
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the interaction between the encapsulated water molecule and the fullerene cages in H2O@C60− and H2O@C59N−
Guo-Zhu
Zhu
 a,
Yuan
Liu
a,
Yoshifumi
Hashikawa
b,
Qian-Fan
Zhang
a,
Yasujiro
Murata
b and
Lai-Sheng
Wang
*a
a,
Yuan
Liu
a,
Yoshifumi
Hashikawa
b,
Qian-Fan
Zhang
a,
Yasujiro
Murata
b and
Lai-Sheng
Wang
*a
aDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: Lai-Sheng_Wang@brown.edu
bInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan
First published on 4th June 2018
Abstract
We report a high-resolution photoelectron imaging study of cryogenically-cooled H2O@C60− and H2O@C59N− endohedral fullerene anions. The electron affinity (EA) of H2O@C60 is measured to be 2.6923 ± 0.0008 eV, which is 0.0088 eV higher than the EA of C60, while the EA of H2O@C59N is measured to be 3.0058 eV ± 0.0007 eV, which is 0.0092 eV lower than the EA of C59N. The opposite shifts are found to be due to the different electrostatic interactions between the encapsulated water molecule and the fullerene cages in the two systems. There is a net coulombic attraction between the guest and host in H2O@C60−, but a repulsive interaction in H2O@C59N−. We have also observed low-frequency features in the photoelectron spectra tentatively attributed to the hindered rotational excitations of the encapsulated H2O molecule, providing further insights into the guest–host interactions in H2O@C60− and H2O@C59N−.
1. Introduction
Endohedral fullerenes with encapsulated atoms, molecules or clusters have attracted wide interest due to their unique electronic, magnetic, and optical properties.1–4 Since the first observation of the endohedral fullerene La@C60 in a mass spectrum in 1985,5 a variety of such novel guest–host complexes containing noble gas atoms,6,7 the N atom and the N2 molecule,8 metal atoms and metal clusters,4,9 have been synthesized using the arc discharge or ion bombardment methods. These harsh production conditions were unable to make endofullerenes containing light molecules.10 A more rational synthetic approach, called molecular surgery on the fullerene surfaces,11,12 was successfully applied to the macroscopic synthesis of H2@C60,13 followed by the syntheses of H2O@C60,14,15 H2O@C59N,16 HF@C60,17 (H2O)2@C70,18 and very recently even (H2O·HF)@C70.19 The H2O@C60 and H2O@C59N species are of special interest because the water molecule is isolated without hydrogen bonds. Many experimental and theoretical studies have been carried out to elucidate the novel properties of H2O@C60, such as its polarity,20–26 quantum dynamics,27–30 magnetic,16,31 mechanical,32 thermal33 and electric properties25,26,34,35 as well as its chemical reactivity.24,36,37One of the most interesting questions about H2O@C60 concerned the nature of the guest–host interactions of the water molecule trapped in the C60 cage. No detectable difference was observed between the UV-Vis absorption spectra of the empty C60 and H2O@C60, suggesting that the water molecule has very weak interactions with the cage.14 This observation was further confirmed by studies of nuclear spin relaxation31 and electric conductance.35 However, theoretical calculations found strong dispersion interactions22,23,38–40 between the free rotating water molecule and C60.14,23,27,40,41 The quantized rotational levels and the nuclear spin-isomerism of ortho- and para-water in H2O@C60 were studied by inelastic neutron scattering, far-infrared spectroscopy, and nuclear magnetic resonance.27–29 These studies revealed a splitting of the ground rotational state of ortho-H2O and a symmetry-breaking of the C60 cage, indicating a quadrupolar interaction between H2O and C60.30 In addition, the dipole moment of H2O@C60 was measured to be around 0.5 D,25,26 in good agreement with theoretical calculations.20–24 The significant reduction of the dipole moment of the encapsulated H2O is a result of the strong shielding effect by the nonpolar C60 cage. A recent study reported that the rotation of the encapsulated water can be electrostatically perturbed by introducing polarized C(C60)–X (X: heteroatom) bonds.42
Unlike the extensive studies on H2O@C60, the H2O@C59N endohedral azafullerene was only synthesized very recently in the dimer form, (H2O@C59N)2.16 The presence of the N atom breaks the symmetry of the fullerene and introduces a polar center. Theoretical calculations suggested an attractive electrostatic interaction between the O atom of H2O and the N atom of C59N.16,43,44 Comparison of the different guest–host interactions in H2O@C60 and H2O@C59N would be very interesting. In particular, the electron affinity (EA) of the endohedral fullerenes can be a good probe of these guest–host interactions, because the extra electron in the C60− and C59N− anions is expected to be sensitive to the encapsulated H2O molecule.
Here, we present a high-resolution photoelectron (PE) imaging study of the H2O@C60− and H2O@C59N− anions cooled in a cryogenic ion trap. The EA of H2O@C60 is accurately measured to be 2.6923 ± 0.0008 eV, which is 0.0088 eV higher than the EA of C60,45 while the EA of H2O@C59N is measured to be 3.0058 eV ± 0.0007 eV, which is 0.0092 eV lower than the EA of C59N.46 The opposite shifts suggest different guest–host interactions between the encapsulated water molecule and the fullerene cages, which are understood by an electrostatic model. A net coulombic attraction between the water molecule and the HOMO electron in H2O@C60− is found to stabilize the anion and enhance the EA of H2O@C60 compared to C60, while a repulsive interaction in H2O@C59N− destabilizes the anion and decreases the EA of H2O@C59N relative to C59N. In addition, low-frequency features in the PE spectra are observed and tentatively attributed to the hindered rotational excitations30 of the encapsulated H2O molecule, providing further insights into the guest–host interactions in H2O@C60− and H2O@C59N−.
