
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible dispersion and release of carbon nanotubes via cooperative clamping interactions with hydrogen-bonded nanorings†
Raquel
Chamorro
a,
Leire
de Juan-Fernández
b,
Belén
Nieto-Ortega
b,
Maria J.
Mayoral
 a,
Santiago
Casado
b,
Luisa
Ruiz-González
c,
Emilio M.
Pérez
*b and
David
González-Rodríguez
*ad
a,
Santiago
Casado
b,
Luisa
Ruiz-González
c,
Emilio M.
Pérez
*b and
David
González-Rodríguez
*ad
aOrganic Chemistry Department, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: david.gonzalez.rodriguez@uam.es
bIMDEA Nanociencia, c/Faraday 9, Campus de Cantoblanco, 28049, Madrid, Spain. E-mail: emilio.perez@imdea.org
cInorganic Chemistry Department, Universidad Complutense de Madrid, 28040, Madrid, Spain
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 4th April 2018
Abstract
Due to their outstanding electronic and mechanical properties, single-walled carbon nanotubes (SWCNTs) are promising nanomaterials for the future generation of optoelectronic devices and composites. However, their scarce solubility limits their application in many technologies that demand solution-processing of high-purity SWCNT samples. Although some non-covalent functionalization approaches have demonstrated their utility in extracting SWCNTs into different media, many of them produce short-lived dispersions or ultimately suffer from contamination by the dispersing agent. Here, we introduce an unprecedented strategy that relies on a cooperative clamping process. When mixing (6,5)SWCNTs with a dinucleoside monomer that is able to self-assemble in nanorings via Watson–Crick base-pairing, a synergistic relationship is established. On one hand, the H-bonded rings are able to associate intimately with SWCNTs by embracing the tube sidewalls, which allows for an efficient SWCNT debundling and for the production of long-lasting SWCNT dispersions of high optical quality along a broad concentration range. On the other, nanoring stability is enhanced in the presence of SWCNTs, which are suitable guests for the ring cavity and contribute to the establishment of multiple cooperative noncovalent interactions. The inhibition of these reversible interactions, by just adding, for instance, a competing solvent for hydrogen-bonding, proved to be a simple and effective method to recover the pristine nanomaterial with no trace of the dispersing agent.
Introduction
Carbon nanotubes (CNTs) are nanomaterials with impressive electronic and mechanical properties and are considered as strong candidates for the next generation of transistors, photovoltaics, and (bio)chemical sensors.1–3 However, they are insoluble in most solvents and infusible at any temperature, due to strong bundling caused by numerous van der Waals interactions, which hampers many of their applications.4,5 Covalent and noncovalent surface modification with molecules, leading to homogeneous CNT dispersions in various solvents or bulk materials, is now common practice to facilitate their processing and to attach specific functions.6–9 The covalent approach grants more stable dispersions, but the modification of the π-conjugated CNT surface with grafted molecules has a detrimental impact on their (semi)conducting and mechanical properties. That is the reason why noncovalent strategies, achieved by promoting strong interactions between CNT sidewalls and π-conjugated or apolar moieties in adequate solvents, are often considered more suitable.9–11However, the weak and dynamic nature of supramolecular interactions frequently produces less durable dispersions of individual CNTs because bundling, which ultimately leads to CNT reprecipitation, remains as a strongly competing supramolecular process in solution. Bundling can be reduced by increasing the number of surface-binding functions in the dispersing agent and, importantly, by inducing wrapping interactions around single CNT cylinders (interaction mode II in Fig. 1a below). A large number of composites with π-conjugated polymers,12,13 DNA,14–16 synthetic peptides,17–20 or, in general, oligomers featuring multiple aromatic units21–24 have been tested to stabilize CNTs selectively in solution by specific helical wrapping conformations around the tube.
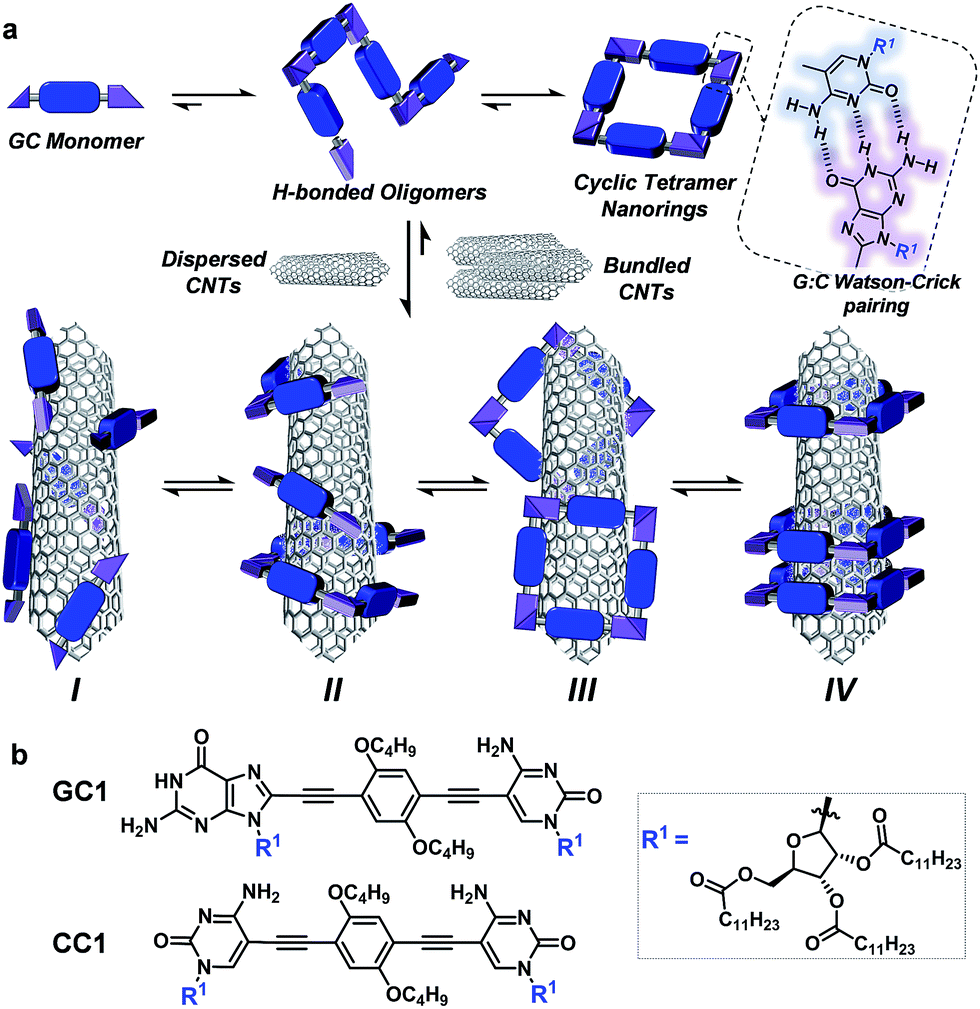 | ||
| Fig. 1 (a) Overall equilibria established between GC monomers and CNTs. Top: GC associates to yield a distribution of H-bonded supramolecular oligomers via Watson–Crick pairing, among which the cyclic tetramer is predominant due to a high chelate cooperativity. These species can in principle stabilize CNT dispersions through (I) monomer–CNT, (II) oligomer–CNT, (III) external nanoring–CNT, or (IV) threaded nanoring–CNT van der Waals interactions. (b) Structure of GC1 and CC1 monomers. | ||
A main drawback that originates from such a robust marriage is that polymer desorption via washing processes is often difficult to achieve, and CNTs purified or processed in this way may remain contaminated with the dispersing agent.25,26 In order to address this issue, recent strategies have been reported that focus mainly on: (1) switching between tightly and loosely bound polymer conformations,27–29 which can be triggered thermally, photochemically27 or by a change in solvent28 or pH,29 and on (2) inducing depolymerization processes,30–34 which can also be made reversible by introducing supramolecular32–34 or dynamic covalent bonds35 along the polymer main chain.
