
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular access to functionalized 5–8–5 fused ring systems via a photoinduced cycloisomerization reaction†
Anna E.
Salvati
,
James A.
Law
,
Josue
Liriano
and
James H.
Frederich
 *
*
Department of Chemistry and Biochemistry, Florida State University, 95 Cheiftan Way, Tallahassee, FL 32306, USA. E-mail: frederich@chem.fsu.edu
First published on 25th May 2018
Abstract
A 5–8–5 carbocyclic ring system forms the core of over 30 distinct natural products. Several members of this family have gained attention for their diverse activity in cell culture. In these cases, biological function is mediated by the arrangement of substituents around a conserved 5–8–5 nucleus. Despite the potential applications of this privileged substructure in medicinal chemistry, modular strategies for its assembly are underdeveloped. Herein, we describe a cycloisomerization reaction that forms the 5–8–5 framework directly. This strategy uniquely allows access to gram quantities of this valuable scaffold in four steps.
Introduction
The dicyclopenta[a,d]cyclooctene (5–8–5) ring system is central to over 30 di- and sesterterpene natural products.1 This molecular framework is produced by an array of plants and microorganisms,2 and several members of this terpene family possess activity in human cell culture (Scheme 1). For example, fusicoccin A (1) functions as an orthosteric stabilizer of 14–3–3 protein–protein interactions (PPIs).3,4 Similarly, ophiobolin A is a potent cytotoxin that modulates calmodulin activity,5 and cyclooctatin inhibits lysophospholipases.6,7 Intriguingly, the avidity of these natural products for their disparate biological targets is tightly linked to both the identity and arrangement of peripheral groups surrounding a common 5–8–5 core (i.e.2). Taken together, these observations indicate that substructure 2 is a privileged scaffold capable of serving as a ligand for a diverse group of receptors.8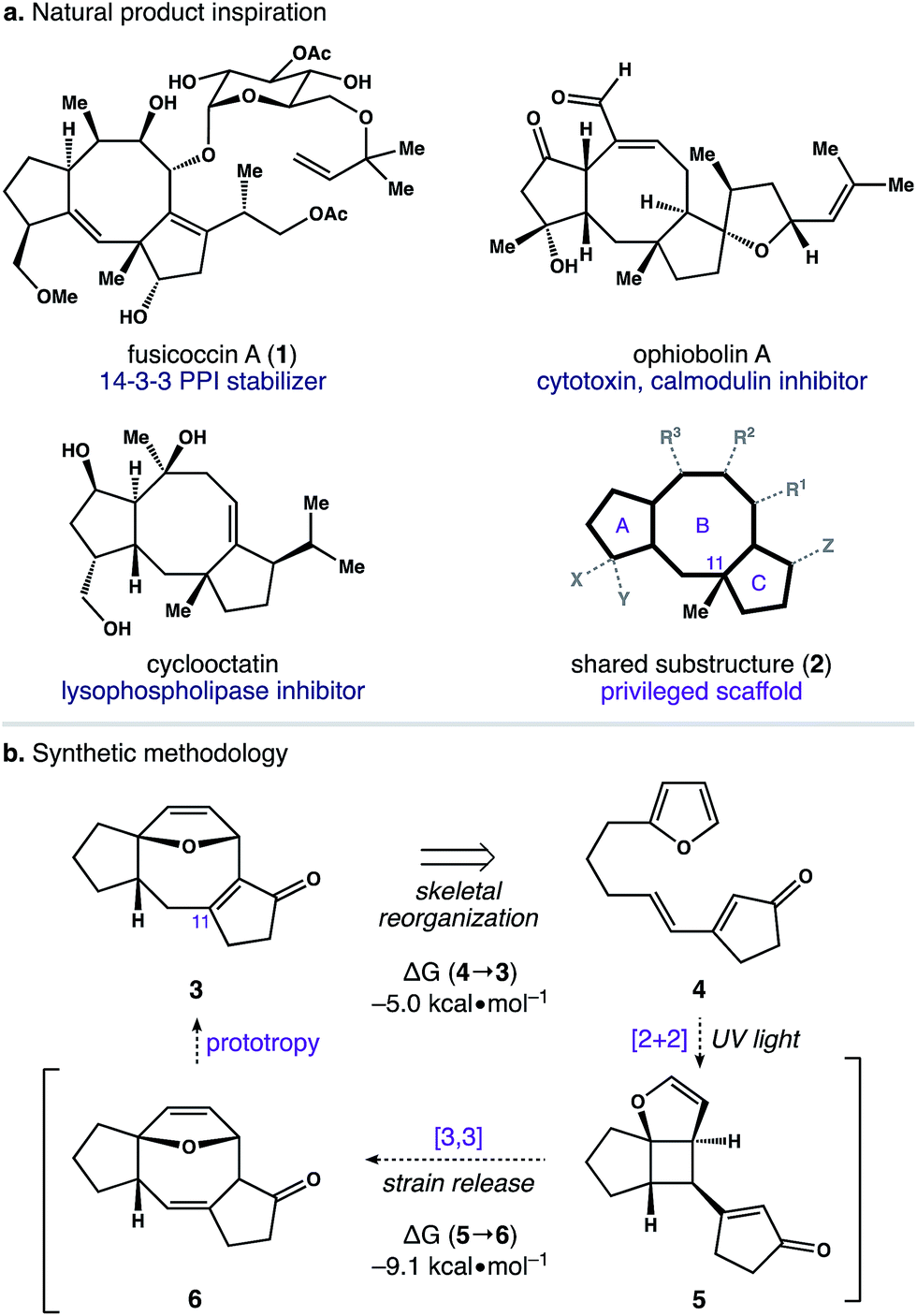 | ||
| Scheme 1 Our design of an efficient and modular entry point to 5–8–5 fused ring systems. | ||