2. Experimental method
The experiment was carried out using our third-generation electrospray PE imaging apparatus,47 equipped with a cryogenically-cooled Paul trap48 and a high-resolution PE imaging lens.49 The electrospray solutions were prepared by dissolving H2O@C60 or (H2O@C59N)2 samples in a mixed solvent of o-dichlorobenzene/CH3CN (1/3 ratio in volume), to which tetrakis(dimethylamino)ethylene50,51 was added as a reducing agent. Anions from the electrospray source were guided into a cryogenically-controlled Paul trap operated at 4.5 K and thermally cooled via collisions with 1 mTorr He/H2 (4/1 in volume) background gas.48 The cold anions were pulsed out of the ion trap at a 10 Hz repetition rate into the extraction zone of a time-of-flight mass spectrometer. The desired anions, H2O@C60− or H2O@C59N−, were selected by a mass gate and photodetached by the third harmonic of a Nd:YAG laser (354.7 nm) and a tunable dye laser in the interaction zone of the imaging lens.49 The PE images were inverse-Abel transformed and reconstructed using both pBasex and BASEX.52,53 The PE spectra were calibrated with the known spectra of Au− at different photon energies. The kinetic energy (KE) resolution was 3.8 cm−1 for electrons with 55 cm−1 KE and about 1.5% (ΔKE/KE) for KE above 1 eV in the current experiment.3. Results and discussions
3.1. The photoelectron images and spectra of H2O@C60− and H2O@C59N− at 354.7 nm
Fig. 1 shows the PE images and spectra of H2O@C60− and H2O@C59N− at 354.7 nm. The first intense peak in each spectrum, labeled as 000, represents the 0–0 transition from the anion to the neutral and defines the EAs for H2O@C60 and H2O@C59N, which are measured more accurately in the low photon energy spectra (vide infra). The peaks at higher binding energies represent transitions from the ground vibrational state of the anion to the excited vibrational levels of the neutral ground electronic state. They are better resolved in the high-resolution PE images at lower photon energies near the detachment threshold to be discussed below. Fig. 1b also shows a weak peak (X′) at ∼1.2 eV, which is derived from the parent dimer dianion, (H2O@C59N)22− with the same m/z as the monoanion. A similar dimer dianion was also observed in the 354.7 nm PE spectrum of C59N− recently.46 The low binding energy for the dianion was due to the strong intramolecular Coulomb repulsion.47,54,55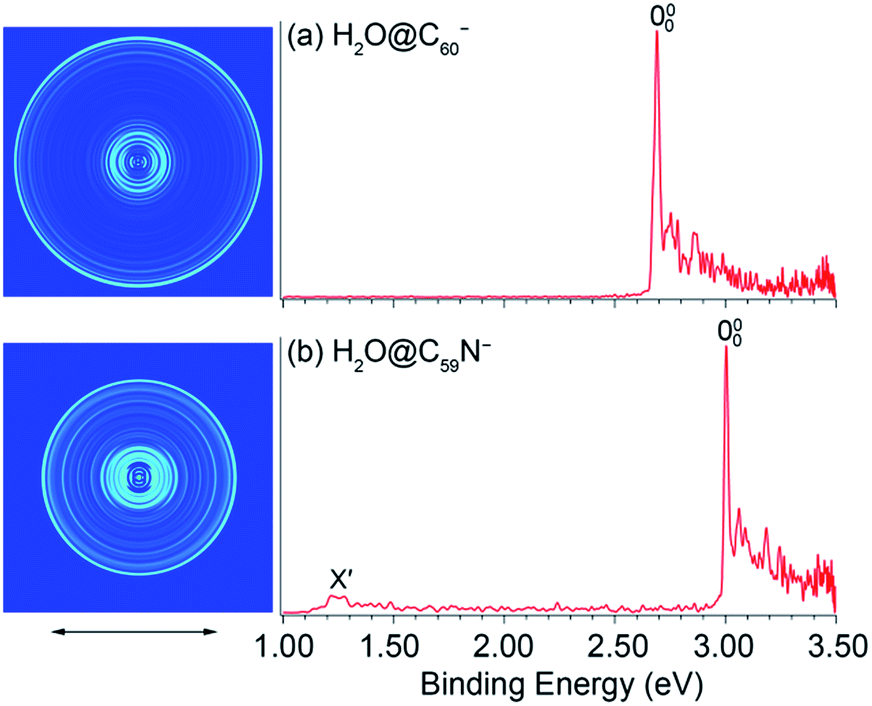 | ||
| Fig. 1 Photoelectron images and spectra of (a) H2O@C60− and (b) H2O@C59N− at 354.7 nm. The double arrow below the images indicates the direction of the laser polarization. Note the image corresponding to peak X′ in (b) is cut off. | ||
The 354.7 nm spectra of the endohedral fullerenes appear to be nearly identical to those of their corresponding parent fullerenes,45,46 as directly compared in Fig. 2. This observation suggests that indeed the encapsulated water molecule has little effect on the electronic and geometrical structures of the fullerene hosts. However, upon closer examination, a small spectral shift was revealed in each case, as shown in the expanded threshold region given in the respective inset of Fig. 2. Surprisingly, the two endohedral fullerenes exhibit opposite shifts. The electron binding energy of H2O@C60− was observed to be shifted slightly higher relative to that of C60− (Fig. 2a), whereas the electron binding energy of H2O@C59N− was shifted slightly lower relative to that of C59N−. The opposite spectral shifts suggest subtle differences in the guest–host interactions of the encapsulated water molecule with the fullerene or azafullerene cages.
