
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTropylium-promoted carbonyl–olefin metathesis reactions†
Uyen P. N.
Tran
,
Giulia
Oss
,
Domenic P.
Pace
,
Junming
Ho
 * and
Thanh V.
Nguyen
*
* and
Thanh V.
Nguyen
*
School of Chemistry, University of New South Wales, Australia. E-mail: t.v.nguyen@unsw.edu.au; junming.ho@unsw.edu.au
First published on 4th May 2018
Abstract
The carbonyl–olefin metathesis (COM) reaction is a highly valuable chemical transformation in a broad range of applications. However, its scope is much less explored compared to analogous olefin–olefin metathesis reactions. Herein we demonstrate the use of tropylium ion as a new effective organic Lewis acid catalyst for both intramolecular and intermolecular COM and new ring-opening metathesis reactions. This represents a significant improvement in substrate scope from recently reported developments in this field.
Introduction
The olefin–olefin metathesis reaction has been extensively studied in the past few decades due to its applicability in direct carbon–carbon bond formation.1 The analogous carbonyl–olefin metathesis (COM) reaction,2 however, is much less investigated, despite the fact that the chemistry of carbonyl compounds has been exploited ubiquitously in organic synthesis.3 There might be several reasons for this, with one linked to the same versatile reactivity of the carbonyl functionality such that other chemical transformations often compete and overshadow the possible metathesis reaction.2,3 Until recently, there were only a small number of stoichiometric Lewis acid-facilitated protocols4 for the COM reaction (Scheme 1a) and relevant stoichiometric olefination reactions of carbonyl moieties.5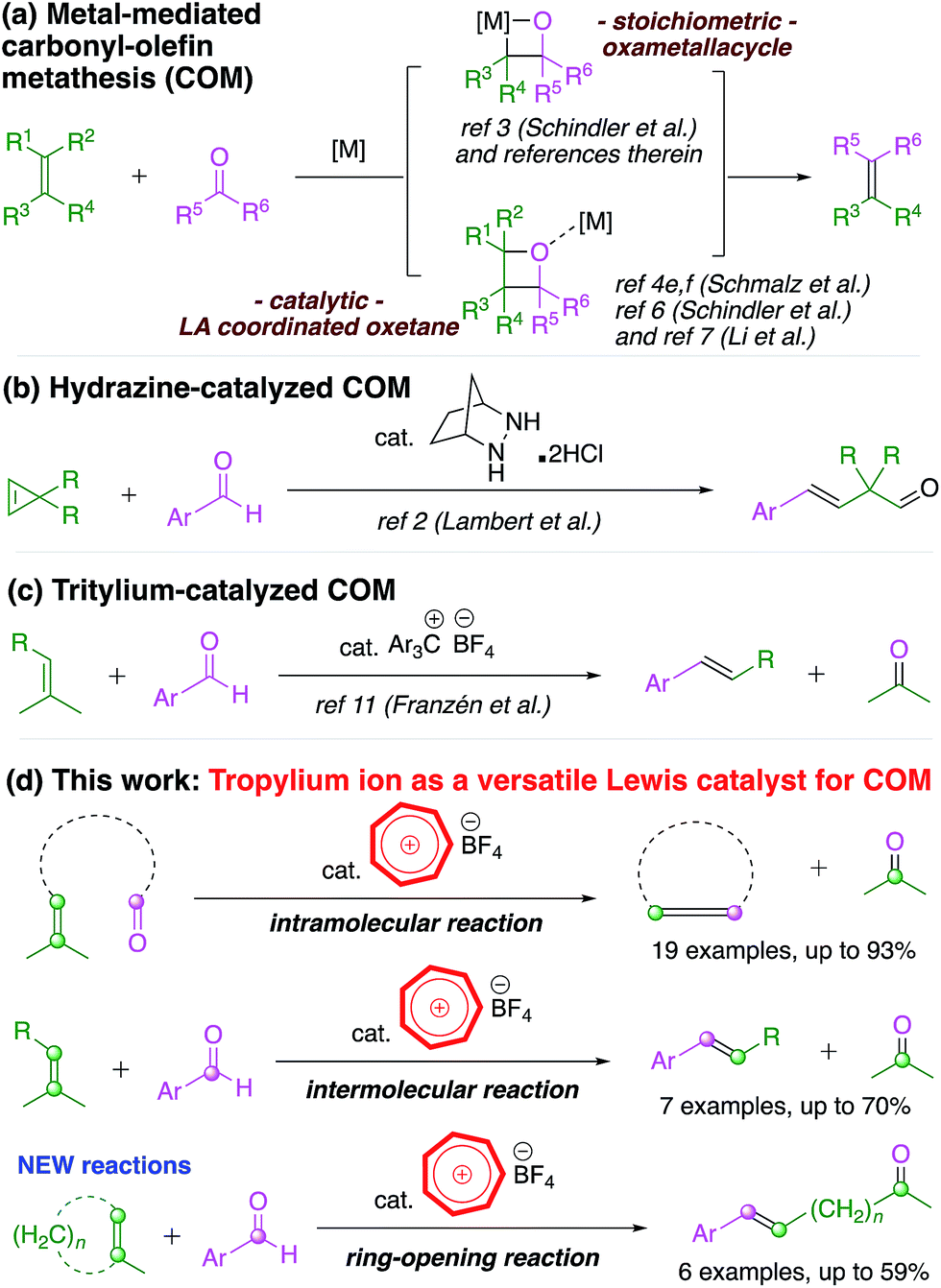 | ||
| Scheme 1 Carbonyl–Olefin Metathesis (COM) reactions. | ||
In the last two years, Schindler's group6 and Li's group7 reported elegant studies in which they utilized salts of iron(III), an abundant transition metal,8 to promote intramolecular cyclization COM reactions (Scheme 1a). However, the full potential of this chemical transformation3,9 has not been adequately studied for ring-opening10 (Scheme 1b) or intermolecular2,11 (Scheme 1c) COM reactions, which are typical variations of the well-studied olefin–olefin metathesis. Therefore, the substrate scope of the COM reaction needs to be expanded beyond intramolecular cyclization to deliver the prospective synthetic applications it invokes.3,9 Although iron(III) catalysts have enjoyed some success, the infancy status of this field beckons further exploratory work in developing a more diverse catalyst pool for the COM reaction.