Here, we introduce a novel approach that is instead based on clamping discrete self-assembled nanorings around the tube cross-section to efficiently and reversibly produce durable dispersions of individual single-walled CNTs (SWCNTs) in organic solvents. The encapsulation of SWCNTs within well-defined macrocycles,19,36–41 forming rotaxane-type ensembles, has been recently explored using covalent cyclization reactions. The strategy followed by some of us relied on two extended aromatic binding sites to promote the supramolecular association of U-shaped molecules to SWCNT, followed by “clipping” through ring-closing metathesis to produce the mechanically interlocked species.36–41 Depending on the chemical nature of the recognition motif, we have also observed the formation of oligomers that wrap around the SWCNTs.38 The approach followed herein, in contrast, profits from a dynamic, strongly cooperative noncovalent macrocyclization process.
Our design focuses on a dinucleoside monomer (GC1; Fig. 1b) that has been rationally synthesized to comply with these objectives (see the ESI† for synthetic details). GC1 structure consists of complementary guanosine (G) and cytidine (C) DNA bases connected by a rigid, linear central block. We demonstrated recently that related molecules self-assembled into cyclic tetramer species42 through Watson–Crick G–C H-bonding interactions43 exhibiting record chelate cooperativities,44–46 which allow the nanorings to be formed quantitatively in nonpolar solvents within a wide concentration range.47–50 The ribose groups in GC1 feature multiple long alkyl chains that, upon cyclic assembly, would point in all directions towards the exterior medium, which should benefit individual CNT debundling and thus afford high solubility to the final composites. The monomer–CNT interaction strength is an additional key factor in our design. A π-conjugated dialkoxyarene central block with modest affinity for the CNT surface has been installed between the lipophilic bases. The idea is that stable dispersions would only be obtained if the rings are able to embrace the tube, so that its sidewalls can interact with several monomers in the nanoring cavity (mode IV in Fig. 1), and not through external binding with sections of the rings (mode III) or with individual monomers or linear oligomers (modes I/II). We also explore herein the special synergy of the GC1–SWCNT marriage, which simultaneously brings an enhanced solubility to the nanotubes (when compared to a related molecule that cannot cyclize: CC1) and an increased stability to the macrocycles. This synergy originates from the cooperative action of G–C H-bonding and monomer–nanotube van der Waals interactions, so we reasoned that the disturbance of any of these distinct noncovalent interactions would cause collapse of the supramolecular ensemble, thus facilitating the recovery of the extracted CNTs.
Results and discussion
Initial experiments and sample preparation
Prior to their combination with CNTs, we confirmed that this novel GC1 monomer displayed a similar self-assembly process to the one already reported by us with closely related dinucleosides (see the ESI† for further details).47,50 NMR and optical spectroscopy experiments indicated that GC1 tetrameric rings are indeed formed close to quantitatively in apolar chlorinated solvents within the 10−1 to 5 × 10−4 M concentration range. These cyclic assemblies are characterized by red-shifted and low intensity emission maxima at ca. 505 nm, and by the presence of a characteristic negative Cotton effect, with maxima at 340 and 387 and a minimum at 428 nm. At lower concentrations, they dissociate gradually into monomeric species, which display distinct emission maxima at 421 and 445 nm and null CD signals (vide infra). The addition of polar cosolvents that can compete for H-bonding, like DMSO or DMF, also results in monomer dissociation. The 1H NMR spectra of GC1 recorded by modifying the CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF-D7 volume ratio (Fig. S1†) revealed a strong all-or-nothing behavior: no significant participation of any other H-bonded oligomer but the tetrameric macrocycle is detected in solution. This is in agreement with the formation of stable ring species with remarkably high chelate cooperativities, as determined in our previous work.
:DMF-D7 volume ratio (Fig. S1†) revealed a strong all-or-nothing behavior: no significant participation of any other H-bonded oligomer but the tetrameric macrocycle is detected in solution. This is in agreement with the formation of stable ring species with remarkably high chelate cooperativities, as determined in our previous work.
An initial theoretical study using DFT calculations (see below and the ESI† for further details) served to properly design the interacting ring–tube system and experimental measurements. We decided to use (6,5)-enriched SWCNTs with a mean diameter of 0.7 nm and very narrow polydispersity (we observe (6,5)SWCNTs exclusively in photoluminescence experiments; see Fig. S2†) because they should fit adequately within the GC1 nanoring cavity. Thus, a dispersion of (6,5)SWCNTs in CHCl3 (0.2 mg mL−1) was produced first by ultrasonication followed by centrifugation, in order to remove large SWCNT bundles and any other carbonaceous impurities. These non-stabilized dispersions are rather short-lived and significant precipitation of (6,5)SWCNTs was clearly observed after a few hours (see central image in Fig. 2a). Immediately after centrifugation, GC1 (or CC1) solutions in CHCl3 (different concentrations were tested, from 5.0 × 10−4 to 10−6 M) were added to the supernatant (6,5)SWCNT dispersion and the mixtures were stirred at room temperature. The (6,5)SWCNT–GC1 suspensions produced (right image in Fig. 2a) were kept under dark and checked at different periods of time. A few spectroscopic changes were observed within the first hours after mixing, but after ca. 12 hours spectral properties remained constant for weeks and are reproducible.