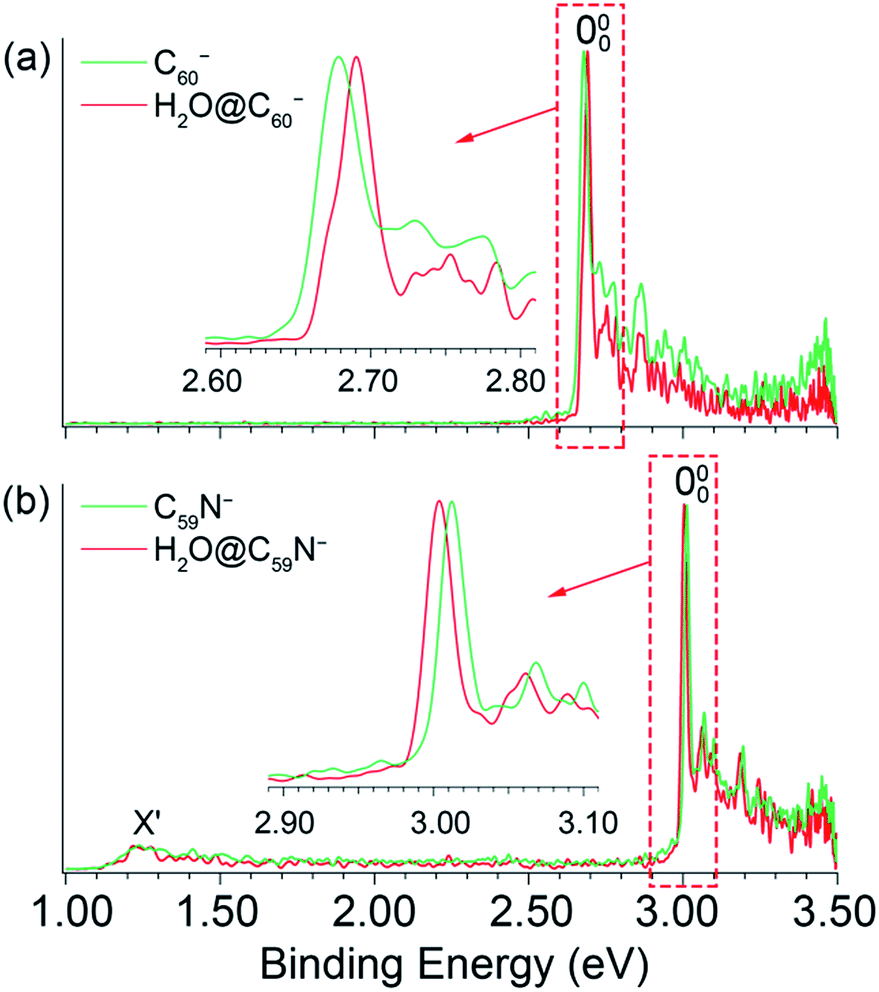 | ||
| Fig. 2 Comparisons of the photoelectron spectra of (a) C60− and H2O@C60−, (b) C59N− and H2O@C59N− at 354.7 nm. | ||
3.2. The high-resolution photoelectron images and spectra of H2O@C60− and H2O@C59N− near detachment thresholds
To measure the EAs more accurately and to resolve low-frequency vibrations, we measured PE images for H2O@C60− and H2O@C59N− at lower photon energies near the detachment thresholds, as shown in Fig. 3. We found that the detachment cross sections for the endohedral fullerenes were weaker than those of the corresponding empty fullerenes,45,46 in particular near the detachment thresholds. The spectra shown in Fig. 3 were averaged from 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 500000 laser shots. At 456.60 nm (Fig. 3a), the 000 peak with a linewidth of 38 cm−1 at an electron kinetic energy of 186 cm−1 defines the most accurate value for the EA of H2O@C60 as 2.6923 ± 0.0008 eV, which is 0.0088 eV higher than the EA of C60.45 The detachment cross section at this wavelength for H2O@C60− was particularly poor. The features below the 000 peak in Fig. 3a were partly due to background noise and partly due to hot band transitions, which were amplified relative to the 000 transition. At 411.12 nm (Fig. 3d), the 000 peak with an electron kinetic energy of 80 cm−1 and linewidth of 15 cm−1 yields the most accurate EA for H2O@C59N to be 3.0058 eV ± 0.0007 eV, which is 0.0092 eV lower than the EA of C59N.46