Based on our previous work on the aromatic tropylium ion,12 we envisage that tropylium could be a suitable organocatalyst for the carbonyl–olefin metathesis reaction. The concept of using an organic compound as a promoter for this type of reaction became attractive after recent developments by Lambert's group using hydrazine2 and Franzén's group using tritylium salts.11 The former system catalyzed the reaction via the formation of covalently bonded hydrazonium intermediates13 while the latter catalyst activated the carbonyl compound via Lewis acid–base coordination. The electrophilicity of the unsubstituted tropylium ion, as reported by Mayr and co-workers, is comparable to that of tritylium ions stabilized by electron-donating substituents such as a methoxy group.14 The tropylium ion15 might therefore be a suitable Lewis acid catalyst with adequate electrophilicity and oxophilicity for the COM reaction.12e Gratifyingly, our study demonstrated that the tropylium ion could indeed be used as an organic Lewis acid catalyst to efficiently promote the carbonyl–olefin metathesis reaction with good to excellent outcomes on a broad range of substrates. This organocatalytic system is of particular interest for future developments in this field as it proves to be a universally versatile promoter for both inter- and intra-molecular reactions as well as the new ring-opening carbonyl–olefin metathesis.
Results and discussion
Intramolecular COM reactions with the tropylium catalyst
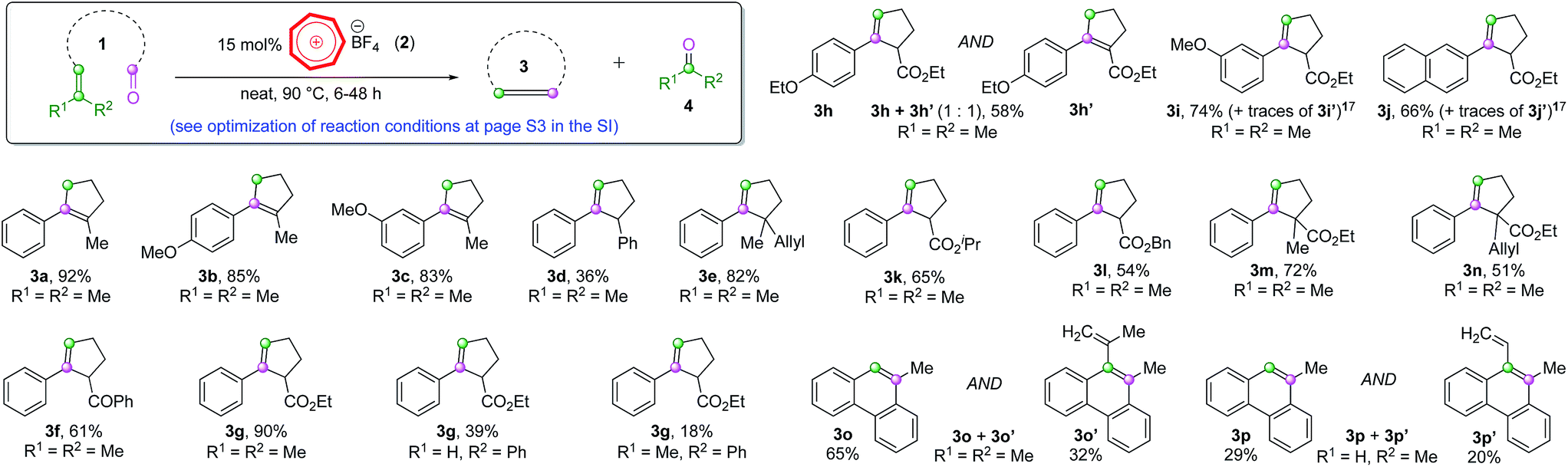
Our proof-of-concept study met with instant success for the cyclization COM reaction of substrate 1a (Scheme 2, also see Table S1 in page S3 in the ESI† for optimization of reaction conditions), a substrate known to work smoothly from Schindler's work employing iron(III) catalysts.6a The non-paramagnetic nature of the tropylium organocatalyst enabled us to follow the progress of this reaction by NMR spectroscopy as illustrated in Fig. 1 (also see Fig. S1 in the ESI for more details†). The conversion of a similar substrate (1b, also see Scheme 2) to 3b and acetone (4a, (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 2.2 ppm) over time was very clean and completed after ca. 48 h at 45 °C. Similar to Schindler's iron(III) catalytic reaction, 3b was formed as the thermodynamically favored olefin product.6a We subsequently applied the intramolecular COM reaction to a broad range of substrates (Scheme 2).16 Most tested precursors went through the tropylium-catalyzed COM reactions smoothly to afford the cyclized products in moderate to excellent yields.
O at 2.2 ppm) over time was very clean and completed after ca. 48 h at 45 °C. Similar to Schindler's iron(III) catalytic reaction, 3b was formed as the thermodynamically favored olefin product.6a We subsequently applied the intramolecular COM reaction to a broad range of substrates (Scheme 2).16 Most tested precursors went through the tropylium-catalyzed COM reactions smoothly to afford the cyclized products in moderate to excellent yields.
 | ||
| Scheme 2 Intramolecular COM reactions. | ||
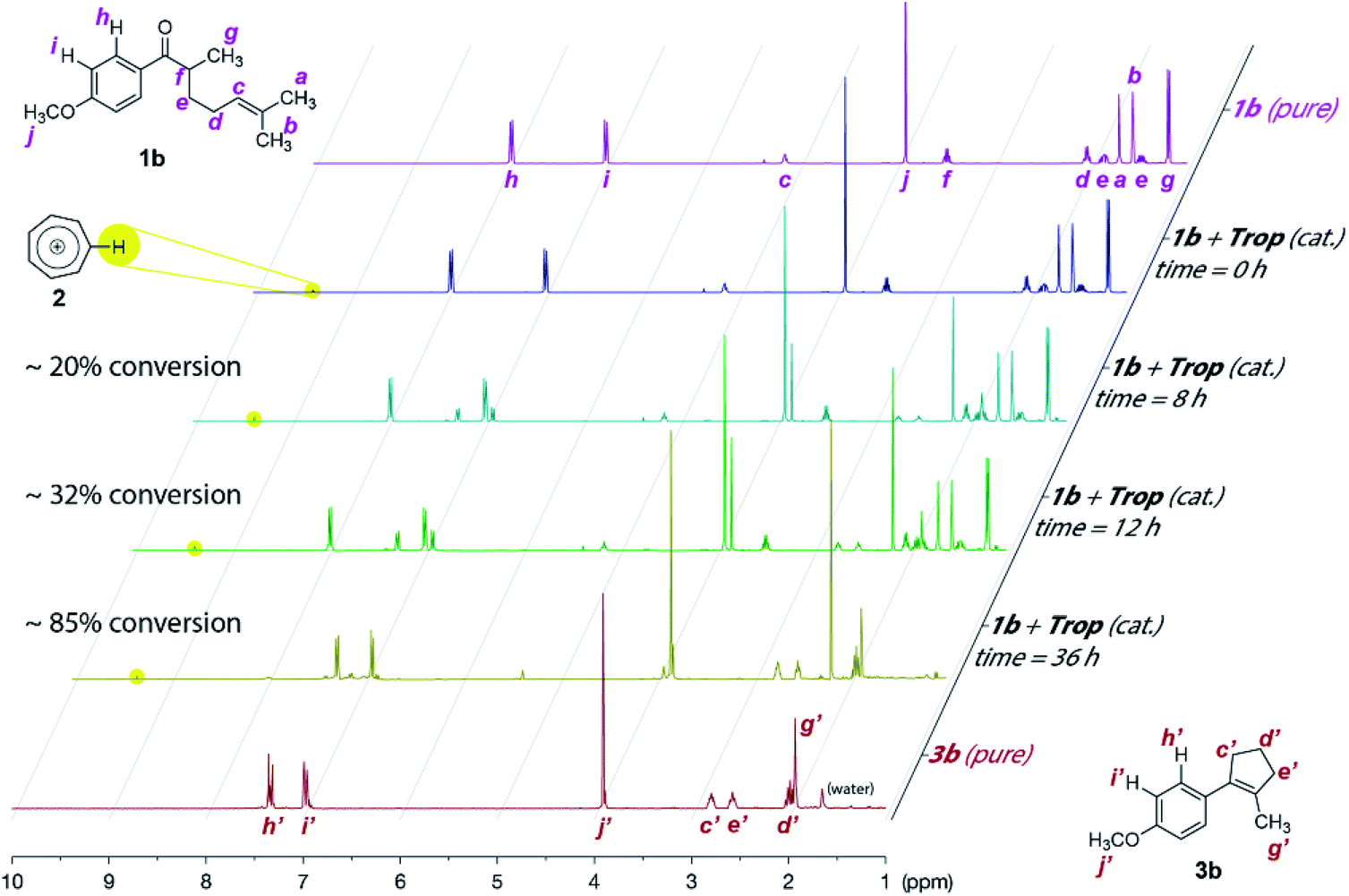 | ||
| Fig. 1 Progress of the tropylium-catalyzed intramolecular carbonyl–olefin metathesis reaction from 1b to 3b by 1H NMR studies (CD2Cl2, 25 °C). | ||
Notably, substrates with methyl substituents at the α-position to the original carbonyl group normally produced rearranged thermodynamically stable olefin products (3a–3c, Scheme 2). On the other hand, the replacement of Me with aryl groups or carbonyl/carboxyl moieties gave the normal ring-closing metathesis products (3d–3n, Scheme 2). Two biphenyl substrates gave the phenanthrene-type products (3o and 3p) with significant formation of the carbonyl-ene products (3o′ and 3p′, Scheme 2).17 These reaction outcomes and observations are comparable to those of iron(III)-catalyzed systems reported by Schindler and co-workers,6 hinting that these intramolecular COM reactions probably proceeded through some analogous pathways, despite being facilitated by two totally different catalysts.