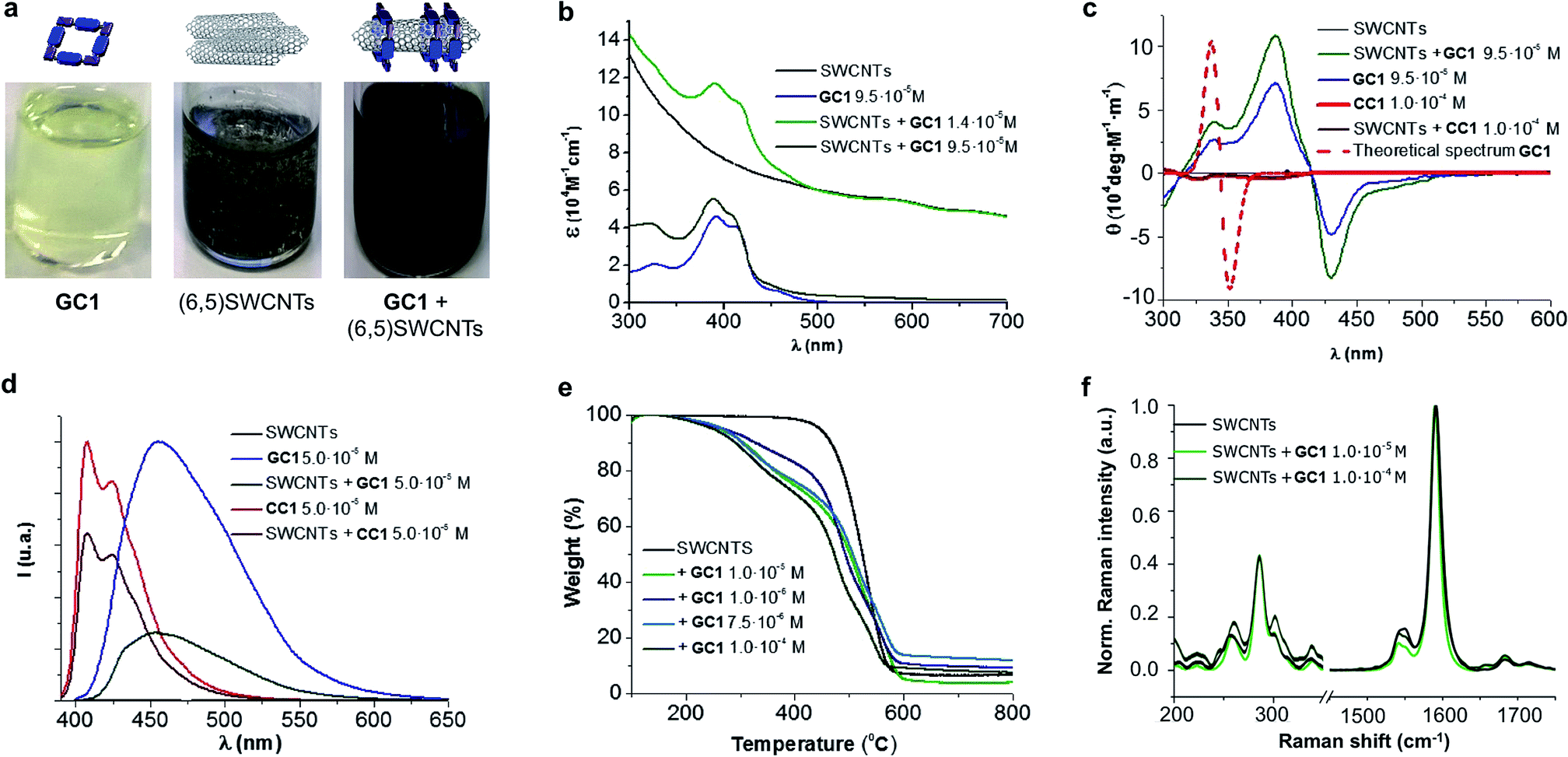 | ||
| Fig. 2 (a) From left to right: image of the solutions/dispersions containing GC1 (9.5 × 10−5 M), (6,5)SWCNTs (0.2 mg mL−1) and their mixture in CHCl3. (b–f) Absorption (b), CD (c), and emission spectra (λexc = 380 nm) (d), TGA analyses (air, 50 °C min−1) (e), and Raman spectra (λexc = 633 nm) (f) of samples containing GC1/CC1, pristine (6,5)SWCNTs, or the products formed by their mixture at different concentrations. | ||
Spectroscopic and thermogravimetric measurements
The (6,5)SWCNT–GC1/CC1 composites were analyzed by absorption, CD, Raman, and emission spectroscopies and by TGA, and compared with pristine (6,5)SWCNTs and GC1/CC1 in the same conditions (Fig. 2). The absorption spectra (Fig. 2b) showed the typical features of both components in the mixture and only minor deviations in absorption maxima were noted. The typical scattering phenomena, evidenced by a baseline rise, was notorious in the original (6,5)SWCNTs dispersions and in mixtures with CC1 or with low GC1 content. However, as the GC1/(6,5)SWCNTs ratio is increased, scattering is concomitantly reduced until it is no longer perceptible in the absorption spectrum, which underlines the high dispersing power of this agent. As a matter of fact, the GC1–(6,5)SWCNTs mixtures produced are clear suspensions (Fig. 2a) that show no evidence for nanotube precipitation along several weeks.CD can be considered as one of the most powerful techniques for stereochemical analysis: it is sensitive to the absolute configuration as well as to conformational features, which are often completely obscured in ordinary absorption spectra.51 Regarding this, the CD spectral shape of GC1–(6,5)SWCNTs composites does not exhibit important differences with GC1 in the same conditions (Fig. 2c). This is a strong indication that the cyclic tetramer structure is preserved in the presence of (6,5)SWCNT, and no dissociation or structural change in the H-bonded assembly was noted. In order to obtain additional proof from a theoretical perspective, we calculated the CD spectrum for the optimized structure of cGC14 (see Fig. 3g below), which is displayed as a dotted red line in Fig. 2c. Our calculations revealed a strong dependency of the CD spectra with the symmetry of the macrocycle. Only when the C4 axis was maintained in the cGC14 ring structure, we obtain a good match between the experimental and the theoretical results (with the exception of the vibronic structure of the positive Cotton effect). If the C4 symmetry axis is lost, due for instance to other binding modes of GC1 to the CNT sidewalls, the CD spectrum is significantly perturbed. As an example, we show in Fig. S3† the structure and calculated spectrum for a cGC14 structure, which is close in energy, but has lost the C4 symmetry. We can therefore conclude that this C4 symmetry is conserved upon the formation of (6,5)SWCNT–cGC14 conjugates, which is a strong indication that the SWCNT is inside the cavity of cGC14 ring.