000 to 500000 laser shots. At 456.60 nm (Fig. 3a), the 000 peak with a linewidth of 38 cm−1 at an electron kinetic energy of 186 cm−1 defines the most accurate value for the EA of H2O@C60 as 2.6923 ± 0.0008 eV, which is 0.0088 eV higher than the EA of C60.45 The detachment cross section at this wavelength for H2O@C60− was particularly poor. The features below the 000 peak in Fig. 3a were partly due to background noise and partly due to hot band transitions, which were amplified relative to the 000 transition. At 411.12 nm (Fig. 3d), the 000 peak with an electron kinetic energy of 80 cm−1 and linewidth of 15 cm−1 yields the most accurate EA for H2O@C59N to be 3.0058 eV ± 0.0007 eV, which is 0.0092 eV lower than the EA of C59N.46
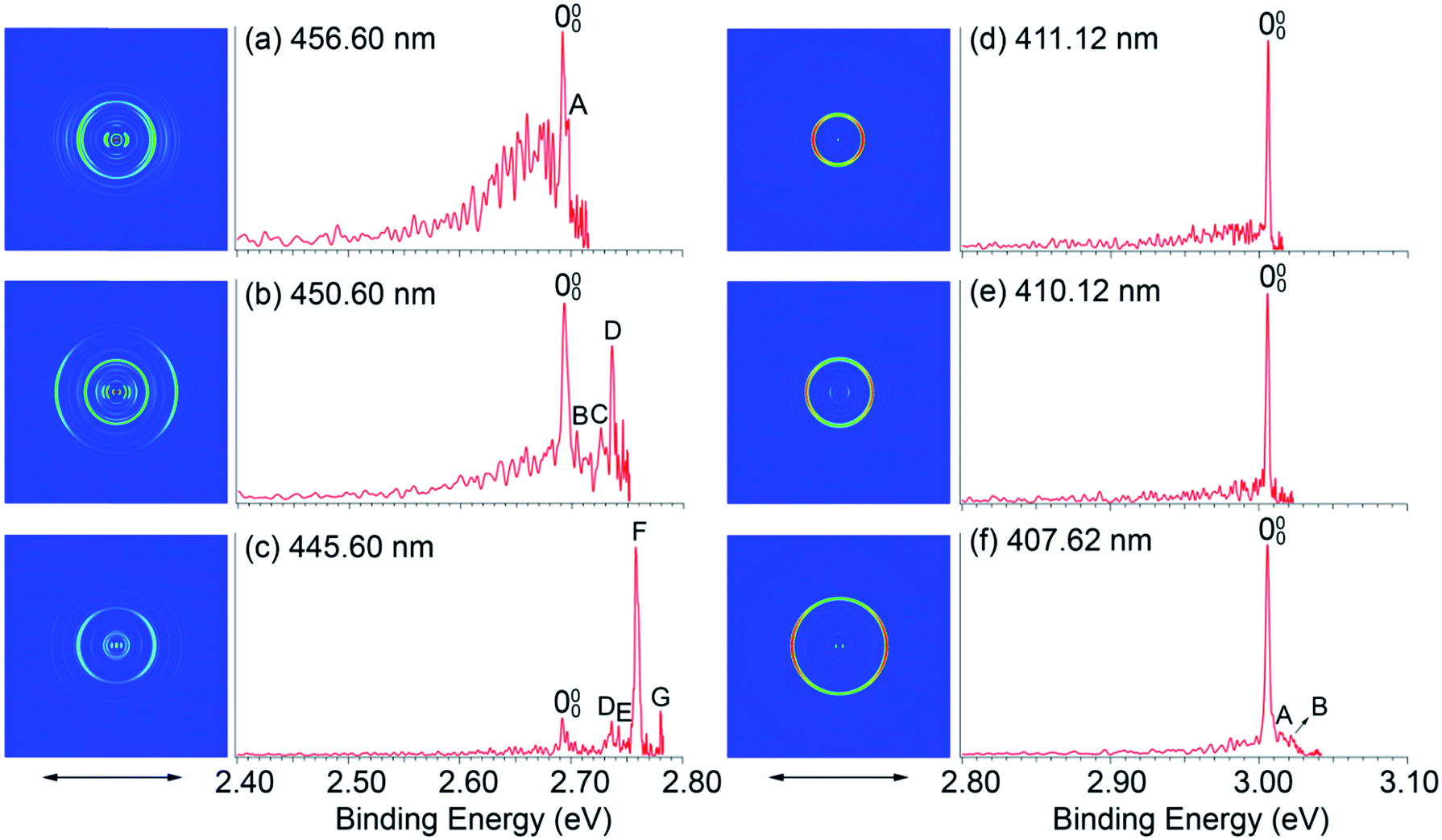 | ||
| Fig. 3 Photoelectron images and spectra of H2O@C60− at (a) 456.60 nm, (b) 450.60 nm, (c) 445.60 nm and H2O@C59N− at (d) 411.12 nm, (e) 410.12 nm, (f) 407.62 nm. The double arrows below the images indicate the direction of the laser polarization. | ||
In addition to the near-threshold spectra, two more spectra were taken to resolve low-frequency vibrational features for H2O@C60 and H2O@C59N, as shown in Fig. 3b, c, e and f, respectively. There are two types of vibrations for the endohedral fullerenes, one involving the fullerene cages and the other involving the encapsulated water molecules including the hindered rotations. The latter should be particularly sensitive to the guest–host interactions in the endohedral fullerenes. Fig. 3a–c resolve seven vibrational peaks, labeled as A–G for H2O@C60, while Fig. 3f resolves two peaks, A and B for H2O@C59N. The relative intensities of the low frequency peaks (A, B) are quite weak for both species, but they seem to be reproducible. The binding energies and shifts relative to the 000 peak for all the vibrational features are summarized in Table 1.
