission band, which was attributed to the anthracene excimer, were observed with decreasing the temperature from 333 to 77 K (Fig. 5b). As shown in Fig. 5c, a clear color change was observed upon cooling the sample. The emission color was dramatically changed from pink to blue by cooling. From 333 to 77 K, the TICT emission peak gradually weakens and the CIE coordinates of the emission change almost linearly from yellow to sky blue. The obvious emission color change induced by a temperature change is clearly visible to the unaided eye. The thermochromic response of heavily ground CAN is fully reversible after 5 cycles (Fig. 5d). This behavior should be useful for the development of pressure triggered temperature-sensing molecular probes.
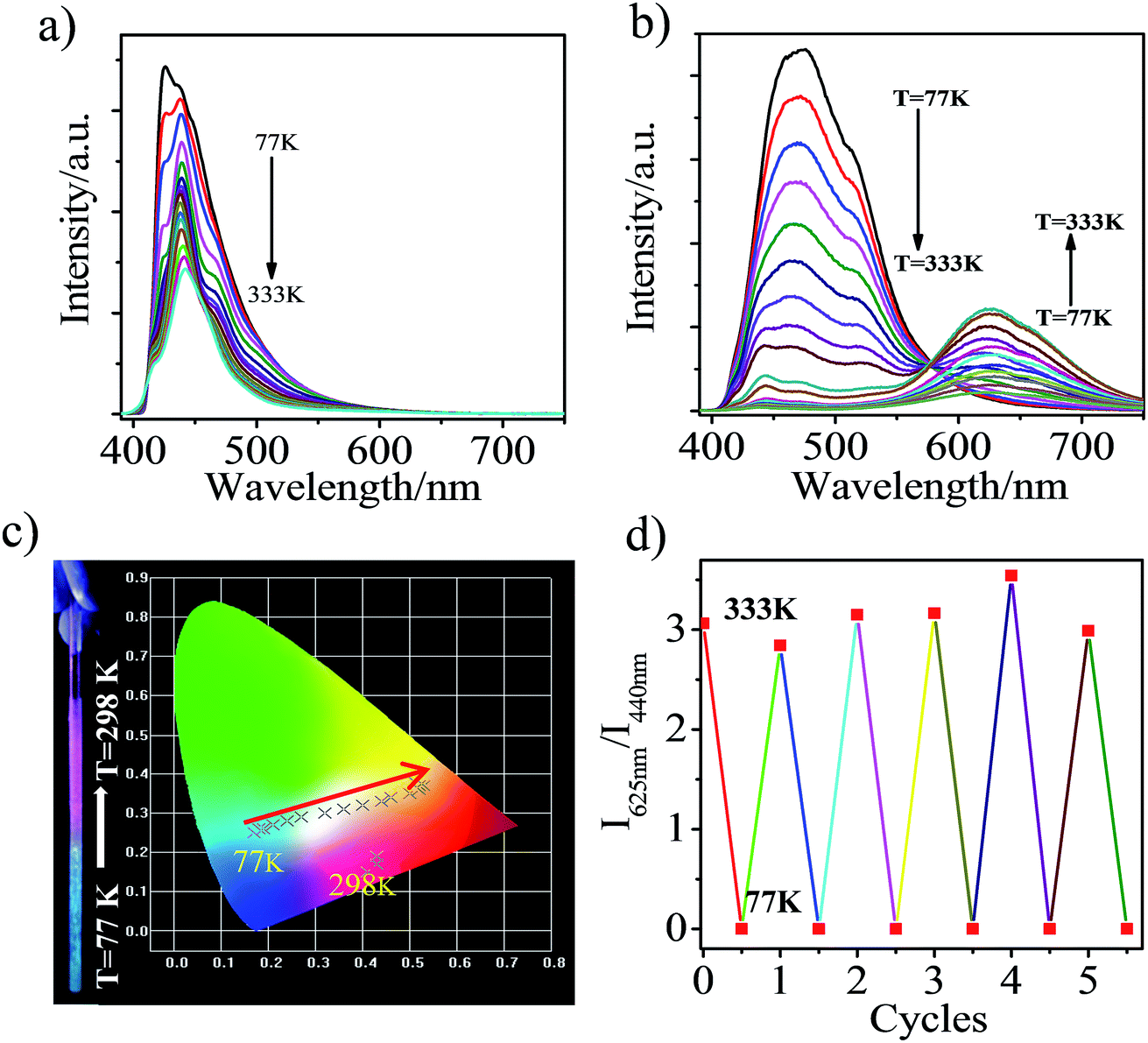 | ||
| Fig. 5 PL spectra of CAN: (a) pristine and (b) after grinding in the solid state during heating from 77 to 333 K; (c) a photograph and diagram showing the color and CIE coordinates (CIE 1931) change of ground CAN with T; (d) a diagram showing the reversibility and reproducibility of the thermochromism of ground CAN over 5 cycles. | ||
Conclusions
In conclusion, we have prepared and discovered a novel organic piezochromic material, CAN, with an anthryl moiety covalently connected with o-carborane, which exhibited reversible tricolor fluorescence switching and pressure triggered temperature dependent TICT emission. It is worth noting that the reversible tricolor piezochromic luminescent mechanism of CAN was attributed to the transition from LE state emission, to force-induced excimer emission and then molecular conformation induced TICT state emission. The external pressure stimuli triggered thermochromic response is caused by TICT in the solid state. The tricolor-switchable and pressure triggered thermochromic attributes of CAN may have potential for application in fluorescent sensors, security inks, and pressure and temperature-sensing logic gates.Experimental
General
The reagents and starting materials were purchased from either Aldrich or Acros Chemical Co. and were used without further purification. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. NMR spectra were recorded on a Bruker ADVANCE III 400 MHz spectrometer (1H NMR: 400 MHz, 13C NMR: 100 MHz). All chemical shifts are reported in δ units with references to the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts. UPLC/MS was recorded on an Ultimate 3000/Q-Exactive. The time of flight mass spectra were recorded using an Orbitrap mass system. DSC experiments were performed on a NETZSCH DSC 200 instrument at a scanning rate of 10 K min−1. The diffuse reflectance Fourier-transform infrared spectroscopy (DRIFTS) was carried out using a Bruker VERTEX-70 Fourier transform infrared (FT-IR) spectrometer in a range of wavenumbers from 400 to 4000 cm−1. Powder X-ray diffraction (XRD) measurement was conducted on a Bruker D8 Advance Diffraction diffractometer in the 2θ range from 5 to 80°, with Cu Kα radiation (θ = 0.15405 nm) at 40 kV and 40 mA. The temperature dependent luminescence spectra were measured with a Janis VPF-100 liquid nitrogen low temperature thermostat (Janis, USA). UV-vis absorption spectra were recorded on a HITACHI U-3900H and the fluorescence spectra were recorded on a HITACHI F-4500 spectrofluorometer and a FluoroLog-3 (Horiba-Jobin-Yvon, Edison, NJ, USA) fluorescence spectrophotometer. Melting points were measured using a Nikon Polarizing Microscope ECLIPSE 50i POL equipped with an INTEC HCS302 heating stage. Luminescence lifetime measurement was carried out using a FluoroLog-3 spectrofluorometer (Horiba-Jobin-Yvon, Edison, NJ, USA). DFT and TD-DFT calculations were performed using the Gaussian 09 suite of programs. Geometry optimizations and the frontier molecular orbitals of the compound were obtained at the B3LYP/6-31G(d, p) level of theory.Synthesis of CAN
2-(1-o-Carboranyl) anthracene, CAN: to a THF solution (5 mL) of o-carborane (288 mg, 2.0 mmol) was slowly added i-PrMgCl (1.2 M in THF, 1.2 mL, 2.4 mmol) at 0 °C under an N2 atmosphere for 3 h, and the mixture was stirred at room temperature for 10 h. After the replacement of THF with toluene (10 mL) and addition of 2-bromoanthracene (617 mg, 2.4 mmol, 1.2 eq.) and NiCl2 (26 mg, 0.2 mmol), the reaction mixture was heated to 105 °C with stirring for 12 h in a closed flask. Then, the reaction was quenched with water (10 mL) and the organic layer was extracted with CH2Cl2 (3 × 30 mL) and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified using silica gel column chromatography using DCM/petroleum ether (1/6, v/v) as the eluent to obtain colorless compound, CAN (541 mg, 84%). M.p.: 147.3–148.4 °C. 1H NMR (400 MHz, CD2Cl2): δ (ppm) 8.47 (d, J = 8.0 Hz, 2H), 8.19 (d, J = 4.0 Hz, 1H), 8.06–8.00 (m, 3H), 7.55–7.51 (m, 3H), 4.25 (s, 1H, carborane C–H), 3.18–1.60 (10H, br, B–H). 