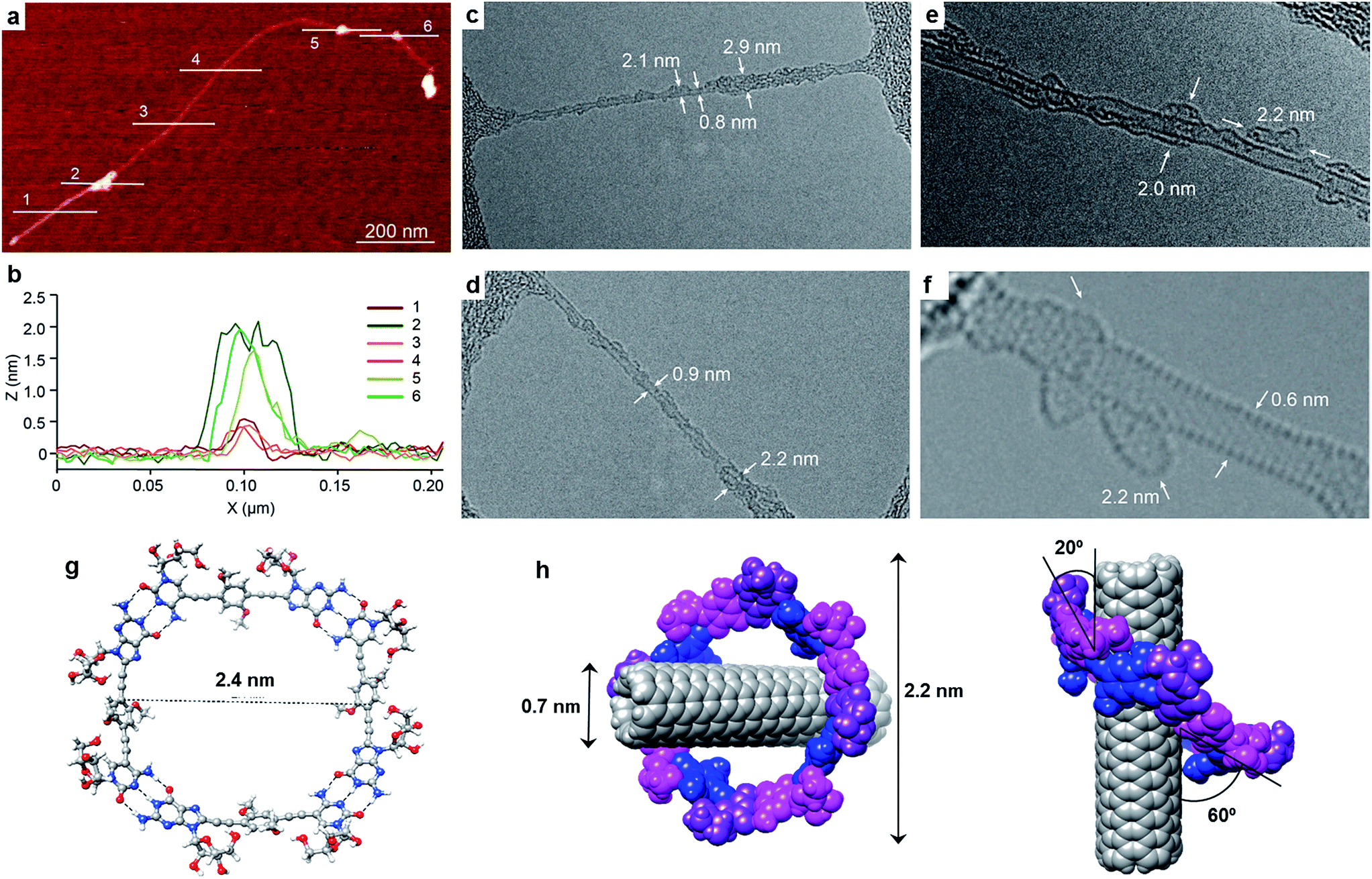 | ||
| Fig. 3 (a) Representative AFM topographic image of a suspension of the product formed by the mixture of (6,5)SWCNTs with GC1 in TCE. (b) Height profiles along the lines marked in (a) show individualized SWCNT (0.6 nm) decorated with objects that are consistently 2 nm high. (c–f) Representative TEM images of an individual SWCNT. Note that the sidewalls are functionalized symmetrically by a low-contrast addend that shows a total a diameter of 2–3 nm, consistent with encapsulation by the GC1 macrocycles. (g) DFT-optimized structure of a cGC14 ring. (h) Lower energy configuration of the (6,5)SWCNT–cGC14 composite, showing diverse geometric parameters. | ||
Another important proof that demonstrates GC1–(6,5)SWCNTs interactions came from comparing the emission spectra (Fig. 2d) with and without CNTs. As a matter of fact GC1 fluorescence is considerably quenched in the presence of (6,5)SWCNTs. This is very characteristic in noncovalent SWCNT assemblies22 and is presumably caused by an energy transfer process between the dinucleoside π-conjugated system and the SWCNT when they are in close contact.
On the other hand, TGA studies showed that macrocycle loading increases with GC1 concentration, to yield 17% and 28% loading at 10−6 and 10−4 M GC1 initial concentration, respectively. Finally, no significant (6,5)SWCNT electronic perturbance was noted in Raman experiments when GC1 or CC1 were added, which is a good indication of the formation of noncovalently bound composites.
AFM, TEM and DFT analysis
Exploration of the GC1–SWCNT suspensions under atomic force microscopy (AFM) was consistent with a picture in which the SWCNTs are encapsulated within GC1 macrocycles. AFM images were obtained upon spin-coating the SWCNT suspensions on mica, and show individualized SWCNTs decorated with objects of about 2 nm in height (Fig. 3a and b), in full accord with the DFT-modelled size of the GC1 tetramer (Fig. 3h). We also explored our samples under transmission electron microscopy (TEM), where we observe mostly re-bundled SWCNTs, most likely due to sample preparation issues. However, wherever individualized SWCNTs were located, they showed heavily functionalized sidewalls (Fig. 3c and d). Unfortunately, and in contrast to covalently linked macrocycles,41 attempts at obtaining higher resolution TEM images were precluded by the instability of the H-bonded organic macrocycle in the conventional transmission electron microscope at 200 kV. High resolution images were obtained in an aberration corrected microscope at 60 kV (Fig. 3e and f). We observe structures of around 2.0 nm surrounding the SWCNTs that are consistent with the picture provided by spectroscopy, but even under low voltage (see also Fig. S5†),52–54 the macrocycles were observed to quickly reorganize and eventually decompose under the electron beam irradiation, probably due to radiolysis damage associated to hydrogen.Further theoretical studies were performed in order to gain a deeper insight into the structure and interactions in the (6,5)SWCNT–cGC14 composites. As explained above, the optimized geometry of cGC14 belongs to the C4 point group of symmetry and is depicted in Fig. 3g. The inner cavity has a mean diameter of 2.1 nm and the central aromatic moieties are bent at an angle of 30°, so that planarity is not conserved, allowing the carbon nanotube to maintain a stronger interaction with the ring. In order to have a second proof of the arrangement between the two moieties, we additionally calculated the intensity of the interaction between SWCNTs and cGC14 ring with close π–π contacts in the range of 3.2–3.5 Å. Among the different possible configurations (see Fig. S4†), we observed that the (6,5)SWCNT and the cGC14 tetramer interact strongly (Eint = −22.7 kcal mol−1) when the macrocycles are clamping the nanotube and the macrocycle tilts slightly with respect to the tube axis, in order to maximize non-covalent interactions (interaction mode IV in Fig. 1). In contrast, all external binding modes investigated (interaction mode III in Fig. 1), result in positive (i.e. disfavorable) interaction energies ranging between +0.7 and +42.0 kcal mol−1. The geometry of the lowest energy configuration is shown in Fig. 3h.