Despite the therapeutic potential of molecules possessing a 5–8–5 nucleus, this motif is unrepresented in drug discovery libraries, primarily because of a lack of modular chemistry to prepare such structures.9 Several target-specific approaches to 5–8–5 ring systems have emerged from total synthesis.10,11 In contrast, more general methods to access this chemotype rely on cycloadditions to construct the eight-membered ring.12,13 Among these, photocycloadditions of pyridones14 and pyrones15 provide direct access to 5–8–5 scaffolds. However, a major limitation of existing strategies is the need for protracted, multi-step synthesis to prepare the requisite cycloaddition precursors. In addition, many reported methods require high temperatures and/or transition-metal catalysis to promote the desired chemistry. Taken together, these features hinder the widespread application of current cycloaddition approaches to 5–8–5 ring systems and the structural diversity they can achieve.
With an eye toward exploiting the potential pharmacology of substructure 2, we set out to develop a route to this molecular architecture that would facilitate straightforward diversification of functional groups surrounding the 5–8–5 nucleus. Along these lines, we considered ring system 3 and the possibility of preparing this motif in one step from more readily accessible isomer 4. A preliminary computational study revealed a thermodynamic driving force of 5.0 kcal mol−1 for the net isomerization of 4 to 3. As such, we envisioned a pathway connecting these isomers initiated by a [2 + 2] photocycloaddition within 4 to give stereodefined cyclobutane 5. We reasoned that ring strain amassed within this polycyclic system might then be used to promote a mild Cope rearrangement to generate cyclooctadiene 6.16 Subsequent isomerization of 6 to conjugated enone 3 could then terminate the transformation and provide a functional handle to install the key C11 quaternary methyl group. Herein, we report the development of this photoinduced cycloisomerization strategy and demonstrate its application as a versatile entry point to scaffold 2.