The double bond isomerization (3a–3c) is presumably catalyzed by trace amounts of Brønsted acid that might form if moisture is present in the reaction mixture. However, when we carried out the reactions under very anhydrous conditions (using a glovebox), the same results were observed, implying that it might not be the case. Unfortunately, further control experiments with hindered proton scavengers (such as 2,6-dimethyl or 2,6-di(t-butyl) pyridines) in addition to our tropylium catalyst only led to low conversion of the starting material to the product. This was exactly the outcome for other control experiments where we used these pyridine additives with Schindler's iron(III) catalyst, which hinted that the pyridines interfered with the general COM reaction. We also found that the tropylium ion reacted directly with these pyridines to form the corresponding N-cycloheptatrienyl pyridinium salts,12g which ruled out the validity of these Brønsted pathway control experiments at this stage.
We observed a clear pattern in the reactivity of the different substrates studied in Scheme 2 that the reactions generally worked more efficiently when they produced acetone as a by-product (R1, R2 = Me). When the by-products were an aldehyde (R1, R2 = H) or an aromatic ketone (R1, R2 = Ph), there were dramatic decreases in product yields (3g and 3p, Scheme 2). We believe that the volatility of acetone might play a role in driving the reaction equilibrium to the productive pathway (also see entry 14, Table S1†). Another possibility is that the formation of acetone is particularly thermodynamically favored for the tropylium-catalyzed ring-closing carbonyl–olefin metathesis reactions, as such a phenomenon was also observed for Schindler's and Li's systems.6,7
Intermolecular COM reactions with the tropylium catalyst
Intermolecular carbonyl–olefin metathesis is arguably a more synthetically valuable or more versatile version of this reaction (also see Scheme 1). Thus far, there has been no report on a practical catalyst system to promote this type of process,3,9b,c including Schindler's6 and Li's7 iron(III) catalysts, except for the moderately successful tritylium-catalyzed reaction reported by Franzén and co-workers.11 We therefore believe that it would be a challenging but suitable reaction scope to probe the catalytic activity of our tropylium catalyst. We started our investigation by looking at the intermolecular carbonyl–olefin metathesis reaction between 2-naphthaldehyde (5a) and amylenes (6a). Pleasingly, a test reaction with 20 mol% tropylium tetrafluoroborate in acetonitrile at 90 °C afforded the desired product in promising yield (see page S16 in the ESI for more details†). Interestingly, the olefin product was formed predominantly as the trans-isomer,17 similar to the selectivity observed from Franzén's tritylium-catalyzed reactions.11 Encouraged by this initial success, further studies were carried out on the effect of solvent, reaction temperature, catalyst loading and ratio of reagents (also see page S16 in the ESI for more details†). The optimal reaction conditions for the intermolecular reaction involved an excess amount of the aldehyde 5a and 10 mol% tropylium catalyst in dichloromethane at 80 °C for 0.5 hour under pressurized microwave irradiation conditions to give the COM product 7a in 52% yield.Thus, we subsequently applied the optimal conditions developed to a family of aromatic aldehydes and isopropylidene-bearing olefin substrates (Scheme 3). Gratifyingly, most of the electron-rich, neutral or weakly electron-poor aldehyde substrates gave the target products in moderate to good yields (7a–7g, Scheme 3), confirming the feasibility and synthetic potential of the intermolecular carbonyl–olefin metathesis reactions with the tropylium catalyst. In contrast to the intramolecular reaction discussed above (see Scheme 2), this intermolecular reaction however did not seem to work with ketone substrates or strongly electron-poor aromatic aldehydes. Indeed, the replacement of 2-naphthaldehyde (5a) with substrates such as acetophenone (5k) or 2-acetonaphthone (5l) did not produce any observable formation of the expected metathesis products. Electron-deficient aryl aldehydes (5j–5l, Scheme 3) did not lead to any productive reaction outcomes either. Heteroaromatic aldehyde 5h formed an adduct with the tropylium ion at the nitrogen-centre and hence deactivated the reaction system. Aliphatic aldehydes also led to complicated reaction mixtures with some aldol and carbonyl-ene18 byproducts.