Analysis of the synergistic interactions
Concentration-dependent experiments, where we recorded absorption, CD and emission spectra, provided a deeper understanding of the mutual interaction between CNTs and H-bonded nanorings. Two parallel dilution measurements were conducted from GC1 CHCl3 solutions: one in the absence (Fig. 4a and c) and the other one in the presence of (6,5)SWCNTs (Fig. 4b and d). The spectral changes with concentration were monitored by CD from 4.0 × 10−4 M (blue lines) down to 3.0 × 10−6 M (CD) or down to 2.0 × 10−7 M (emission) (red lines). The trends obtained from both techniques are compared in Fig. 4i.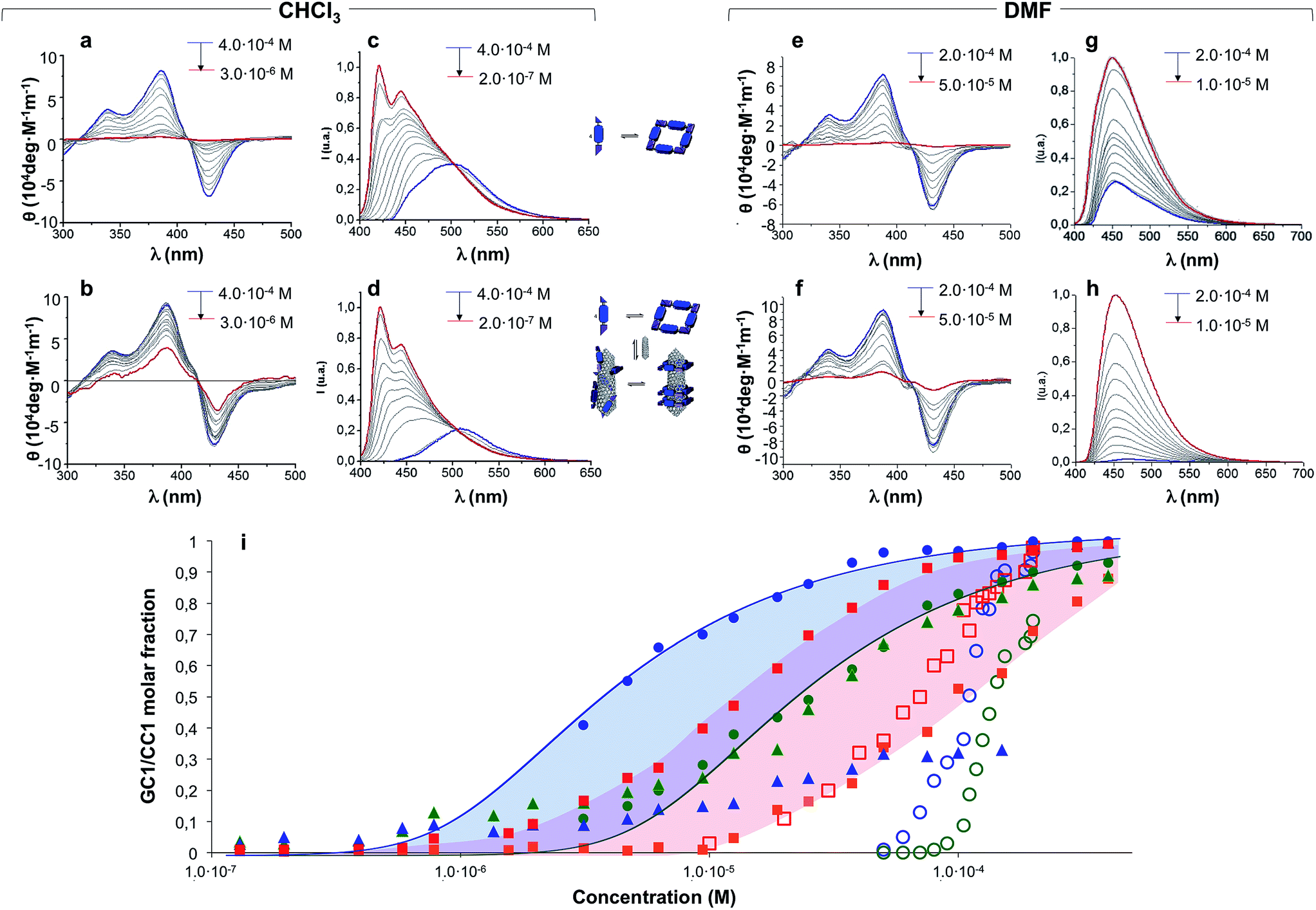 | ||
| Fig. 4 CD (a, b, e and f) and normalized emission (c, d, g and h) spectra of GC1 in the absence (a, c, e and g) or presence (b, d, f and h) of (6,5)SWCNTs recorded at different concentrations in dilution experiments with CHCl3 (left panel) or DMF (right panel). Initial and final concentrations are indicated in blue and red, respectively. (i) Representation of: (1) the molar fraction of GC1 molecules that are assembled as cyclic tetramers, as determined by CD (circles) or emission spectroscopy (triangles) in the absence (green) or presence (blue) of (6,5)SWCNTs, or (2) the molar fraction of GC1 (red squares) or CC1 (orange squares) molecules that are bound to (6,5)SWCNTs, as estimated from the degree of emission quenching. Dilution experiments were made in CHCl3 (solid shapes) or DMF (open shapes). The CD trends in CHCl3 were fitted to cyclotetramerization models (blue and green lines). | ||
A first experimental observation that came from comparison of both dilution experiments is that the presence of (6,5)SWCNTs preserved the characteristic cyclic tetramer features along a wider concentration range. This is unambiguously observed in the evolution of the CD spectra as a function of concentration. While GC1 CD features readily disappear below ca. 10−5 M (Fig. 4a), indicating cyclic tetramer dissociation,47–50 they remain distinct down to 3 × 10−6 M in the presence of (6,5)SWCNTs (Fig. 4b). The degree of cyclotetramerization, that is, the molar fraction of GC1 molecules associated as cyclic tetramers (horizontal equilibria in Fig. 1a), can be calculated from each dilution experiment by integrating CD intensity. The comparison of both trends, represented with solid blue and green circles in Fig. 4i, suggests that GC1 nanorings are significantly stabilized when mixed with (6,5)SWCNTs. Fitting these data to cyclotetramerization processes (blue and green lines in Fig. 4i) afforded cyclotetramerization constants (KT) in the order of KT = 3.2 × 1014 M−3 for GC1 and KT = 4.0 × 1016 M−3 for GC1 + (6,5)SWCNTs. That is, chelate cooperativity greatly benefits from the presence of SWCNTs and nanoring stability is considerably increased.
We then turned our attention to the evolution of the fluorescence features in these dilutions experiments. The emission spectra were analyzed in two different ways, attending to: (1) their relative intensity, which can be correlated to the fraction of CNT-bound molecules (vertical equilibrium in Fig. 1a), or (2) the spectral shape and emission wavelength, which reports again on the degree of cyclotetramerization (horizontal equilibria in Fig. 1a). In the first case we compared GC1 emission intensity in the 400–650 nm range in the absence (IGC) or presence (IGC–CNT) of (6,5)SWCNTs at each concentration (see Fig. S6A†). The degree of emission quenching, defined as 1 − IGC–CNT/IGC, was then calculated and plotted as red squares in Fig. 4i. Since energy transfer from the photoexcited dinucleoside molecules to the CNT is close to quantitative when they are closely interacting, as determined from the fully quenched emission of the samples at high concentration, these curves actually reveal the molar fraction of GC1 that is bound to (6,5)SWCNTs at each concentration. Our results show that GC1–(6,5)SWCNTs association is virtually quantitative at concentrations above 10−4 M and then falls in the 10−4 to 10−6 M range. This nonlinear trend suggests a cooperative GC1–(6,5)SWCNT interaction that seems to be coupled to the cyclotetramerization equilibrium. In order to compare our GC monomer with a related dispersing agent that does not associate in cyclic systems, we carried out another set of parallel dilution experiments with CC1 in the absence and presence of (6,5)SWCNTs. The degree of emission quenching was likewise calculated at each concentration and the results are shown as orange squares in Fig. 4i (see also Fig. S6B†). The CC1–(6,5)SWCNTs association is clearly weaker and no longer detected below 10−5 M, a concentration where ca. half of the GC1 molecules are still bound to CNTs.