Results and discussion
Our first objective was to develop a concise route to photosubstrate 4 that was flexible and amenable to scale. As shown in Scheme 2, we began by establishing a large-scale protocol for the alkylation of 5-iodopentyne (7) with 2-furyllithium to give 8 in 91% yield. Subsequent exposure of 8 to Schwartz's reagent formed vinyl zirconium 9. This species was then added to a solution of cyclopentenone and catalytic amounts of [Rh(cod)Cl]2 and (±)-BINAP in THF.17 Warming the resulting solution to 30 °C for 2 h formed zirconium enolate 10, which was cooled to 0 °C and exposed to N-tert-butylbenzenesulfinimidoyl chloride18 to afford 4 in 82% yield. This two-operation sequence allowed us to prepare gram quantities of 4 in a single pass. Moreover, the components within this assembly scheme could be readily varied to generate derivatives of 4 for subsequent studies. | ||
| Scheme 2 Assembly of photosubstrate 4. | ||
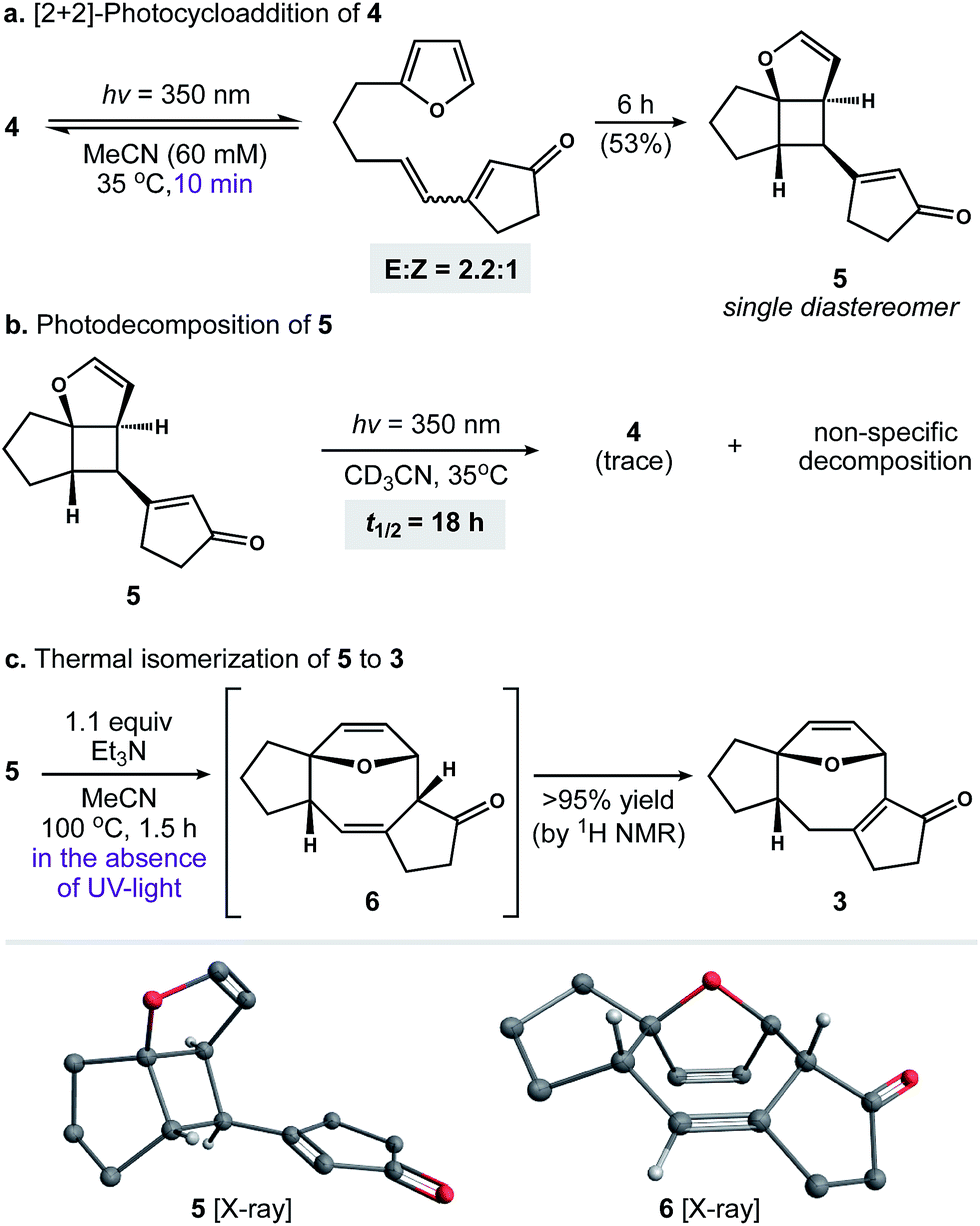
With photosubstrate 4 in hand, we turned our attention toward implementing the chemistry shown in Scheme 1b. We quickly found that exposing solutions of 4 to UV-light (hν = 350 nm) in a Rayonet photoreactor generated cyclobutane 5.19 After extensive experimentation, we identified MeCN and n-BuOH as optimal solvents for the [2 + 2] photocycloaddition. To our delight, irradiation of 4 in MeCN (60 mM, 35 °C) furnished 5 as a single diastereomer (Scheme 3a). This reactive species was isolated as a crystalline solid in 53% yield. The remaining mass balance of the reaction was largely an isomeric mixture of E- and Z-4. Further analysis revealed that UV-light promoted isomerization of the acyclic alkene within 4. This reaction reached equilibrium within 10 min (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio = 2.2:1, see the ESI for details†).20 Importantly, increasing the photoreaction time to 12 h markedly increased the extent of decomposition, resulting in a low yield of 5. Similarly, the addition of triplet sensitizers (e.g. benzophenone) to the reaction media had no beneficial effect.