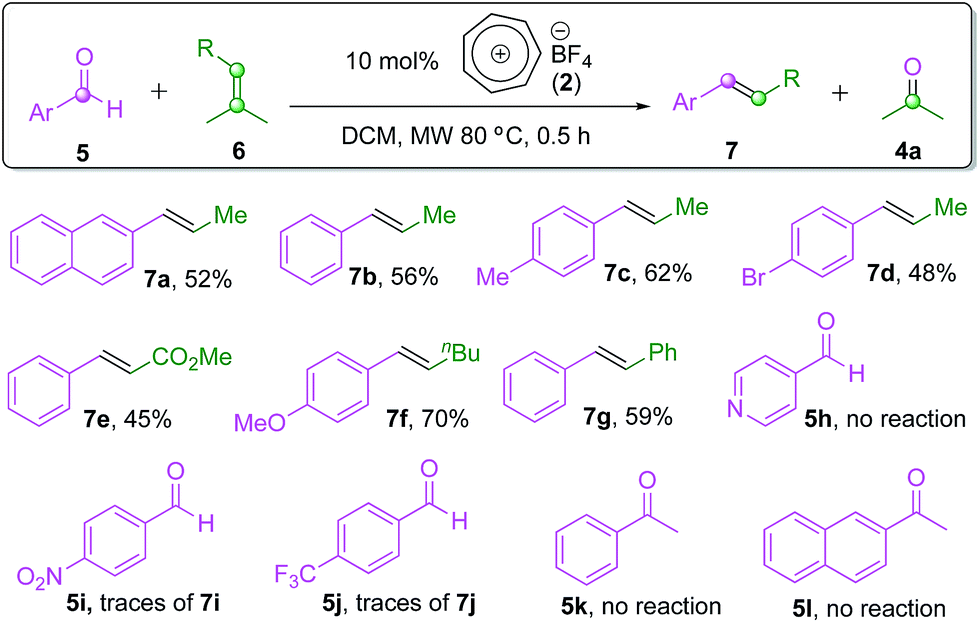 | ||
| Scheme 3 Intermolecular COM reactions. | ||
The vast difference between the inter- and intramolecular reactions is presumably due to the different coordinating affinity of the weakly Lewis acidic tropylium ion to these substrate systems, but a conclusive explanation cannot be easily derived. There are also several other factors limiting the efficiency of this reaction, such as the low boiling point of amylenes and their tendency to polymerize in the presence of the tropylium catalyst. Although these intermolecular COM reactions only give moderate success, they still provide an important benchmark for this research field, as this is only the second time11 the intermolecular COM reaction was investigated in a catalytic sense. Both the first study by the Franzén group with tritylium catalysts11 and our study with the tropylium catalyst have proven that it might be problematic to adapt the COM reaction to bimolecular systems. In terms of entropy change, the intermolecular COM reaction (two molecules react to form another two molecules) is presumably less favourable than the intramolecular version (one molecule cyclizes to form two products).
Intermolecular ring-opening COM reactions
While both catalytic intramolecular and intermolecular COM reactions have been recently realized by seminal contributions from the Schindler and Li groups6,7 and the Franzén group11 respectively, the ‘intermolecular’ ring-opening carbonyl–olefin metathesis reaction10a has been rather neglected. An elegant study by Lambert and co-workers exploited a hydrazine organocatalytic system to promote this type of reaction between aromatic aldehydes and ring-strained cyclopropene substrates.2 These reactions, however, followed a totally different metathesis paradigm involving hydrazonium intermediates and subsequent [2 + 3] cycloaddition followed by rearrangement to form the products. Therefore, it is of great interest to examine the potential of our tropylium catalyst as an organo-Lewis acid promoter for the ring-opening COM reaction in a similar manner to intra- and intermolecular reactions discussed earlier in this work.We focus the preliminary study on the tropylium-catalyzed intermolecular ring-opening reactions (Scheme 4) of some readily accessible cycloalkenes with aromatic aldehyde 5c, which proved to be a good substrate for the intermolecular COM reaction (Scheme 3). Interestingly, six-membered and seven-membered cycloalkenes (8h–8k, n = 4 or 5, Scheme 4) did not metathesize to the target products while 1-methyl cyclopentene 8g reacted smoothly to give the product 9g2 in good yield (Scheme 4). It seems that six- and seven-membered cycloalkenes probably do not possess the ring-strain necessary for the ring-opening reaction. Furthermore, a methyl substituent on the C–C double bond also helps to promote the metathesis reaction in a similar way to how acetone formation is favored for the normal intra- and inter-molecular reactions. This phenomenon is clearly demonstrated in Scheme 4 with n = 3, where other substituted or non-substituted cyclopentenes 8d–8f did not react. We used this knowledge to prepare and test the ring-opening COM reaction with 1-methylcyclobutene 8c (n = 2) and 1-methylcyclopropene 8a (n = 1). These two substrates gave promising results, however the efficiency of the reactions was severely affected by their high volatility. Pressurized reaction conditions to prevent substrate evaporation led to other issues with side oligomerization reactions, which need to be addressed by further work in reaction design. Highly substituted cyclopropene 8b, a substrate that we had access to from another project in our group, did not show any reactivity.