In the second case, the degree of cyclotetramerization was estimated by analysing the shape of each normalized fluorescence spectra during the dilution measurements shown in Fig. 4c and d, as we described in our previous work (see also the ESI†).47,50 As commented above, when the monomeric species, showing emission maxima at 421 and 445 nm in CHCl3 (see red spectra in Fig. 4c and d), associate in cyclic tetramers, a red-shift to >500 nm and a reduction in emission intensity is noted. The results obtained at each concentration are also included in Fig. 4i as solid green and blue triangles for the GC1 samples without and with CNTs, respectively. The cyclic tetramer association trends calculated by CD and fluorescence spectroscopy (green circles and triangles) display a quite decent correlation for GC1. That is not the case when (6,5)SWCNTs are present (please compare blue circles and triangles), and this is because CD and emission spectroscopy are not reporting on the same GC1 population. Whereas the measured CD spectra is representative of all GC1 molecules in solution, the emission spectra primarily provide information of the fraction of molecules that are not bound to the CNTs, since the emission of the GC1 molecules that are bound is strongly quenched. As a consequence, the shape evolution of the emission spectra when the CNTs are present exhibits again a strong coupling between the horizontal cyclotetramerization and vertical GC1–(6,5)SWCNT association equilibria in Fig. 1a. At concentrations above 10−4 M most GC1 molecules are bound to the CNTs and the residual emission recorded is extremely weak in intensity and representative of the cyclic tetramer in shape and emission wavelength (see blue spectrum in Fig. 4d as an example). However, when GC1–(6,5)SWCNT association is no longer quantitative in the 10−4 to 10−6 M range, the emission spectra becomes rapidly dominated by the fraction of GC1 molecules that are not bound to CNTs, which have stronger monomer-like features (red spectrum in Fig. 4d) because their actual concentration is lower. This is reflected in a very sharp transition around 10−4 M in the blue-triangle trend in Fig. 4i. In other words, in the 10−4 to 10−6 M range, the shape of the emission spectrum is more shifted to the monomer features in the presence of CNTs, since the actual concentration of emissive GC1 molecules (i.e. not bound to CNTs) is lower than in the absence of CNTs. Only when the GC1–(6,5)SWCNT interactions become no longer important, below 10−6 M, samples with and without CNTs display similar emission intensity and shape (see also Fig. S6A†).
We believe the graph in Fig. 4i provides a rather faithful description of the self-assembly of this two-component mixture in solution as a function of concentration. The results gathered from these dilution experiments in CHCl3 clearly indicate that each species benefits synergistically from the presence of the other, as it is schematically represented in Fig. 5. On one hand, the nanorings are more stable in the presence of nanotubes, a gain that is represented by the blue area in Fig. 4i. On the other, the nanotubes can host a higher number of dispersing agent molecules and thus enjoy enhanced solubility along a broader concentration range when the monomer cyclizes, a gain that is represented by the red area in Fig. 4i when comparing GC1 and CC1. In short, H-bonding between complementary bases and van der Waals dispersion forces between monomer and the π-conjugated CNT sidewalls are noncovalent interactions that work here cooperatively to build strongly-associated GC1–(6,5)SWCNTs composites. It is interesting to note that smaller nanorings, where the nucleobases are directly connected by a triple bond (see GC3 in the ESI†), whose cavities cannot host (6,5)SWCNTs, do not exhibit the same synergistic effects as GC1, but rather behave as a regular dispersing agent, like CC1. On the other hand, GC1 cannot solubilize as efficiently multi-walled nanotubes of larger diameter. Both observations are in agreement with the clamping interaction mode IV, shown in Fig. 1a and 5, being the most likely and abundant in CHCl3 solutions (Fig. 1a). We should nonetheless consider that, when the nanorings are assembled around the tube, local concentration effects may occur, so that a rearrangement into linear polymers may take place when several rings coincide locally. However, these ring-to-chain rearrangements should lead to a reduction in CD intensity at high concentrations that we did not observe, so we can discard this is a relevant situation. As we reduce concentration, these effects are even less likely to occur, because the rings are favoured entropically.
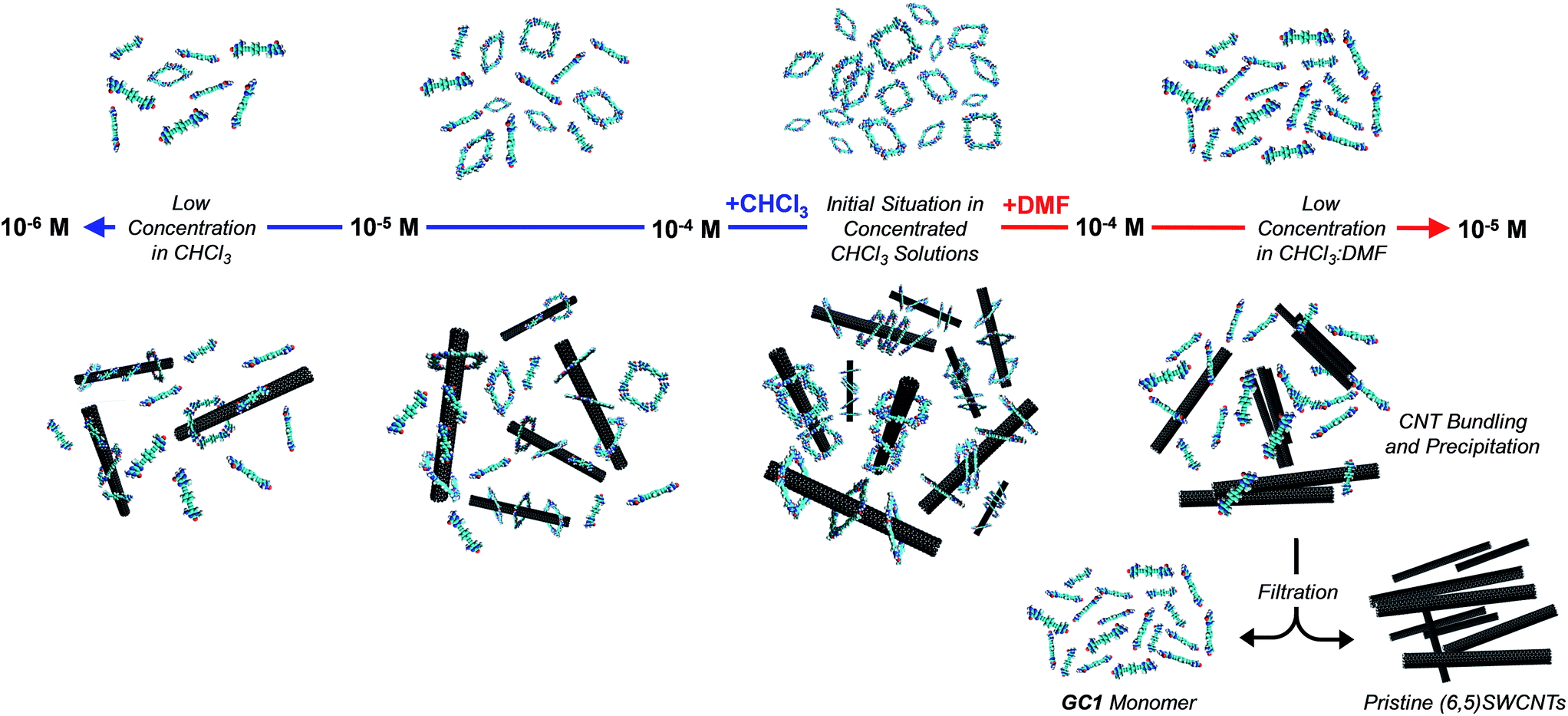 | ||
| Fig. 5 Schematic representation of the dilution processes in CHCl3 (left) and DMF (right) without (top) and with (6,5)SWCNTs (bottom). At high concentrations in CHCl3, GC1 ring assemblies are formed quantitatively and establish strong interactions with the CNTs. Upon dilution with CHCl3 the cyclic tetramers gradually dissociate, but such dissociation occurs to a lower extent when CNTs are present, due to the stronger (6,5)SWCNT–cGC14 clamping interactions. On the contrary, upon dilution with DMF, the cGC14 rings are fully dissociated at relatively high concentrations, which eventually produces CNT precipitation due to the weaker interaction of GC1 monomers with the (6,5)SWCNTs. In this last situation, a simple filtration and washing protocol allows to separate efficiently the (6,5)SWCNTs from the GC1 monomer. | ||
Recovery of the pristine SWCNTs