:Z ratio = 2.2:1, see the ESI for details†).20 Importantly, increasing the photoreaction time to 12 h markedly increased the extent of decomposition, resulting in a low yield of 5. Similarly, the addition of triplet sensitizers (e.g. benzophenone) to the reaction media had no beneficial effect.
 | ||
| Scheme 3 Mechanistic experiments. | ||
These observations prompted us to examine the stability of 5. Accordingly, exposure of 5 to optimized photochemical conditions (hν = 350 nm, 60 mM CD3CN, 35 °C) resulted in slow decomposition and trace cycloreversion to 4 over 24 h (Scheme 3b, t1/2 = 18 h). Alternatively, when 5 was protected from light and heated above 35 °C we observed gradual formation of cyclooctadiene isomers 6 and 3. After further experimentation, we found that 5 rearranged cleanly to 6 when heated to 100 °C for 1.5 h.21 Subsequent isomerization of 6 to enone 3 occurred slowly under neutral conditions, allowing us to crystallize 6 from toluene. In contrast, exposure of 6 to Et3N or silica gel promoted the rapid formation of 3. Capitalizing on this observation, we found that 5 rearranged to 3 in quantitative yield when heated to 100 °C in the presence of Et3N (Scheme 3c).22 Taken together, these experiments establish a stereocontrolled mechanism for the cycloisomerization of 4 to fused 5–8–5 carbotricycle 3 by way of cyclobutane 5.
Our next objective was to develop a procedure to convert 4 to ring system 3 in a single operation. Results from our optimization studies are summarized in Table 1. We began by comparing reactions carried out on 0.5 mmol scale (108 mg of 4). The best results were achieved using a two-stage protocol, wherein a solution of 4 in MeCN (60 mM) was reacted at 35 °C in the Rayonet. After 12 h, the reaction mixture was moved to the bench-top, treated with Et3N (1.1 equiv.), and warmed to 100 °C for 6 h. This procedure afforded 3 in 51% yield along with 26% of unreacted 4 (Table 1, entry 1). We found that n-BuOH was a practical alternative for MeCN, providing 3 in 67% yield (entry 2). Importantly, addition of Et3N to the reaction was required to suppress formation of side product 11 (entry 3). We speculate that 11 is formed from intermediate 6via competitive ring-opening of the dihydrofuran and 6π electrocyclization within the resultant cyclooctatriene.23
|
|
||||
|---|---|---|---|---|
| Entry | Modificationa,b,c | Timed (h) | 3 (%) | Recovered 4e (%) |
| a Photochemistry was carried out using 24 W UV-lamps. b Reactions performed in a 50 mL quartz flask on 0.5 mmol scale, unless otherwise noted. c Gram-scale reactions were carried out a 100 mL quartz flask. d Reaction times are reported as follows: time exposed to UV-light/time of conventional heating on the bench-top (see the ESI for a detailed description of reaction step). e Isolated yield after purification. f Side product 11 was formed in 10–15% yield. | ||||
| 1 | None | 12/6 | 51 | 26 |
| 2 | n-BuOH | 12/2 | 67 | 9 |
| 3 | n-BuOH, no Et3N | 12/2 | 50f | 14 |
| 4 | hν at 65 °C | 21/0 | 18 | 0 |
| 5 | hν at 65 °C, n-BuOH | 8/0 | 36 | 0 |
| 6 | hν at 65 °C with Et3N | 24/0 | 24 | 0 |
| 7 | None, gram-scale | 55/6 | 61 | 24 |
| 8 | n-BuOH, gram-scale | 34/2 | 59 | 22 |
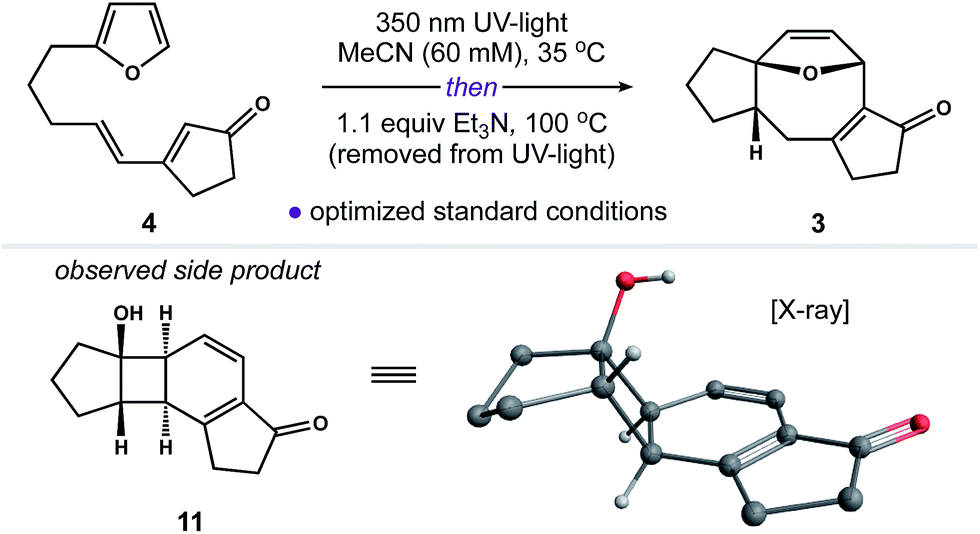
The utility of this two-stage procedure notwithstanding, we also explored conditions to carry out both the [2 + 2] cycloaddition and Cope rearrangement in tandem at 65 °C in the Rayonet (entries 4–6). This modification invariably gave 3 in poor yield. Subsequent control experiments revealed that both 6 and 3 decompose at 65 °C when exposed to UV-light. Alternatively, we found that our two-stage process could be readily scaled without loss of reaction efficiency (entries 7–8). Increased reaction times were required for photoreactions carried out on scale (55 h in MeCN); however, gram quantities of 4 were processed to 3 in 61% yield (85% yield based on recovered 4) employing these otherwise mild reaction conditions.
Having developed a scalable route to 3, we focused on exploring the scope of this process. Utilizing the two-step assembly described in Scheme 2,24 we prepared a library of 10 representative photoprecursors 12 designed to probe the scope of substitution patterns around the 5–8–5 core of 2. As shown in Fig. 1, substrates with modifications to the furan subunit (R1–R3) reacted smoothly to give B-ring variants 13a–f in 41–60% yield. In each case, we observed formation of a single diastereomer. The only exception was 12g, which afforded 13g in 25% isolated yield (50% yield by NMR) along with small amounts of the corresponding C2 diastereomer (d.r. = 10:1). Similarly, photosubstrates possessing changes to the hydrocarbon linker (i.e. X and Y) gave A-ring variants 13h and 13i as single diastereomers in 22% and 40% yield, respectively. In general, these products were challenging to isolate from impurities; however, the photoreaction of substrate 12h was also notably inefficient, proceeding to only 27% conversion after 24 h. Finally, we examined the isomerization of chiral tether (±)-12j (Z = OH), which afforded C-ring derivative (±)-13j as a mixture of diastereomers (d.r. = 1.3:1 at C12) in 64% combined yield.
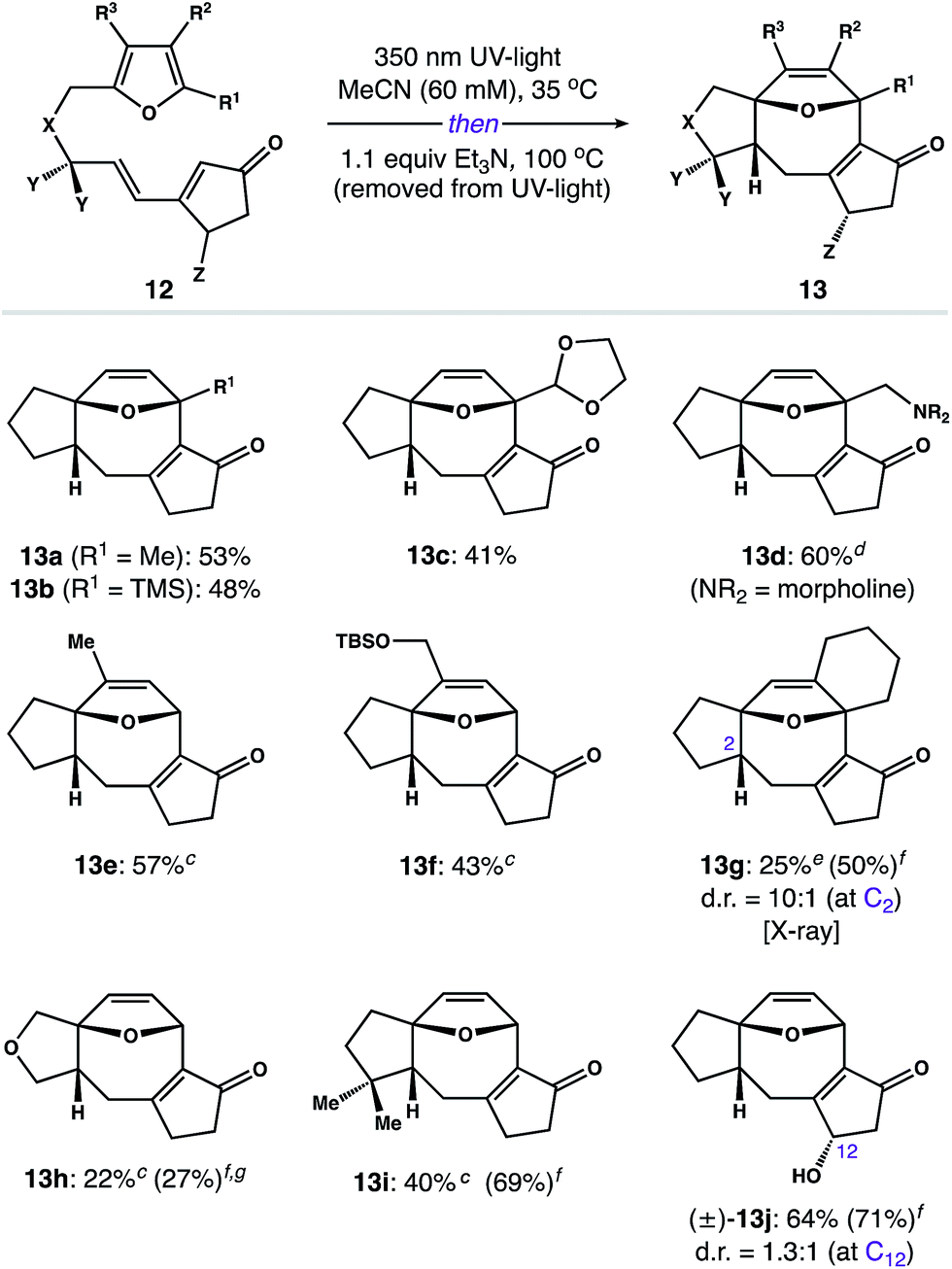 | ||
| Fig. 1 Reaction scopea,b – aIsolated yields of experiments on 0.2–0.5 mmol scale, unless otherwise noted. bReactions afforded 13 as a single diastereomer, unless otherwise noted. cn-BuOH was used in place of MeCN. dYield of a gram-scale reaction. eThe C2 epimer was isolated in 26% yield and >20:1 d.r. when n-BuOH was used as the solvent. fYield was determined by 1H NMR using N-benzylbenzamide as an internal standard. gSubstrate 12h required 24 h in the photoreactor to achieve a 27% conversion to the corresponding [2 + 2] photoadduct (see the ESI for details†). | ||
With a rapid and general entry to the 5–8–5 ring system in place, we set out to identify conditions to install the quaternary methyl group at C11. To our delight, exposure of enone 3 to lithium dimethylcuprate and TMSCl at −78 °C generated silyl enol ether 14 as a single diastereomer (Scheme 4). This reaction was carried out on gram-scale, without the need for chromatography, to afford 14 in >95% yield. Subsequent hydrolysis of 14 with 1 M aq. HCl gave ketone 15 as a crystalline solid, allowing us to establish the relative stereochemistry of 1,4-addition by single-crystal X-ray diffraction.25 Alternatively, we found that addition of BCl3 to a cooled solution of 14 resulted in ring-opening of the oxabicycle to afford conjugated dienone 16.26 Notably, this chemistry establishes a scalable entry point to variations of 2 in four steps from readily available starting materials.
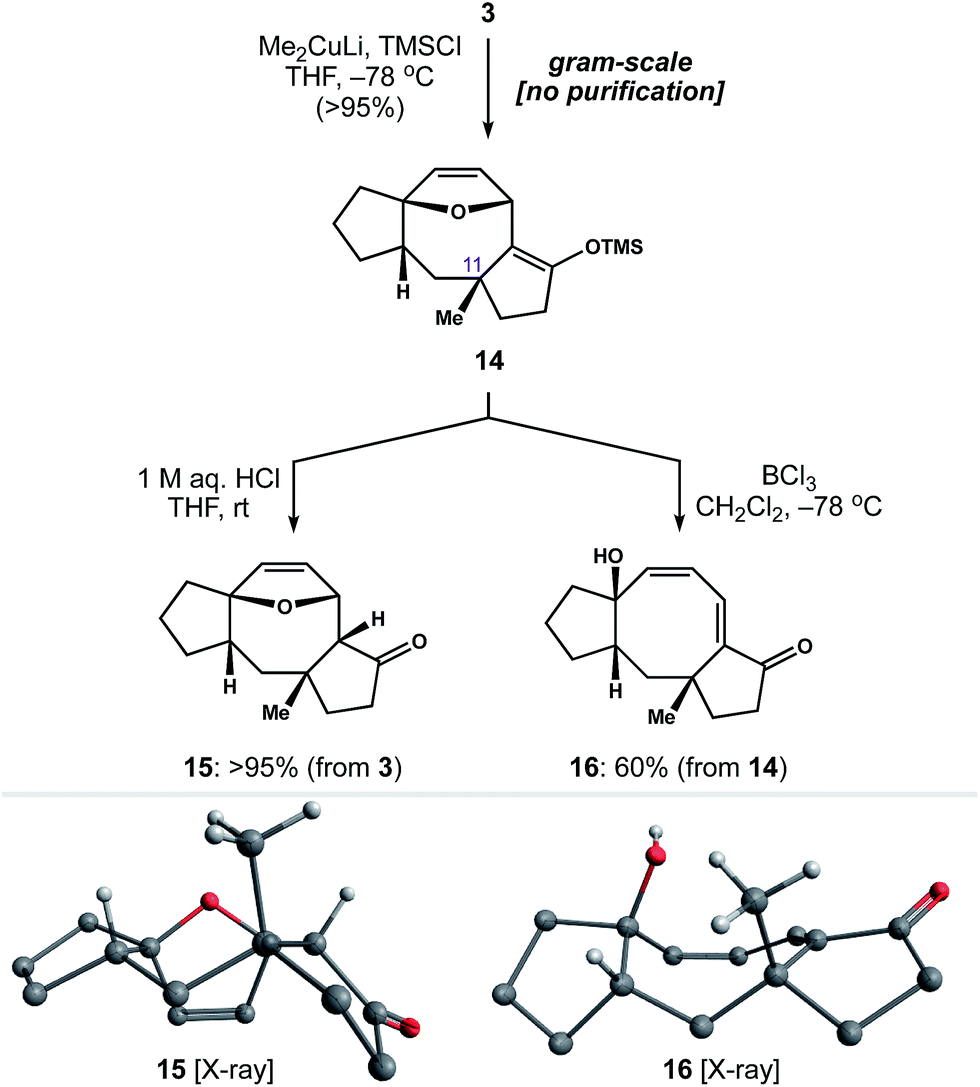 | ||
| Scheme 4 Installation of the C11 quaternary methyl group. | ||
Conclusions
In summary, this study describes a modular strategy for the synthesis of 5–8–5 scaffolds. Conceptually, the method described here is appealing because it harnesses strain amassed within stereodefined cyclobutanes 5 to achieve an otherwise challenging ring formation. This approach also enables installation of the C11 quaternary stereocenter, and thus, establishes a four-step entry point to the conserved core of multiple bioactive natural products. This chemistry is expected to facilitate structure–function studies of peripheral groups attached to privileged scaffold 2. Work along these lines is currently underway in our laboratory.