13C NMR (100 MHz, CD2Cl2): δ (ppm) 132.31, 133.02, 130.40, 129.82, 129.49, 128.77, 127.83, 127.77, 126.98, 126.05, 125.83, 125.75, 123.28, 76.73, 60.42. HRMS: m/z calcd for [C16H20B10–H]−: m/z = 321.2417. Found: m/z = 321.2424.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21571152 and U1703251), the Scientific Research Program of the Higher Education Institution of Xinjiang (XJEDU2017A001), and the Doctoral Innovation Program of Xinjiang University (XJUBSCX-2015009).Notes and references
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef PubMed.
- N. Eddingsaas and K. Suslick, J. Am. Chem. Soc., 2007, 129, 6718–6719 CrossRef PubMed.
- P. Chen, H. Zhang, L. Niu, Y. Zhang, Y. Chen, H. Fu and Q. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef PubMed.
- (a) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef PubMed; (b) M. Teng, X. Jia, X. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398–6401 CrossRef PubMed; (c) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., 2013, 125, 12494–12498 CrossRef; (d) Z. Ma, Z. Wang, Z. Xu, X. Jia and Y. Wei, J. Mater. Chem. C, 2015, 3, 3399–3405 RSC; (e) Z. Ma, F. Yang, Z. Wang and X. Jia, Tetrahedron Lett., 2015, 56, 393–396 CrossRef; (f) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef PubMed.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef.
- Z. Zhang, Y. Gao, H. Liu, Q. Bai, J. Li, L. Liu, C. Wu, B. Yang, K. Wang, B. Zou, Y. Wang and P. Lu, Dyes Pigments, 2017, 145, 294–300 CrossRef.
- X. Luo, J. Li, C. Li, L. Heng, Y. Dong, Z. Liu, Z. Bo and B. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef PubMed.
- W. Miao, S. Wang and M. Liu, Adv. Funct. Mater., 2017, 1701368 CrossRef.
- B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC.
- L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160–9164 CrossRef PubMed.
- X. Wang, Q. Liu, H. Yan, Z. Liu, M. Yao, Q. Zhang, S. Gong and W. He, Chem. Commun., 2015, 51, 7497–7500 RSC.
- K. Ohno, S. Yamaguchi, A. Nagasawa and T. Fujihara, Dalton Trans., 2016, 45, 5492–5503 RSC.
- H. Yu, W. Ren, H. Lu, Y. Liang and Q. Wang, Chem. Commun., 2016, 52, 7387–7389 RSC.
- Q. Qi, J. Zhang, B. Xu, B. Li, X. Zhang and W. Tian, J. Phys. Chem. C, 2013, 117, 24997–25003 Search PubMed.
- J. Yang, Q. Guo, X. Wen, X. Gao, Q. Peng, Q. Li, D. Ma and Z. Li, J. Mater. Chem. C, 2016, 4, 8506–8513 RSC.
- D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Zhang, H. Yan and Q. Zhao, Chem. Commun., 2016, 52, 12494–12497 RSC.
- Y. Matsunaga and J. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985–7989 CrossRef PubMed.
- C. Tang and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 7662–7665 CrossRef PubMed.
- Y. Chen, J. Guo, X. Wu, D. Jia and F. Tong, Dyes Pigments, 2018, 148, 180–188 CrossRef.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef PubMed.
- (a) K. Tanaka, K. Nishino, S. Ito, H. Yamane, K. Suenaga, K. Hashimoto and Y. Chujo, Faraday Discuss., 2017, 196, 31–42 RSC; (b) K. Kokado and Y. Chujo, Dalton Trans., 2011, 40, 1919–1923 RSC; (c) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef PubMed.
- M. Teng, X. Jia, X. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 124, 6504–6507 CrossRef.
- K. Wee, Y. Cho, J. Song and S. Kang, Angew. Chem., Int. Ed., 2013, 125, 9864–9867 CrossRef.
- M. Teng, X. Jia, S. Yang, X. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255–1261 CrossRef PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Asian J. Org. Chem., 2017, 6, 1818–1822 CrossRef.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef PubMed.
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef PubMed.
- E. Kwon, J. Kim, K. Lee and T. Kim, Inorg. Chem., 2017, 56, 943–949 CrossRef PubMed.
- F. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef PubMed.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef PubMed.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Boston, MA, 2006 Search PubMed.
- P. Zhang, W. Dou, Z. Ju, X. Tang, W. Liu, C. Chen, B. Wang and W. Liu, Adv. Mater., 2013, 25, 6112–6116 CrossRef PubMed.
- G. Barberà, A. Vaca, F. Teixidor, R. Sillanpää, R. Kivekäs and C. Viñas, Inorg. Chem., 2008, 47, 7309–7316 CrossRef PubMed.
- J. Yue, Y. Li, H. Li, Y. Zhao, C. Zhao and X. Wang, RSC Adv., 2015, 5, 98010–98019 RSC.
- F. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef.
- S. Kim, A. Lee, G. Jin, Y. Cho, H. Son, W. Han and S. Kang, J. Org. Chem., 2015, 80, 4573–4580 CrossRef PubMed.
- K. Wee, Y. Cho, J. Song and S. Kang, Angew. Chem., Int. Ed., 2013, 52, 9682–9685 CrossRef PubMed.
- S. Kim, Y. Cho, G. Jin, W. Han, H. Son, D. Cho and S. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679–15682 RSC.
- M. Son, Y. Cho, S. Kim, H. Son, D. Cho and S. Kang, Phys. Chem. Chem. Phys., 2017, 19, 24485–24492 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. H. Petersson, M. Nakatsuji, X. Caricato, H. P. F. Li, A. Hratchian, J. Izmaylov, G. Bloino, J. L. Zheng, M. Sonnenberg, M. Hada, K. Ehara, R. Toyota, J. Fukuda, M. Hasegawa, T. Ishida, Y. Nakajima, O. Honda, H. Kitao, T. Nakai, J. A. Vreven, J. E. Montgomery Jr, F. Peralta, M. Ogliaro, J. J. Bearpark, E. Heyd, K. N. Brothers, V. N. Kudin, T. Staroverov, R. Keith, J. Kobayashi, K. Normand, A. Raghavachari, J. C. Rendell, S. S. Burant, J. Iyengar, M. Tomasi, N. Cossi, J. M. Rega, M. Millam, J. E. Klene, J. B. Knox, V. Cross, C. Bakken, J. Adamo, R. Jaramillo, R. E. Gomperts, O. Stratmann, A. J. Yazyev, R. Austin, C. Cammi, J. W. Pomelli, R. L. Ochterski, K. Martin, V. G. Morokuma, G. A. Zakrzewski, P. Voth, J. J. Salvador, S. Dannenberg, A. D. Dapprich, O. Daniels, J. B. Farkas, J. V. Foresman, J. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09W (Revision A.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption and fluorescence spectra, DFT calculations. See DOI: 10.1039/c8sc00833g |
| This journal is © The Royal Society of Chemistry 2018 |