At this point we reasoned that the whole assembly could be demolished by addressing just one of the supramolecular interactions, which could be a simple and straightforward strategy for pristine (6,5)SWCNTs recovery. For instance, an increase in solvent polarity should disrupt H-bonding interactions without strongly affecting monomer–(6,5)SWCNTs van der Waals interactions. In order to prove this, we performed now the same dilution experiments from GC1 samples with and without CNTs by gradual addition of DMF instead of CHCl3, and recorded again nanoring dissociation by CD (Fig. 4e and f; open blue and green circles, respectively, in Fig. 4i), and the degree of emission quenching by fluorescence spectroscopy (Fig. 4g and h; open red squares in Fig. 4i).In line with the previous observations, the cycles are more stable and the CD features resist a higher amount of DMF when (6,5)SWCNTs are present (please compare open blue and green circles). Together with the disappearance of the CD signals, GC1 monomer emission was recovered in a narrower concentration range when the parent GC1–(6,5)SWCNTs dispersion was diluted with DMF instead of CHCl3 (please compare solid and open red squares), which suggests much weaker interactions with the CNTs in this polar solvent. Interestingly, several minutes after DMF addition a clear precipitate emerged from the original dispersion that, after washing, showed no residual GC1 spectroscopic features. Despite the strong association and remarkable endurance of the GC1–(6,5)SWCNTs solutions in CHCl3, which can last for several weeks maintaining the original optical quality, CNT rebundling and precipitation occurred rapidly in DMF, likely due to the lower efficiency of the dissociated monomer as a dispersing agent (see Fig. 5). It is also interesting to note that cGC14 dissociation can also be induced by increasing the temperature in CHCl3 or CHCl2CHCl2 solutions. After being subjected to a heating–cooling cycle the spectra of the initial and final samples differed considerably and clear precipitation of the CNTs was noted. However, the results are highly dependent on the concentration and not as reproducible and efficient as the addition of DMF to release and recover the pristine (6,5)SWCNTs.
Conclusions
In conclusion, we have explored herein an unprecedented approach to solubilize SWCNTs in apolar solvents that relies on dynamic macrocycle clamping around the nanotube sidewalls, which allows for efficient SWCNT debundling, and on cooperative noncovalent interactions, which supplies the required reversibility to simply and effectively recover the pristine material. The combination of theoretical DFT-based methodologies, spectroscopic techniques, as well as AFM and TEM microscopies, provide solid evidence for a preferred association mode where the H-bonded nanorings are embracing the tube (mode IV in Fig. 1). Furthermore, a comparison between dilution experiments performed on GC1 in the absence or presence of (6,5)SWCNTs provided a deep insight into the mutual benefits offered by the combination of these two species. On one hand, nanoring stability is unambiguously enhanced in the presence of the CNTs, likely due to the interaction of more than one GC1 monomer with the tube sidewalls. On the other, the GC1 molecule exhibited an extraordinary solubilizing power, in comparison with a regular dispersing agent that cannot cyclize, like CC1. When comparing these two monomers, we observe dramatic differences in terms of the quality of the dispersions produced (that only show very low scattering in the case of GC1; see Fig. 2b), their durability (the dispersions produced from CC1 show a CNT precipitate after a few days, while those from GC1 were seen to resist for many weeks), and the resistance of the monomer–SWCNT interaction to dilution (as determined from the experiments shown in Fig. 4). We believe that these remarkable differences, which are translated in the generation of clear, long-lasting (6,5)SWCNTs dispersions along a broad concentration range, can only be explained if the dispersing agent is able to surround individual nanotubes, as in mode IV in Fig. 1.Future efforts will be directed to study if our self-assembled nanorings can selectively extract CNTs as a function of diameter and chirality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Union (ERC-Starting Grants: 279548 (DGR) and 307609 (EMP)), MINECO (Grants: CTQ2014-57729-P (DGR) and CTQ2014-60541-P (EMP), FPU13/03371 (LdJ), JdC-2015-23531 postdoctoral fellowship (BNO)) and the Comunidad de Madrid (Grant: MAD2D-CM program S2013/MIT-3007 (EMP)) is gratefully acknowledged. IMDEA Nanociencia acknowledges support from the 'Severo Ochoa' Programme for Centres of Excellence in R&D (MINECO, Grant SEV-2016-0686). The computational work was supported by the Campus de Excelencia Internacional UAM + CSIC. Additionally, we express our gratitude to the Supercomputing and Bioinnovation Center (SCBI) of the University of Málaga (Spain) for their support and resources. We thank the National Centre for Electron Microscopy (ICTS-CNME, Universidad Complutense) for electron microscopy facilities.Notes and references
- E. S. Snow, F. K. Perkins, E. J. Houser, S. C. Badescu and T. L. Reinecke, Science, 2005, 307, 1942 CrossRef CAS PubMed.
- Q. Cao, H.-S. Kim, N. Pimparkar, J. P. Kulkarni, C. Wang, M. Shim, K. Roy, M. A. Alam and J. A. Rogers, Nature, 2008, 454, 495 CrossRef CAS PubMed.
- A. D. Franklin, Science, 2015, 349, aab2750 CrossRef PubMed.
- S. H. Munson-McGee, Phys. Rev. B, 1991, 43, 3331 CrossRef.
- M. Cadek, J. N. Coleman, K. P. Ryan, V. Nicolosi, G. Bister, A. Fonseca, J. B. Nagy, K. Szostak, F. Beguin and W. J. Blau, Nano Lett., 2004, 4, 353 CrossRef CAS.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS PubMed.
- P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214 RSC.
- N. Karousis and N. Tagmatarchis, Chem. Rev., 2010, 110, 5366 CrossRef CAS PubMed.
- D. Britz and A. Khlobystov, Chem. Soc. Rev., 2006, 35, 637 RSC.
- Y.-L. Zhao and J. F. Stoddart, Acc. Chem. Res., 2009, 42, 1161 CrossRef CAS PubMed.
- D. Tuncel, Nanoscale, 2011, 3, 3545 RSC.
- P. Bilalis, D. Katsigiannopoulos, A. Avgeropoulos and G. Sakellariou, RSC Adv., 2014, 4, 2911 RSC.
- M. Zheng, A. Jagota, M. S. Strano, A. P. Santos, P. Barone, S. G. Chou, B. A. Diner, M. S. Dresselhaus, R. S. McLean, G. B. Onoa, G. G. Samsonidze, E. D. Semke, M. Usrey and D. Walls, Science, 2003, 302, 1545 CrossRef CAS PubMed.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. McLean, S. R. S. Lustig, S. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338 CrossRef CAS PubMed.
- H. Cathcart, V. Nicolosi, J. M. Hughes, W. J. Blau, J. M. Kelly, S. J. Quinn and J. N. Coleman, J. Am. Chem. Soc., 2008, 130, 12734 CrossRef CAS PubMed.
- G. R. Dieckmann, A. B. Dalton, P. A. Johnson, J. Razal, J. Chen, G. M. Giordano, E. Muñoz, I. H. Musselman, R. H. Baughman and R. K. Draper, J. Am. Chem. Soc., 2003, 125, 1770 CrossRef CAS PubMed.
- A. B. Dalton, A. Ortiz-Acevedo, V. Zorbas, E. Brunner, W. M. Sampson, S. Collins, J. M. Razal, M. M. Yoshida, R. H. Baughman, R. K. Draper, I. H. Musselman, M. Jose-Yacaman and G. R. Dieckmann, Adv. Funct. Mater., 2004, 14, 1147 CrossRef CAS.
- A. Ortiz-Acevedo, H. Xie, V. Zorbas, W. M. Sampson, A. B. Dalton, R. H. Baughman, R. K. Draper, I. H. Musselman and G. R. Dieckmann, J. Am. Chem. Soc., 2005, 127, 9512 CrossRef CAS PubMed.
- J. Montenegro, C. Vázquez-Vázquez, A. Kalinin, K. E. Geckeler and J. R. Granja, J. Am. Chem. Soc., 2014, 136, 2484 CrossRef CAS PubMed.
- J. K. Sprafke, S. D. Stranks, J. H. Warner, R. J. Nicholas and H. L. Anderson, Angew. Chem., Int. Ed., 2011, 50, 2313 CrossRef CAS PubMed.
- G. Liu, F. Wang, S. Chaunchaiyakul, Y. Saito, A. K. Bauri, T. Kimura, Y. Kuwahara and N. Komatsu, J. Am. Chem. Soc., 2013, 135, 4805 CrossRef CAS PubMed.
- G. Clavé, G. Delport, C. Roquelet, J.-S. Lauret, E. Deleporte, F. Vialla, B. Langlois, R. Parret, C. Voisin, P. Roussignol, B. Jousselme, A. Gloter, O. Stephan, A. Filoramo, V. Derycke and S. Campidelli, Chem. Mater., 2013, 25, 2700 CrossRef.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348 CrossRef CAS PubMed.
- A. Nish, J. Hwang, J. Doig and R. Nicholas, Nat. Nanotechnol., 2007, 2, 640 CrossRef CAS PubMed.
- P. Imin, M. Imit and A. Adronov, Macromolecules, 2012, 45, 5045 CrossRef CAS.
- T. Umeyama, K. Kawabata, N. Tezuka, Y. Matano, Y. Miyato, K. Matsushige, M. Tsujimoto, S. Isoda, M. Takano and H. Imahori, Chem. Commun., 2010, 46, 5969 RSC.
- Z. Zhang, Y. Che, R. A. Smaldone, M. Xu, B. R. Bunes, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2010, 132, 14113 CrossRef CAS PubMed.
- S. Liang, Y. Zhao and A. Adronov, J. Am. Chem. Soc., 2014, 136, 970 CrossRef CAS PubMed.
- W. Z. Wang, W. F. Li, X. Y. Pan, C. M. Li, L.-J. Li, Y. G. Mu, J. A. Rogers and M. B. Chan-Park, Adv. Funct. Mater., 2011, 21, 1643 CrossRef CAS.
- F. Lemasson, J. Tittmann, F. Hennrich, N. Sturzl, S. Malik, M. M. Kappes and M. Mayor, Chem. Commun., 2011, 47, 7428 RSC.
- A. Llanes-Pallas, K. Yoosaf, H. Traboulsi, J. Mohanraj, T. Seldrum, J. Dumont, A. Minoia, R. Lazzaroni, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2011, 133, 15412 CrossRef CAS PubMed.
- I. Pochorovski, H. Wang, J. I. Feldblyum, X. Zhang, A. L. Antaris and Z. Bao, J. Am. Chem. Soc., 2015, 137, 4328 CrossRef CAS PubMed.
- F. Toshimitsu and N. Nakashima, Nat. Commun., 2014, 5, 5041 CrossRef CAS PubMed.
- T. Lei, X. Chen, G. Pitner, H. S. P. Wong and Z. Bao, J. Am. Chem. Soc., 2016, 138, 802 CrossRef CAS PubMed.
- E. Martínez-Periñán, A. de Juan, Y. Pouillon, C. Schierl, V. Strauss, N. Martín, A. Rubio, D. M. Guldi, E. Lorenzo and E. M. Pérez, Nanoscale, 2016, 8, 9254 RSC.
- A. López-Moreno, B. Nieto-Ortega, M. Moffa, A. de Juan, M. M. Bernal, J. P. Fernández-Blázquez, J. J. Vilatela, D. Pisignano and E. M. Pérez, ACS Nano, 2016, 10, 8012 CrossRef PubMed.
- S. Leret, Y. Pouillon, S. Casado, C. Navio, A. Rubio and E. M. Pérez, Chem. Sci., 2017, 8, 1927 RSC.
- A. López-Moreno and E. M. Pérez, Chem. Commun., 2015, 51, 5421 RSC.
- A. de Juan, M. M. Bernal and E. M. Pérez, ChemPlusChem, 2015, 80, 1153 CrossRef CAS.
- A. de Juan, Y. Pouillon, L. Ruiz-González, A. Torres-Pardo, S. Casado, N. Marín, A. Rubio and E. M. Pérez, Angew. Chem., Int. Ed., 2014, 53, 5394 CrossRef CAS PubMed.
- N. Bilbao, I. Destoop, S. De Feyter and D. González-Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 659 CrossRef CAS PubMed.
- J. Camacho-García, C. Montoro-García, A. M. López-Pérez, N. Bilbao, S. Romero-Pérez and D. González-Rodríguez, Org. Biomol. Chem., 2015, 13, 4506 Search PubMed.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762 CrossRef CAS PubMed.
- M. J. Mayoral, N. Bilbao and D. González-Rodríguez, ChemistryOpen, 2016, 5, 10 CrossRef CAS PubMed.
- C. Montoro-García, J. Camacho-García, A. M. López-Pérez, N. Bilbao, S. Romero-Pérez, M. J. Mayoral and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 6780 CrossRef PubMed.
- S. Romero-Pérez, J. Camacho-García, C. Montoro-García, A. M. López-Pérez, A. Sanz, M. J. Mayoral and D. González-Rodríguez, Org. Lett., 2015, 17, 2664 CrossRef PubMed.
- C. Montoro-García, J. Camacho-García, A. M. López-Pérez, M. J. Mayoral, N. Bilbao and D. González-Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 223 CrossRef PubMed.
- C. Montoro-García, M. J. Mayoral, R. Chamorro and D. González-Rodríguez, Angew. Chem., Int. Ed., 2017, 56, 15649 CrossRef PubMed.
- N. Berova, K. Nakanishi and R. Woody, Circular Dichroism: Principles and Applications, Wiley, 2000 Search PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622 CrossRef CAS PubMed.
- E. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 236 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, A. V. Markevich, S. Kurasch, O. Reimer, K. E. Walker, G. A. Rance, X. Feng, K. Müllen, A. Turchanin, M. A. Lebedeva, A. G. Majouga, V. G. Nenajdenko, U. Kaiser, E. Besley and A. N. Khlobystov, ACS Nano, 2017, 11, 2509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical procedures and compound characterization data, along with Fig. S1–S6. See DOI: 10.1039/c8sc00843d |
| This journal is © The Royal Society of Chemistry 2018 |