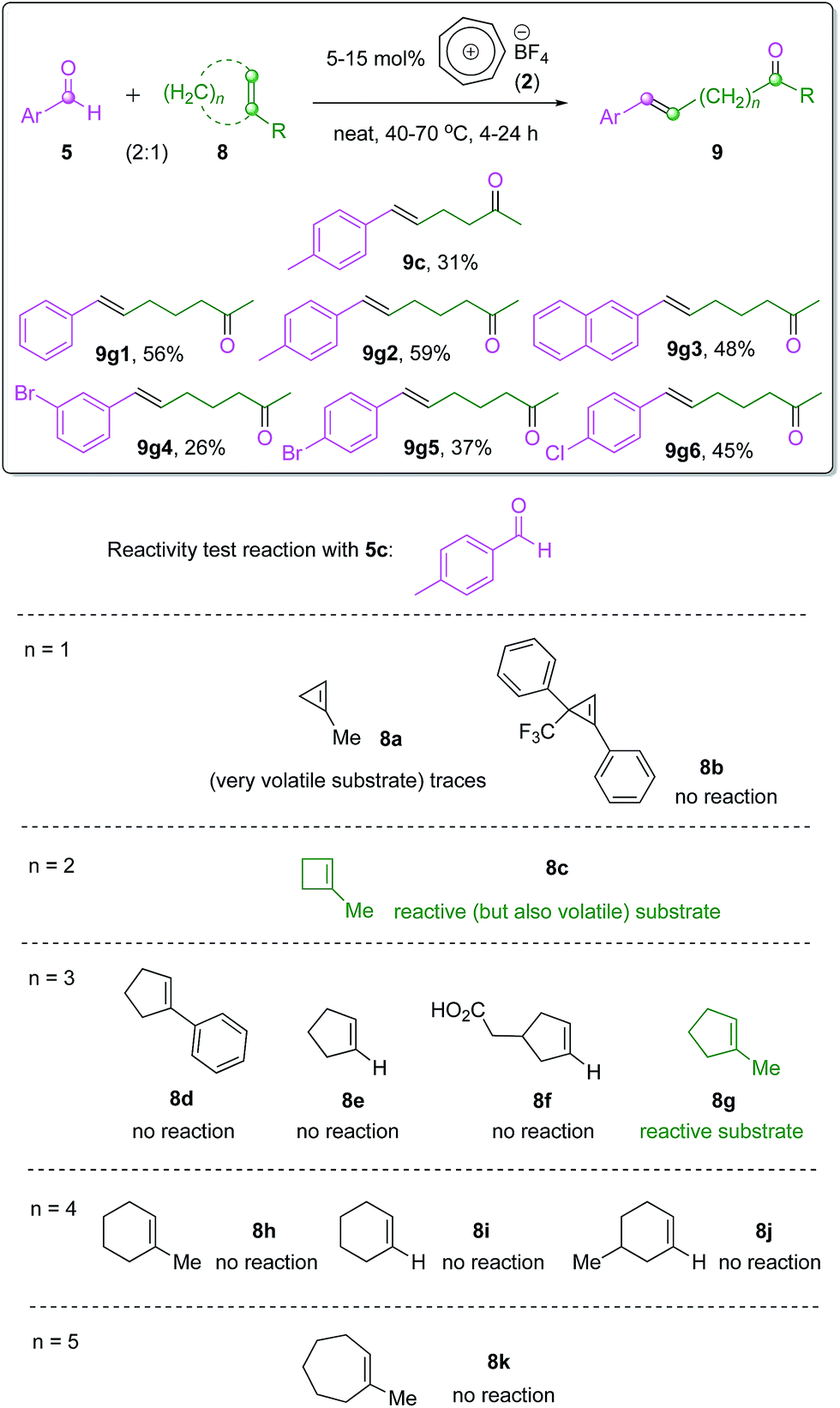 | ||
| Scheme 4 Intermolecular ring-opening COM reactions. | ||
We subsequently used substrate 8c and substrate 8g to prepare a range of ω-enone products with moderate to good yields via these newly developed ring-opening intermolecular COM reactions (Scheme 4) with aromatic aldehydes. Notably, all the products were formed with excellent trans-stereoselectivity. A full investigation on the ring-opening metathesis reactions with other types of cycloalkene substrates is currently underway.
Comparison of catalytic activity between tropylium, tritylium and iron(III) Lewis acid catalysts – role of Brønsted acids
As discussed above for Scheme 3, the intermolecular COM reaction seems to be the bottleneck of development in this field. We were curious to see if iron(III) catalysts from the Schindler6 and Li7 groups, so far only known to catalyze the intramolecular version, could address this issue. At the same time, there is also a question of whether or not Franzén's tritylium catalysts11 can catalyze the ring-closing intramolecular COM reaction. To examine these questions and also to probe the efficiency of our tropylium catalytic system (2) compared to the known Schindler's iron(III)6 and Franzén's tritylium11 catalysts, we carried out a comparative study on the intramolecular COM reaction of substrate 1g (Scheme 5a) and the intermolecular COM reaction of substrate 5a (Scheme 5b). Under the optimal reaction conditions developed by Schindler and Franzén for their catalytic systems6,11 against our catalyst, it was interesting to find out that the tropylium ion can act efficiently for both intra/inter-molecular reactions while iron(III) chloride and the tritylium ion seem to be good catalysts for only one type of reaction but not the other (Scheme 5).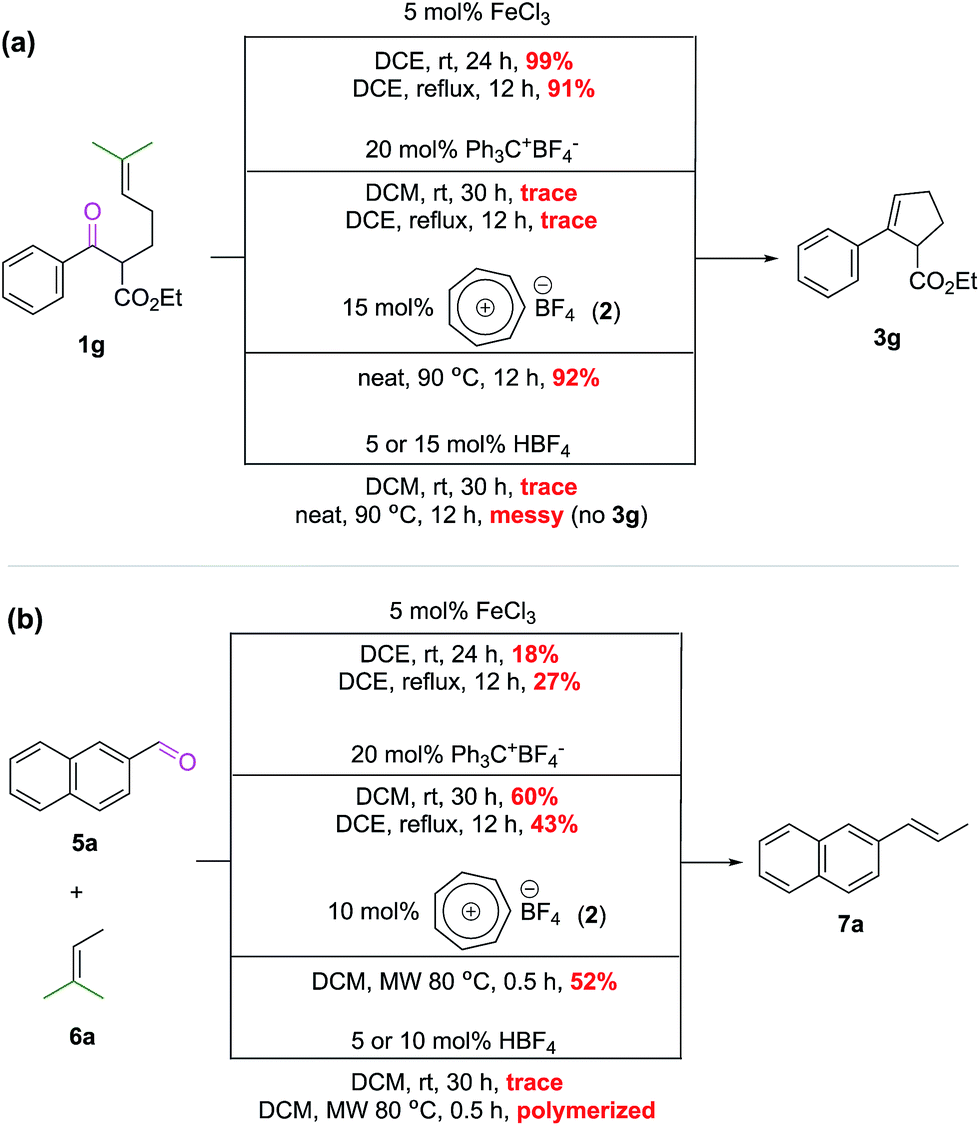 | ||
| Scheme 5 Catalytic activity comparison. | ||
It is not surprising to see that the bulky tritylium ion failed to facilitate the sterically demanding intramolecular reaction; but to why the iron(III) catalyst performed poorer than expectation for the intermolecular reaction9b would require further investigation. This comparison is obviously imperfect, as it does not take into account the reaction temperature and reaction time as well as catalyst loading. It is, however, indicative of the versatility of the tropylium catalyst where its electronic and steric properties in combination with oxophilicity/Lewis acidity are suitable for both inter- and intramolecular COM reactions.