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Institutes of Health (R01-GM125926) and Florida State University (FSU). Dr Ron Clark (FSU) is gratefully acknowledged for his assistance with single-crystal X-ray diffraction. We also thank Gabriel dos Passos Gomes (FSU) for his assistance with computations described in Scheme 1b. Mass spectra were collected at the University of Florida Mass Spectrometry and Education Center using NIH-supported equipment (S10-OD021758A1).Notes and references
- A. H. de Boer and I. J. de Vries van Leeuwen, Trends Plant Sci., 2012, 17, 360 CrossRef PubMed.
- For select examples, see: from plants, (a) G. V. Subbararo, K. Nakahara, M. P. Hartado, H. Ono, D. E. Moreta, A. F. Salcedo, A. T. Yoshihashi, T. Ishikawa, M. Ishitani, M. Yoshia, I. M. Rao, C. E. Lascano, W. L. Berry and O. Ito, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17302 CrossRef PubMed; from fungi, (b) S. Kim, D. S. Shin, T. Lee and K. B. Oh, J. Nat. Prod., 2004, 67, 448 CrossRef PubMed; from bacteria, (c) D. Zheng, L. Han, H. Qu, X. Chen, J. Zhong, X. Bi, J. Liu, Y. Jiang and X. Huang, J. Nat. Prod., 2017, 80, 837 CrossRef PubMed.
- M. Würtele, C. Jelich-Ottmann, A. Wittinghofer and C. Oecking, EMBO J., 2003, 22, 987 CrossRef PubMed.
- (a) M. Skwarczynska, M. Molzan and C. Ottmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 377 CrossRef PubMed; (b) I. J. de Vries van Leeuwen, D. da Costa Pereira, K. D. Flach, S. R. Piersma, C. Hasse, D. Bier, Z. Yaicin, R. Michalides, K. A. Feenstra, C. R. Koemex, L. Brunsveld, C. Ottmann, W. Zwart and A. H. de Boer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8894 CrossRef PubMed.
- R. Dasari, M. Masi, R. Lisy, M. Ferderin, L. R. English, A. Cimmino, V. Mathieu, A. J. Brenner, J. G. Kuhn, S. T. Witten, A. Evidente, R. Kiss and A. Kornienko, Bioorg. Med. Chem. Lett., 2012, 22, 579 CrossRef PubMed.
- T. Aoagi, T. Aoyama, F. Fojima, S. Hattori, Y. Honma, M. Hamada and T. Takeuchi, J. Antibiot., 1992, 45, 1587 CrossRef.
- G. B. Mills and W. H. Moolenaar, Nat. Rev. Cancer, 2003, 3, 582 CrossRef PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef PubMed.
- A. Hussain, S. K. Yousuf and D. Mukherjee, RSC Adv., 2014, 4, 43241 RSC.
- D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207 CrossRef PubMed.
- For examples, see: (a) Z. G. Brill, H. K. Grover and T. J. Maimone, Science, 2016, 352, 1078 CrossRef PubMed; (b) K. Tsuna, N. Noguchi and M. Nakada, Angew. Chem., Int. Ed., 2011, 50, 9452 CrossRef PubMed; (c) N. Kato, H. Okamoto and H. Takshita, Tetrahedron, 1996, 52, 3921 CrossRef; (d) T. F. Jamison, S. Shambayati, W. E. Crowe and S. L. Schreiber, J. Am. Chem. Soc., 1994, 116, 5505 CrossRef; (e) M. Rowley, M. Tsukamoto and Y. Kishi, J. Am. Chem. Soc., 1989, 111, 2735 CrossRef.
- For a review, see: S. M. Seiburth and N. T. Cunard, Tetrahedron, 1996, 52, 6251 CrossRef.
- For select examples, see: (a) F. Huang, Z.-K. Yao, Y. Wang, J. Zhang and Z.-X. Yu, Chem.–Asian J., 2010, 5, 1555 CrossRef PubMed; (b) S. J. Bader and M. L. Snapper, J. Am. Chem. Soc., 2005, 127, 1201 CrossRef PubMed; (c) C. P. Lo and M. L. Snapper, Org. Lett., 2001, 3, 2819 CrossRef PubMed; (d) P. A. Wender, J. M. Nuss, D. B. Smith, A. Surez-Sobrino, J. Vagberg, D. DeCosta and J. Bordner, J. Org. Chem., 1997, 62, 4908 CrossRef.
- (a) S. M. Sieburth, K. F. McGee and T. H. Al-Tel, J. Am. Chem. Soc., 1998, 120, 587 CrossRef; (b) K. F. McGee, T. H. Al-Tel and S. M. Sieburth, Synthesis, 2001, 8, 1185 Search PubMed.
- D. Song, R. McDonald and F. G. West, Org. Lett., 2006, 8, 4075 CrossRef PubMed.
- For conceptually related cascade reactions targeting fused 5,8-cyclooctadienes, see: (a) P. A. Wender and C. R. D. Correia, J. Am. Chem. Soc., 1987, 109, 2523 CrossRef; (b) M. R. Krout, C. E. Henry, T. Jensen, K.-L. Wu, S. C. Virgil, B. M. Stoltz, J. Org. Chem. DOI:10.1021/acs/joc.7b02972.
- J. Westmeier, C. Pfaff, J. Siewert and P. von Zezschwitz, Adv. Synth. Catal., 2013, 355, 2651 CrossRef.
- T. Mukiayama, J. Matsuo and H. Kitagawa, Chem. Lett., 2000, 29, 1250 CrossRef.
- Photoreactions were carried out in a Rayonet photochemical chamber reactor at 35 °C utilizing 24 W UV-lamps wherein ∼90% of emission is 350 nm. In general, the photochemistry reported here was carried out in quartz glassware under an atmosphere of N2; however, identical results (±3% yield) can be obtained using standard Pyrex glassware. See the ESI† for details regarding reaction setup and glassware performance.
- Z-4 was independently prepared and found to react identically to E-4 under the reported conditions. See the ESI for details.†.
- Solutions of photoproduct 5 in MeCN also cleanly rearranged to 6 at 23 °C in over the course of 36 h.
- Yield was determined by 1H NMR spectroscopy using 1,2,3-timethoxybenzene as an internal standard. See the ESI for details.†.
- We suspect that 11 is formed via the enol tautomer of 6 as shown below:
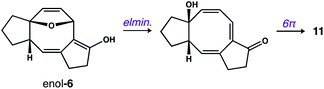 .
. - X-ray data for structures 5–6, 11, 13g, and 15–16. See the ESI for CCDC entry numbers.†.
- Photoprecursor 12i could not be prepared via the route described in Scheme 2 and was synthesized using a related three-step strategy. See the ESI for details.†.
- For a related ring-opening reaction of a similar dihydrofuran, see: L. Li, R. McDonald and F. G. West, Org. Lett., 2008, 10, 3733 CrossRef PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures at characterization data. CCDC 1816383–1816390. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00999f |
| This journal is © The Royal Society of Chemistry 2018 |