| Anions | Peaks | BEa (eV) | Shifts (cm−1) | Vib. freq.a (cm−1) |
|---|---|---|---|---|
| a Ref. 45. b Ref. 46. | ||||
| C60− | 2.6835(6)a | |||
| H2O@C60− | 000 | 2.6923(8) | 0 | |
| A | 2.6967(7) | 35 | ||
| B | 2.7041(10) | 95 | ||
| C | 2.7259(10) | 271 | 262 | |
| D | 2.7361(7) | 353 | 348 | |
| E | 2.7427(10) | 406 | ||
| F | 2.7582(7) | 531 | 531 | |
| G | 2.7803(10) | 710 | 717 | |
| C59N− | 3.0150(7)b | |||
| H2O@C59N− | 000 | 3.0058(7) | 0 | |
| A | 3.0151(12) | 74 | ||
| B | 3.0217(12) | 128 | ||
Peaks C, D, F, G with shifts of 271, 353, 531, and 710 cm−1, are similar to those observed in the PE spectra of C60− and they should correspond to vibrational modes involving the C60 cage.45 The strong and highly non-Franck–Condon peak F observed in the 445.60 nm spectrum (Fig. 3c) is also observed for C60−, which was attributed to strong Hertzberg–Teller coupling.45 The weak peak E with a shift of 406 cm−1 corresponds to a Hu(1) vibrational mode of C60 also observed by inelastic neutron scattering.56 These observations suggest that the H2O molecule has little effect on the geometrical and electronic structure of the C60 host. Additionally, two weak peaks A and B with small shifts of 35 and 95 cm−1, corresponding to very low-frequency transitions, are also tentatively identified. The lowest vibrational frequency of C60 is around 260 cm−1.45,56 Hence, these features should correspond to the hindered rotational excitations of the encapsulated water molecule, as revealed by the rigorous full-dimensional quantum calculations of the coupled translation-rotation eigenstates of the water molecule in H2O@C60.30 In Fig. 3f, similarly the two weak peaks A and B with shifts of 74 and 128 cm−1 were tentatively identified as the hindered rotational excitations of the encapsulated water molecule. The observation of hindered rotational transitions indicates weak interactions between the encapsulated water molecule and the fullerene cages. The relatively high frequencies observed for the hindered rotational transitions in H2O@C59N suggest stronger guest–host interactions in this system.
The PE images of H2O@C60− and H2O@C59N− in Fig. 3 all exhibit distinct p-wave character with the photoelectron angular distributions parallel to the direction of the laser polarization, similar to those for C60− and C59N−.45,46 These observations indicate that the encapsulated water molecule does not affect the s-like HOMO of the fullerene cages. The p-wave nature of the outgoing electron is partly responsible for the low detachment cross sections near threshold according to the Wigner threshold law.57
3.3. The opposite shifts of the EAs in H2O@C60 and H2O@C59N relative to the empty fullerenes: an electrostatic model
The opposite shifts of the EAs of H2O@C60 and H2O@C59N relative to their corresponding empty cages are consistent with previous theoretical calculations.24,43 The different effects of the encapsulated water on the EAs can be glimpsed from the electrostatic potential maps of the HOMO of the fullerene anions, as presented in Fig. 4. The extra charge in the half-filled HOMO of C60− is evenly distributed on the surface (Fig. 4a). Even though the encapsulated water molecule was known to have no preferred directions,14,23,27,40,41 it breaks the symmetry and dynamically induces a slightly higher charge density on the cage surface, where the H atoms point to (Fig. 4b). On the contrary, the HOMO of C59N− is partially localized on the N atom and the C atoms around the N atom (Fig. 4c).46 The water molecule in H2O@C59N− has been shown to adopt a global minimal structure with the O atom pointing to the N atom of the cage due to a weak N⋯O attractive interaction.43,44 Despite its orientation preference, the water encapsulation has relatively little effect on the HOMO of C59N− (Fig. 4d). However, this orientation of the water molecule brings the electronegative O atom closer to the extra charge, inducing a repulsive interaction.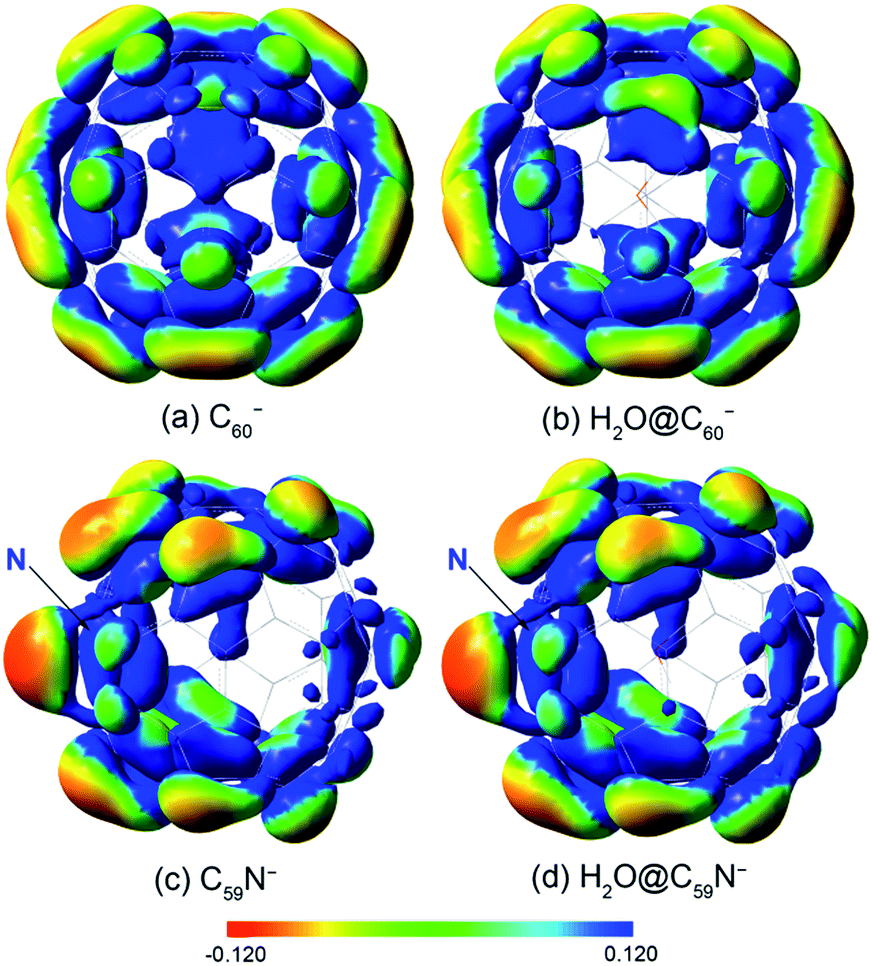 | ||