As tropylium salts and many other Lewis acids (including FeCl3) might react with moisture present in the reaction system to produce strong Brønsted acids, it is necessary to confirm that Brønsted acids, if any, are not interfering with the COM reactions. Thus, we also carried out some control studies in which HBF4 was used as a potential catalyst at different loadings. These reactions were performed under various conditions for both inter- and intramolecular COM reactions (Scheme 5) but all led to non-productive outcomes. A simple Brønsted acid catalytic pathway is unlikely to be productive for the COM reaction as the Tiefenbacher group have recently discovered in their interesting supramolecule-assisted COM study.19 Therefore, it can be concluded that the Lewis acidity of tropylium catalyst 2 indeed plays a crucial role in promoting the COM reactions.
Mechanistic studies
Mechanistically, these metathesis reactions involve a non-photochemical [2 + 2] cycloaddition and [2 + 2] cycloreversion, each of which could proceed via a concerted or stepwise pathway. The former is forbidden by orbital symmetry rules and would normally entail a barrier that is too high to overcome through thermal activation. In recent studies by Schlinder and co-workers,6a–c the authors demonstrated that Lewis acidic FeCl3 could stabilize the zwitterionic intermediate (through coordination to the carbonyl oxygen) and provided compelling experimental and computational evidence to support an asynchronous concerted pathway.To better understand the catalytic role of tropylium, we have carried out high-level ab initio calculation (G3(MP2)-RAD)20 in conjunction with the SMD implicit solvent model21 to compare the energetics of three COM pathways: (1) in the absence of the tropylium ion, (2) aldehyde hydrogen bonded to the tropylium ion, and (3) coordination of aldehyde to the tropylium ion (see page S26 in the ESI† for 1H NMR spectroscopic evidence and computational studies of tropylium–carbonyl interactions). For pathways (1) and (2), the reactants and products are connected by two concerted cycloaddition transition states and a cycloaddition intermediate (Fig. 2), whilst pathway (3) involves four stepwise transition states and additionally two zwitterionic intermediates (Fig. 3). Consistent with orbital symmetry rules, both pathways (1) and (2) are accompanied by very high barriers (TS1 and TS2) exceeding 200 kJ mol−1 and are unlikely to occur during thermal activation. As shown in Fig. 2, it is also interesting to note that H-bonding to the tropylium ion does not provide any stabilization of the transition states. Presumably, the concerted nature of these transition states (no charged intermediates) also means that any electrostatic stabilization from tropylium is likely to be minimal.
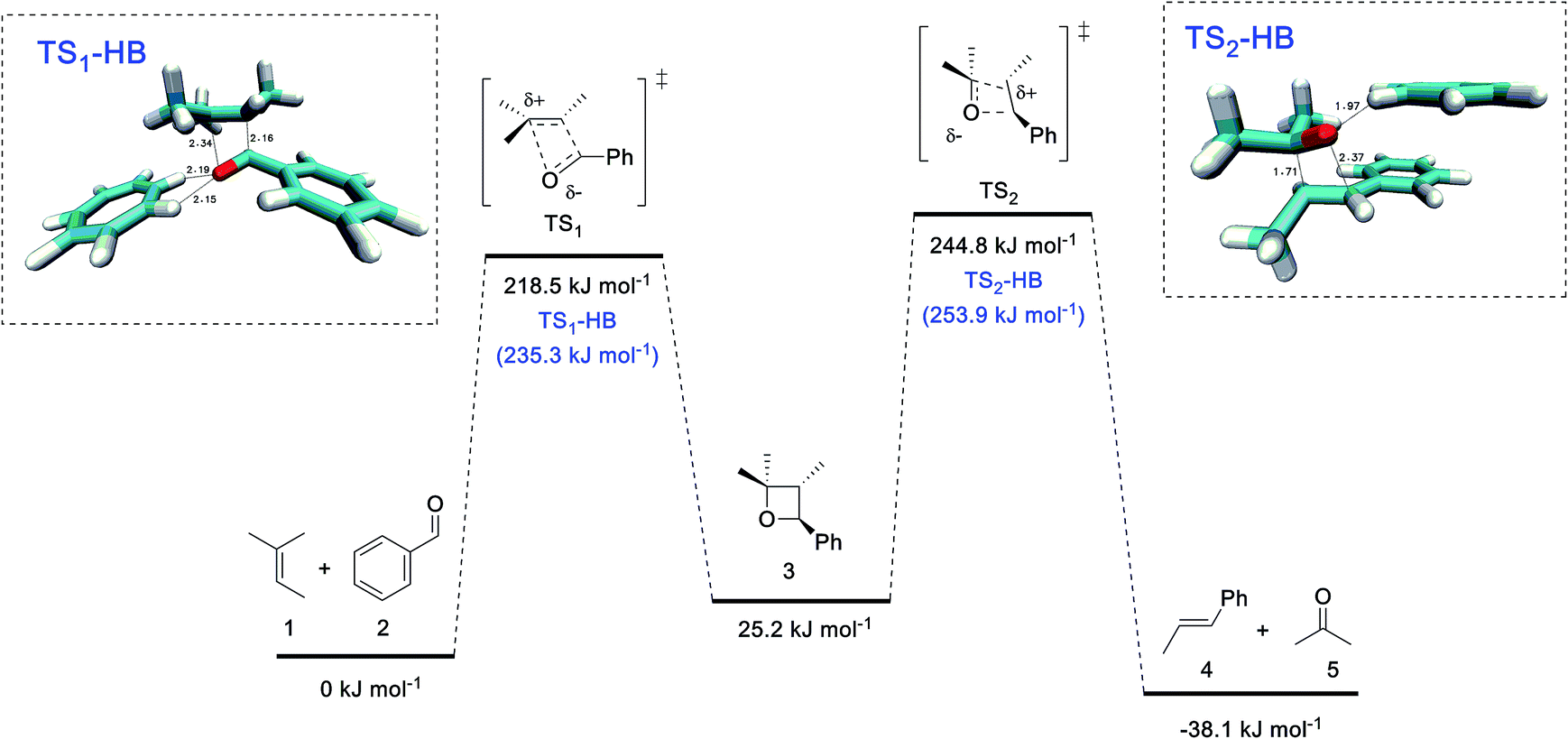 | ||
| Fig. 2 G3(MP2)-RAD + SMD(DCM) free energies for reactions in the absence of tropylium and with H-bonding to tropylium. The barriers for the latter are shown in parentheses. | ||
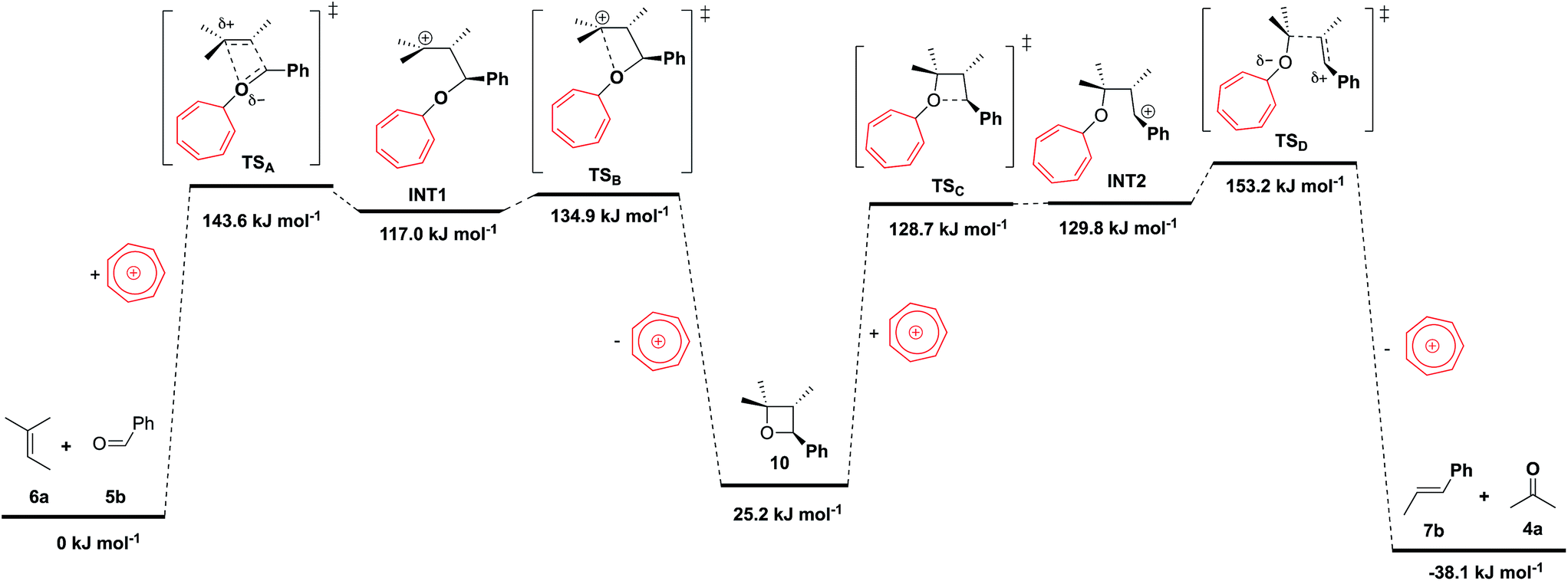 | ||
| Fig. 3 G3(MP2)-RAD + SMD(DCM) free energies (at 298 K) for reactions catalyzed by coordination of CO oxygen to tropylium. | ||
Fig. 3 shows the free energy profile for the stepwise pathway, and it is evident that coordination of the anionic oxygen to the tropylium ion lowers the barriers significantly. Specifically, the rate-limiting step for this pathway is about 90 kJ mol−1 lower compared to that of the reaction in the absence of the tropylium ion (153 cf. 245 kJ mol−1). This result is somewhat surprising because coordination to oxygen to form the heptatriene adduct inevitably disrupts the aromaticity of the tropylium ring. Presumably, this enthalpic cost is more than compensated when the anionic oxygen is neutralized through coordination to tropylium.
The structures of the stepwise transition states hint at a termolecular mechanism, although intrinsic reaction coordinate (IRC) simulations of these transition states show that they relax to reactants and products where the tropylium ion remains coordinated. On the other hand, potential energy scans indicate that addition of CO oxygen to tropylium (while the remaining atoms are constrained to positions at the transition state geometry) is approximately barrier-less (see ESI†). A plausible mechanistic picture is that tropylium exists as a π-stacked complex with aldehyde 5b (cf. Table S3† configuration C, page S28 in the ESI†), which spontaneously coordinates to the C–O oxygen as the anionic charge develops upon nucleophilic addition. Indeed, our kinetic experiments show that the rate of metathesis is first-order with respect to the concentration of the tropylium ion.17 It is also worth pointing out that the computed barriers in Fig. 3 are likely to represent the upper bound estimates of the actual values. This is because these reactions involve the consumption of an aromatic cation (tropylium) and the generation of a localized carbocation, so the solvation contribution is likely to be under-estimated by quantum chemical implicit solvation models.21b Regardless, it is clear from the calculations that the reaction is significantly enhanced only when tropylium acts as a Lewis acid to stabilize the zwitterionic intermediate formed in the stepwise pathway.
Conclusions
We have developed a novel catalytic system employing the tropylium ion as an organo-Lewis acid to promote the carbonyl–olefin metathesis reaction. The carbonyl–olefin metathesis reaction has always been considered a highly valuable chemical transformation but much less explored than its analogous olefin–olefin metathesis. We have demonstrated that the tropylium ion can efficiently catalyze this type of reaction on a broad range of substrates, which are applicable to both intramolecular and intermolecular reactions as well as the ring-opening metathesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Australian Research Council (grant DE150100517 awarded to T.V.N. and grant DE160100807 awarded to J.H.). J.H. acknowledges the Australian NCI and Intersect NSW for generous allocation of supercomputer resources.Notes and references
- (a) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef; (b) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef; (c) Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed; (d) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef PubMed; (e) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef PubMed; (f) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef PubMed.
- A. K. Griffith, C. M. Vanos and T. H. Lambert, J. Am. Chem. Soc., 2012, 134, 18581–18584 CrossRef PubMed.
- J. R. Ludwig and C. S. Schindler, Synlett, 2017, 28, 1501–1509 CrossRef.
- (a) H. A. J. Carless and H. S. Trivedi, J. Chem. Soc., Chem. Commun., 1979, 382–383 RSC; (b) I. Schopov and C. Jossifov, Makromol. Chem., Rapid Commun., 1983, 4, 659–662 CrossRef; (c) A. C. Jackson, B. E. Goldman and B. B. Snider, J. Org. Chem., 1984, 49, 3988–3994 CrossRef; (d) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 3800–3801 CrossRef; (e) H.-P. van Schaik, R.-J. Vijn and F. Bickelhaupt, Angew. Chem., Int. Ed., 1994, 33, 1611–1612 CrossRef; (f) V. A. Khripach, V. N. Zhabinskii, A. I. Kuchto, Y. Y. Zhiburtovich, V. V. Gromak, M. B. Groen, J. van der Louw and A. de Groot, Tetrahedron Lett., 2006, 47, 6715–6718 CrossRef; (g) N. Slavov, J. Cvengroš, J.-M. Neudörfl and H.-G. Schmalz, Angew. Chem., Int. Ed., 2010, 49, 7588–7591 CrossRef PubMed; (h) A. Soicke, N. Slavov, J.-M. Neudörfl and H.-G. Schmalz, Synlett, 2011, 2011, 2487–2490 CrossRef.
- (a) J. R. Stille and R. H. Grubbs, J. Am. Chem. Soc., 1986, 108, 855–856 CrossRef; (b) J. R. Stille, B. D. Santarsiero and R. H. Grubbs, J. Org. Chem., 1990, 55, 843–862 CrossRef; (c) K. C. Nicolaou, M. H. D. Postema and C. F. Claiborne, J. Am. Chem. Soc., 1996, 118, 1565–1566 CrossRef; (d) J. D. Rainier, S. P. Allwein and J. M. Cox, J. Org. Chem., 2001, 66, 1380–1386 CrossRef PubMed; (e) U. Majumder and J. D. Rainier, Tetrahedron Lett., 2005, 46, 7209–7211 CrossRef; (f) K. Iyer and J. D. Rainier, J. Am. Chem. Soc., 2007, 129, 12604–12605 CrossRef PubMed; (g) S. T. Heller, T. Kiho, A. R. H. Narayan and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 11129–11133 CrossRef PubMed; (h) B. Hong, H. Li, J. Wu, J. Zhang and X. Lei, Angew. Chem., Int. Ed., 2015, 54, 1011–1015 CrossRef PubMed.
- (a) J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef PubMed; (b) J. R. Ludwig, S. Phan, C. C. McAtee, P. M. Zimmerman, J. J. Devery and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 10832–10842 CrossRef PubMed; (c) C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef PubMed; (d) E. J. Groso, A. N. Golonka, R. A. Harding, B. W. Alexander, T. M. Sodano and C. S. Schindler, ACS Catal., 2018, 8, 2006–2011 CrossRef.
- L. Ma, W. Li, H. Xi, X. Bai, E. Ma, X. Yan and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 10410–10413 CrossRef PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- (a) A.-L. Lee, Angew. Chem., Int. Ed., 2013, 52, 4524–4525 CrossRef PubMed; (b) E. T. Hennessy and E. N. Jacobsen, Nat. Chem., 2016, 8, 741–742 CrossRef PubMed; (c) C. Saá, Angew. Chem., Int. Ed., 2016, 55, 10960–10961 CrossRef PubMed.
- (a) G. Jones, M. A. Acquadro and M. A. Carmody, J. Chem. Soc., Chem. Commun., 1975, 206–207 RSC; (b) C. Jossifov and R. Kalinova, in Green Metathesis Chemistry, ed. V. Dragutan, A. Demonceau, I. Dragutan and E. S. Finkelshtein, Springer Netherlands, Dordrecht, 2010, pp. 305–314 Search PubMed.
- N. Veluru Ramesh, J. Bah and J. Franzén, Eur. J. Org. Chem., 2015, 1834–1839 CrossRef.
- (a) T. V. Nguyen and A. Bekensir, Org. Lett., 2014, 16, 1720–1723 CrossRef PubMed; (b) T. V. Nguyen and M. Hall, Tetrahedron Lett., 2014, 55, 6895–6898 CrossRef; (c) T. V. Nguyen and D. J. M. Lyons, Chem. Commun., 2015, 51, 3131–3134 RSC; (d) D. J. M. Lyons, R. D. Crocker, M. Blümel and T. V. Nguyen, Angew. Chem., Int. Ed., 2017, 56, 1466–1484 CrossRef PubMed; (e) D. J. M. Lyons, R. D. Crocker, D. Enders and T. V. Nguyen, Green Chem., 2017, 19, 3993–3996 RSC; (f) G. Oss, S. D. de Vos, K. N. H. Luc, J. B. Harper and T. V. Nguyen, J. Org. Chem., 2018, 83, 1000–1010 CrossRef PubMed; (g) G. Oss, J. Ho and T. V. Nguyen, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201800579.
- X. Hong, Y. Liang, A. K. Griffith, T. H. Lambert and K. N. Houk, Chem. Sci., 2014, 5, 471–475 RSC.
- (a) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef PubMed; (b) S. Minegishi and H. Mayr, J. Am. Chem. Soc., 2003, 125, 286–295 CrossRef PubMed; (c) M. Horn and H. Mayr, Chem.–Eur. J., 2010, 16, 7478–7487 CrossRef PubMed; (d) M. Horn and H. Mayr, Eur. J. Org. Chem., 2011, 6470–6475 CrossRef; (e) M. Horn and H. Mayr, J. Phys. Org. Chem., 2012, 25, 979–988 CrossRef; (f) M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem.–Eur. J., 2013, 19, 249–263 CrossRef PubMed.
- (a) C. Jandl, D. C. Mayer and A. Pöthig, Eur. J. Org. Chem., 2017, 2017, 4255–4259 CrossRef; (b) C. Jandl and A. Pothig, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2017, 73, 810–813 Search PubMed.
- Intramolecular reactions involved substrates with aldehyde groups gave poor conversion and complicated reaction mixtures.
- See the ESI for more details†.
- J. Lv, Q. Zhang, X. Zhong and S. Luo, J. Am. Chem. Soc., 2015, 137, 15576–15583 CrossRef PubMed.
- L. Catti and K. Tiefenbacher, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201712141.
- D. J. Henry, M. B. Sullivan and L. Radom, J. Chem. Phys., 2003, 118, 4849–4860 CrossRef.
- (a) J. Ho and M. Z. Ertem, J. Phys. Chem. B, 2016, 120, 1319–1329 CrossRef PubMed; (b) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra and Gaussian coordinates. See DOI: 10.1039/c8sc00907d |
| This journal is © The Royal Society of Chemistry 2018 |