| Fig. 4 The electrostatic potential maps for the HOMO of (a) C60−, (b) H2O@C60−, (c) C59N−, (d) H2O@C59N−, calculated at B3LYP/6-311++G(d,p) level using the GAUSSIAN 09 package.58 | ||
A simple electrostatic model is used to understand the interactions between the water molecule and the extra charge in the HOMO of the fullerene cages and to obtain insights about the observed different EA shifts in the two systems. In the model, partial charges on the water molecule are represented by point charges with −2q located on the oxygen atom and +q on each H atom, where q is obtained from a Mulliken population analysis of the total wavefunction of the water molecule. The Coulomb interaction can be expressed as:
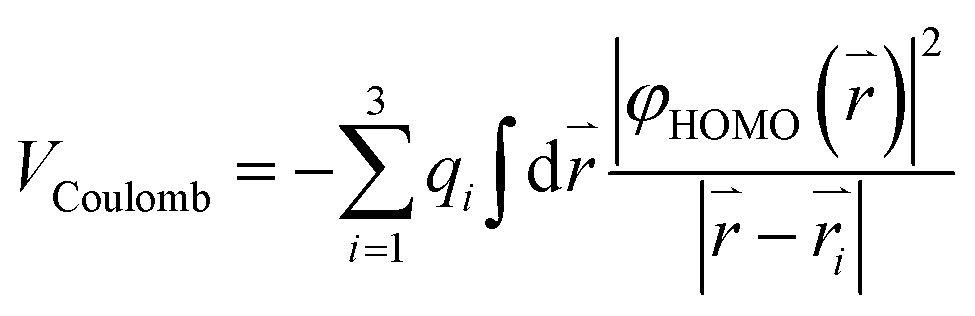
The Coulomb interaction in H2O@C60− was calculated to be −23 meV, indicating an attractive interaction between the encapsulated water molecule and the HOMO electron in H2O@C60−. This weak attraction, which is in good agreement with previous calculations,22,38–40 stabilizes the H2O@C60− anion and increases the EA of H2O@C60 relative to C60. On the other hand, the simple electrostatic calculation on the H2O@C59N− anion yields a repulsive interaction of 64 meV. Hence, the water encapsulation destabilized the HOMO of the C59N− anion, reducing the EA of H2O@C59N relative to C59N. This repulsive interaction is expected from the orientation of the H2O molecule in C59N− and its asymmetric electron density distribution (Fig. 4d). Even though the electrostatic model is rather crude, it correctly predicts the directions of the EA shifts in the two endohedral fullerenes. The interactions between the encapsulated water molecule and the fullerene cages are so weak that they were not detectable in the UV-Vis absorption spectra14 or the electrical conductance experiment.35
4. Conclusions
In conclusion, we report a high-resolution photoelectron imaging study of two endohedral fullerene anions, H2O@C60− and H2O@C59N−. Accurate electron affinities are obtained for H2O@C60 (2.6923 ± 0.0008 eV) and H2O@C59N (3.0058 ± 0.0007 eV) for the first time. The EA of H2O@C60 is found to be higher than that of C60 by 0.0088 eV, whereas the EA of H2O@C59N is found to be lower than that of C59N by 0.0092 eV. These small EA shifts reflect the weak guest–host interactions in the endohedral fullerenes and the opposite shifts are understood using a simple electrostatic model between the encapsulated H2O molecule and the HOMO of the fullerene anions. Low-frequency features due to the hindered rotational transitions of the encapsulated water molecule are also tentatively identified, providing further insight into the weak guest–host interactions in the two endohedral fullerenes.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Y. L. would like to thank Prof. Chuan-Gang Ning from Tsinghua University for providing the program to extract Kohn–Sham orbitals from DFT calculations. This work was supported by the Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division of the U.S. Department of Energy.References
- H. Shinohara, Rep. Prog. Phys., 2000, 63, 843–892 CrossRef.
- S. Guha and K. Nakamoto, Coord. Chem. Rev., 2005, 249, 1111–1132 CrossRef.
- N. J. Turro, J. Y. C. Chen, E. Sartori, M. Ruzzi, A. Marti, R. Lawler, S. Jockusch, J. Lopez-Gejo, K. Komatsu and Y. Murata, Acc. Chem. Res., 2010, 43, 335–345 CrossRef PubMed.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef PubMed.
- J. R. Heath, S. C. O'Brien, Q. Zhang, Y. Liu, R. F. Curl, H. W. Kroto, F. K. Tittel and R. E. Smalley, J. Am. Chem. Soc., 1985, 107, 7779–7780 CrossRef.
- T. Weiske, D. K. Bohme, J. Hrusak, W. Kratschmer and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1991, 30, 884–886 CrossRef.
- M. Saunders, H. A. Jimenez–Vazquez, R. J. Cross and R. J. Poreda, Science, 1993, 259, 1428–1430 Search PubMed.
- (a) T. A. Murphy, T. Pawlik, A. Weidinger, M. Hohne, R. Alcala and J.-M. Spaeth, Phys. Rev. Lett., 1996, 77, 1075–1078 CrossRef PubMed; (b) T. Suetsuna, N. Dragoe, W. Harneit, A. Weidinger, H. Shimotani, S. Ito, H. Takagi and K. Kitazawa, Chem.–Eur. J., 2002, 8, 5079–5083 CrossRef PubMed.
- (a) L. S. Wang, J. M. Alford, Y. Chai, M. Diener and R. E. Smalley, J. Phys. D, 1993, 26, 297–299 Search PubMed; (b) L. S. Wang, J. M. Alford, Y. Chai, M. Diener, J. Zhang, S. M. McClure, T. Guo, G. E. Scuseria and R. E. Smalley, Chem. Phys. Lett., 1993, 207, 354–359 CrossRef.
- M. H. Levitt, Philos. Trans. R. Soc., A, 2013, 371, 20120429 CrossRef PubMed.
- Y. Rubin, Chem.–Eur. J., 1997, 3, 1009–1016 CrossRef.
- M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2008, 6083–6094 RSC.
- K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238–240 CrossRef PubMed.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613–642 CrossRef PubMed.
- A. Krachmalnicoff, M. H. Levitt and R. J. Whitby, Chem. Commun., 2014, 50, 13037 RSC.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2016, 138, 4096–4104 CrossRef PubMed.
- A. Krachmalnicoff, R. Bounds, S. Mamone, S. Alom, M. Concistrè, B. Meier, K. Kouřil, M. E. Light, M. R. Johnson, S. Rols, A. J. Horsewill, A. Shugai, U. Nagel, T. Rõõm, M. Carravetta, M. H. Levitt and R. J. Whitby, Nat. Chem., 2016, 8, 953–957 CrossRef PubMed.
- R. Zhang, M. Murata, T. Aharen, A. Wakamiya, T. Shimoaka, T. Hasegawa and Y. Murata, Nat. Chem., 2016, 8, 435–441 CrossRef PubMed.
- R. Zhang, M. Murata, A. Wakamiya, T. Shimoaka, T. Hasegawa and Y. Murata, Sci. Adv., 2017, 3, e1602833 CrossRef PubMed.
- C. N. Ramachandran and N. Sathyamurthy, Chem. Phys. Lett., 2005, 410, 348–351 CrossRef.
- K. Yagi and D. Watanabe, Int. J. Quantum Chem., 2009, 109, 2080–2090 CrossRef.
- A. Varadwaj and P. R. Varadwaj, Chem.–Eur. J., 2012, 18, 15345–15360 CrossRef PubMed.
- B. Ensing, F. Costanzo and P. L. Silvestrelli, J. Phys. Chem. A, 2012, 116, 12184–12188 CrossRef PubMed.
- A. Galano, A. Pérez-González, L. del Olmo, M. Francisco-Marquez and J. R. León-Carmona, J. Mol. Model., 2014, 20, 1–12 CrossRef PubMed.
- S. Aoyagi, N. Hoshino, T. Akutagawa, Y. Sado, R. Kitaura, H. Shinohara, K. Sugimoto, R. Zhang and Y. Murata, Chem. Commun., 2014, 50, 524–526 RSC.
- B. Meier, S. Mamone, S. Concistre, J. Alonso-Valdesueiro, A. Krachmalnicoff, R. J. Whitby and M. H. Levitt, Nat. Commun., 2015, 6, 8112 CrossRef PubMed.
- C. Beduz, M. Carravetta, J. Y.-C. Chen, M. Concistrè, M. Denning, M. Frunzi, A. J. Horsewill, O. G. Johannessen, R. Lawler, X. Lei, M. H. Levitt, Y. Li, S. Mamone, Y. Murata, U. Nagel, T. Nishida, J. Ollivier, S. Rols, T. Rõõm, R. Sarkar, N. J. Turro and Y. Yanga, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12894 CrossRef PubMed.
- S. Mamone, M. Concistrè, E. Carignani, B. Meier, A. Krachmalnicoff, O. G. Johannessen, X. Lei, Y. Li, M. Denning and M. Carravetta, J. Chem. Phys., 2014, 140, 194306 CrossRef PubMed.
- K. S. K. Goh, M. Jiménez-Ruiz, M. R. Johnson, S. Rols, J. Ollivier, M. S. Denning, S. Mamone, M. H. Levitt, X. Lei, Y. Li, N. J. Turro, Y. Murata and A. J. Horsewill, Phys. Chem. Chem. Phys., 2014, 16, 21330 RSC.
- (a) P. M. Felker, V. Vlcek, I. Hietanen, S. FitzGerald, D. Neuhauser and Z. Bacic, Phys. Chem. Chem. Phys., 2017, 19, 31274–31283 RSC; (b) P. M. Felker and Z. Bacic, J. Chem. Phys., 2016, 144, 201101 CrossRef PubMed; (c) Z. Bacic, V. Vlcek, D. Neuhauser and P. M. Felker, Faraday Discuss., 2018 10.1039/c8fd00082d.
- Y. Li, J. Y.-C. Chen, X. Lei, R. G. Lawler, Y. Murata, K. Komatsu and N. J. Turro, J. Phys. Chem. Lett., 2012, 3, 1165–1168 CrossRef PubMed.
- K. Min, A. B. Farimani and N. R. Aluru, Appl. Phys. Lett., 2013, 103, 263112 CrossRef.
- Y. Gao and B. Xu, J. Phys. Chem. C, 2015, 119, 20466–20473 Search PubMed.
- C. Zhu and X. Wang, Sci. Rep., 2015, 5, 17932 CrossRef PubMed.
- S. Kaneko, Y. Hashikawa, S. Fujii, Y. Murata and M. Kiguchi, ChemPhysChem, 2017, 18, 1229–1233 CrossRef PubMed.
- E. E. Maroto, J. Mateos, M. Garcia-Borràs, S. Osuna, S. Filippone, M. Á. Herranz, Y. Murata, M. Solà and N. Martín, J. Am. Chem. Soc., 2015, 137, 1190–1197 CrossRef PubMed.
- J. U. Reveles, G. KC, T. Baruah and R. R. Zope, Chem. Phys. Lett., 2017, 685, 198–204 CrossRef.
- T. Korona and H. Dodziuk, J. Chem. Theory Comput., 2011, 7, 1476–1483 CrossRef PubMed.
- C. I. Williams, M. A. Whitehead and L. Pang, J. Phys. Chem., 1993, 97, 11652–11656 CrossRef.
- D. Bucher, Chem. Phys. Lett., 2012, 534, 38–42 CrossRef.
- A. B. Farimani, Y. Wu and N. R. Aluru, Phys. Chem. Chem. Phys., 2013, 15, 17993 RSC.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, Angew. Chem., Int. Ed., 2016, 55, 13109–13113 CrossRef PubMed.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Org. Chem., 2017, 82, 4465–4469 CrossRef PubMed.
- Y. Hashikawa and Y. Murata, J. Am. Chem. Soc., 2017, 139, 18468–18471 CrossRef PubMed.
- D. L. Huang, P. D. Dau, H. T. Liu and L. S. Wang, J. Chem. Phys., 2014, 140, 224315 CrossRef PubMed.
- G. Z. Zhu, Y. Hashikawa, Y. Liu, Q. F. Zhang, Y. Murata and L. S. Wang, J. Phys. Chem. Lett., 2017, 8, 6220–6225 CrossRef PubMed.
- L. S. Wang, J. Chem. Phys., 2015, 143, 040901 CrossRef PubMed.
- X. B. Wang and L. S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- I. León, Z. Yang, H. T. Liu and L. S. Wang, Rev. Sci. Instrum., 2014, 85, 083106 CrossRef PubMed.
- J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef PubMed.
- M. Mahesh, J. A. Murphy, F. LeStrat and H. P. Wessel, Beilstein J. Org. Chem., 2009, 5, 1–12 Search PubMed.
- G. A. Garcia, L. Nahon and I. Powis, Rev. Sci. Instrum., 2004, 75, 4989 CrossRef.
- V. Dribinski, A. Ossadtchi, V. A. Mandelshtam and H. Reisler, Rev. Sci. Instrum., 2002, 73, 2634 CrossRef.
- L. S. Wang, C. F. Ding, X. B. Wang and J. B. Nicholas, Phys. Rev. Lett., 1998, 81, 2667–2670 CrossRef.
- L. S. Wang and X. B. Wang, J. Phys. Chem. A, 2000, 104, 1978–1990 CrossRef.
- S. F. Parker, S. M. Bennington, J. W. Taylor, H. Herman, I. Silverwood, P. Albers and K. Refson, Phys. Chem. Chem. Phys., 2011, 13, 7789–7804 RSC.
- E. P. Wigner, Phys. Rev., 1948, 73, 1002–1009 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. J. Knox, B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- C. G. Ning, B. Hajgato, Y. R. Huang, S. F. Zhang, K. Liu, Z. H. Luo, S. Knippenberg, J. K. Deng and M. S. Deleuze, Chem. Phys., 2008, 343, 19–30 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |