
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid state frustrated Lewis pair chemistry†
Long
Wang
a,
Gerald
Kehr
 a,
Constantin G.
Daniliuc
a,
Melanie
Brinkkötter
b,
Thomas
Wiegand
c,
Anna-Lena
Wübker
b,
Hellmut
Eckert
*bd,
Lei
Liu
f,
Jan Gerit
Brandenburg
ef,
Stefan
Grimme
*f and
Gerhard
Erker
*a
a,
Constantin G.
Daniliuc
a,
Melanie
Brinkkötter
b,
Thomas
Wiegand
c,
Anna-Lena
Wübker
b,
Hellmut
Eckert
*bd,
Lei
Liu
f,
Jan Gerit
Brandenburg
ef,
Stefan
Grimme
*f and
Gerhard
Erker
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
bInstitut für Physikalische Chemie, Graduate School of Chemistry, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, 48149 Münster, Germany. E-mail: eckerth@uni-muenster.de
cLaboratorium für Physikalische Chemie, ETH Zürich, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland
dInstitute of Physics in Sao Carlos, University of Sao Paulo, CEP 369, Sao Carlos SP 13566-590, Brazil
eLondon Centre for Nanotechnology, University College London, 17-19 Gordon Street, London, WC1H 0AH, UK
fMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Universität Bonn, Beringstraße 4, 53115 Bonn, Germany. E-mail: grimme@thch.uni-bonn.de
First published on 23rd April 2018
Abstract
In solution the PCy3/B(C6F5)3 pair is rapidly deactivated by nucleophilic aromatic substitution. In the solid state deactivation is effectively suppressed and the active frustrated phosphane/borane Lewis pair splits dihydrogen or adds to sulfur dioxide. A variety of phosphane/B(C6F5)3 pairs have been used to carry out active FLP reactions in the solid state. The reactions were analyzed by DFT calculations and by solid state NMR spectroscopy. The solid state dihydrogen splitting reaction was also carried out under near to ambient conditions with suspensions of the non-quenched phosphane/borane mixtures in the fluorous liquid perfluoromethylcyclohexane.
Introduction
Lewis acids and bases when brought together in solution typically undergo rapid formation of strong adducts. Similar to the neutralization reaction of Brønsted acids and bases this leads to an annihilation of the typical Lewis acid and Lewis base properties.1,2 This situation can be changed if one effectively hinders the Lewis pair from the neutralizing adduct formation, e.g. by electronic means3 or, more commonly, by attaching very bulky substituents at the core atoms of the pair.4–6 This invariably leads to situations where active Lewis acids and active Lewis bases are present in a solution at the same time, opening possibilities for cooperative reactions with added substrates. Such “frustrated Lewis pairs” (FLPs)7 have very successfully been used within the last decade for small molecule binding and activation, most notably among this the metal-free splitting and activation of dihydrogen.8,9 A number of new reaction types have been found in this way10,11 and a great variety of different FLPs have been devised, characterized and their reactions reported.12–23 The vast majority of them relies on avoiding the effective neutralizing Lewis acid/Lewis base adduct formation by steric hindrance, and this has led to the discovery and development of a great variety of interesting reactions in solution.24–26We thought that we should not confine ourselves to searching for FLP reactions in the liquid phase. One may safely assume that large molecules in the solid state are more or less rigidly confined to their positions in the crystal lattice. Therefore, Lewis acids and Lewis bases should effectively be hindered from adduct formation or other deactivating reaction pathways even in the absence of efficient steric hindrance by their substituents as long as we keep them in the solid state. This may actually set the scene for possibly finding new frustrated Lewis pairs and, consequently, new FLP reactions, by exposing such Lewis pairs to suitable reagents in the solid state. We have tried this principle and found that FLP chemistry can be done in this way.
There had been some reports about frustrated Lewis pair related behavior at certain heterogeneous catalysts. In these cases the active sites were part of the catalytic solid. Activation of small molecules at such systems had been achieved either thermally or by photolysis.27–29 There have also been a few reports about heterogenized Lewis acids, bonded to suitable supports, that have been employed in FLP type reactions.30 Our here reported case is distinctly different: we have employed solid physical mixtures of phosphane Lewis bases with the strong B(C6F5)3 boron Lewis acid and reacted them under suitable conditions with selected small molecules. Activation occurred and the phosphane/borane pair became consumed with selective formation of the FLP reaction products. First examples of this FLP development will be described below in this account.
Results and discussion
Tricyclohexylphosphane (1a) and tris(pentafluorophenyl)-borane (2) represent a Lewis base/Lewis acid pair that is known to react rapidly in solution. It undergoes a nucleophilic aromatic substitution by the phosphane at the para-position of a C6F5 group of the borane4,31 to eventually yield the reaction product 3a.7,32 This favorable SNAr reaction eliminates any FLP reactivity of the 1a/2 pair very effectively in solution. Even in the presence of 50 bar H2 no significant hydrogen splitting is observed, the formation of 3a even prevails under these conditions. We isolated this compound in 86% yield and confirmed its formation by X-ray diffraction and by NMR spectroscopy [Scheme 1a (H2); for details see the ESI†].32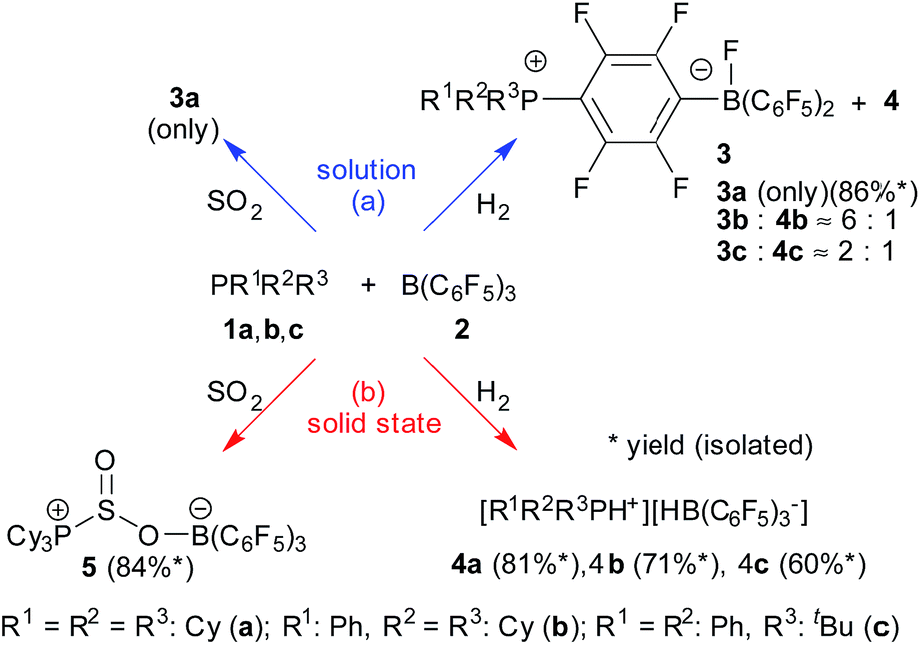 | ||
| Scheme 1 Reactions of the PR1R2R3/B(C6F5)3 FLP systems in a dihydrogen and SO2 atmosphere, respectively, in solution (a) and in the solid state (b). [Cy: cyclohexyl]. | ||
The situation is drastically different in the solid state: we mixed equimolar quantities of PCy3 (1a) and B(C6F5)3 (2) and exposed it in a glass vial inside a steel autoclave to 50 bar of dihydrogen for a total of 10 days with constant agitation by a Teflon coated magnetic stirring bar. After this time a sample was taken and dissolved in D2-dichloromethane. The NMR analysis revealed the hydrogen splitting product [HPCy3+][HB(C6F5)3−] (4a) had been formed as the by far major product [Scheme 1b (H2)]. Only negligible if any amounts of the SNAr product 3a, which would have been formed readily from any residual PCy3/B(C6F5)3 upon dissolving in dichloromethane, were present in the in situ samples. The phosphonium/hydridoborate product 4a was identified by its typical 31P (δ 33.2, 1JPH ∼443 Hz) and 11B (δ −25.3, 1JBH ∼92 Hz) NMR signals with correlated 1H NMR features at δ 5.15 (dq, 1JPH = 444.0 Hz, 3JHH = 4.1 Hz) and δ 3.59 (br 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 q, [B]H), respectively. Workup of a representative sample eventually furnished the salt 4a isolated in 81% yield on a 100 mg scale. Crystallization from dichloromethane/pentane gave single crystals which were used to confirm the formation of the FLP H2 splitting product under these special conditions by X-ray diffraction (for details see the ESI†).33 The salt 4a is an active reducing agent. Its reaction with the bulky N-phenyl-4-methylacetophenon-imine gave the respective secondary amine reduction product (24 h at 70 °C, 86% conversion, for details see the ESI†).
:1:1:1 q, [B]H), respectively. Workup of a representative sample eventually furnished the salt 4a isolated in 81% yield on a 100 mg scale. Crystallization from dichloromethane/pentane gave single crystals which were used to confirm the formation of the FLP H2 splitting product under these special conditions by X-ray diffraction (for details see the ESI†).33 The salt 4a is an active reducing agent. Its reaction with the bulky N-phenyl-4-methylacetophenon-imine gave the respective secondary amine reduction product (24 h at 70 °C, 86% conversion, for details see the ESI†).
We carried out an ample characterization of the solid product material directly (i.e. without ever dissolving it) by solid state NMR spectroscopy. Evidence for the solid state hydrogenation of the PCy3/B(C6F5)3 mixture comes from the 31P and 11B MAS NMR data (Fig. 1). In the 31P NMR spectrum we recognize the starting material at 7.1 ppm, whereas the phosphonium ion gives a broad signal at 30.1 ppm. In the 11B MAS NMR spectrum the signal of the free B(C6F5)3 gives rise to the previously documented second-order quadrupolar lineshape34 whereas after the hydrogenation a much narrower signal appears at the isotropic chemical shift of −24.9 ppm after applying the correction for the second-order quadrupolar shift (see ESI†). In addition, the spectrum reveals the presence of a minor amount of the substitution product at −2.4 ppm. The latter is the only product formed when the reaction is carried out in solution. The complete NMR characterization of the substitution product both in solution and the solid state is given in the ESI.† In a control experiment, the formation of only a minor amount of the substitution product 3a was observed in the solid state NMR spectra of a PCy3/B(C6F5)3 mixture subjected to identical reaction conditions in the absence of H2.
 | ||
| Fig. 1 11B{1H} MAS (left) and 31P{1H} CPMAS NMR spectra (right) of the B(C6F5)3/PCy3 control mixture sample (grey trace) and the hydrogenation sample (black trace) from the solid state reaction with H2. Minor side products are labelled by +. | ||
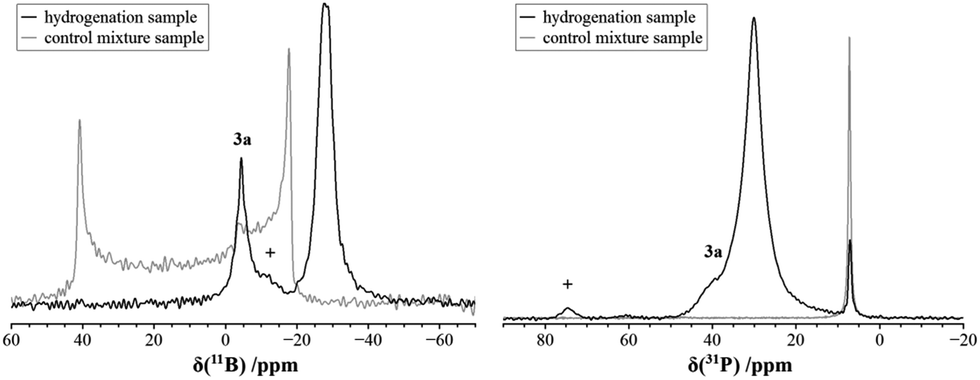
Fig. 2 shows the 1H MAS NMR spectrum of the FLP–H2 adduct 4a acquired at 20.0 T and a MAS spinning frequency of 60.0 kHz with the EASY scheme for suppression of background signals from the MAS probe and the MAS rotor cap.35 Under these conditions, the strong 1H–1H dipolar couplings are sufficiently suppressed, even though residual line broadening is still detected owing to higher order terms in the Hamiltonian which are not fully eliminated even at 60.0 kHz.36 A distinct doublet (J(1H–31P) ∼ 430 Hz) can be identified at 5.5 ppm which is assigned to P-bound hydrogen, whereas the singlet at 4.0 ppm is assigned to the B-bound hydrogen (the expected multiplet is not resolved in this case).
 | ||
| Fig. 2 1H MAS NMR spectrum of a reaction mixture after the solid state reaction of B(C6F5)3/PCy3 with H2, acquired using the EASY scheme for probe and rotor cap background suppression measured at 20.0 T and a MAS frequency of 60.0 kHz (a) and corresponding line shape simulation (b). + marks a small signal remaining from the rotor cap. | ||
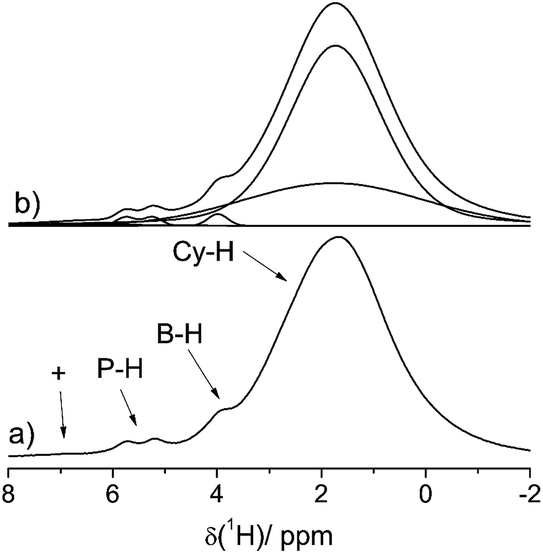
This assignment is supported by 11B{1H} and 31P{1H} heteronuclear correlation experiments (Fig. 3) which show intense cross-peaks linking these resonances to the corresponding 31P and 11B NMR signals of the solid state hydrogenation product. Further support for this assignment comes from 1H{11B} REAPDOR experiments. The obtained peak assignments are in agreement with solution state NMR data (vide supra), as well as DFT computations of 1H NMR chemical shifts for the isolated cationic H–PCy3+ and anionic H–B(C6F5)3− species (5.4 and 4.5 ppm on a B3-LYP/def2-TZVP level of theory, respectively). The absence of an encounter complex is proven by 31P{11B} REAPDOR and 11B{31P} REDOR experiments (see the ESI†). As previously discussed, such experiments can probe the B⋯P distance by measuring the strength of the heteronuclear 11B–31P dipole–dipole interactions in both FLPs and their reaction products.37 In the present material, no dephasing was observed over a dipolar mixing time of ∼5 ms. Comparing these experimental data with corresponding two-spin simulations we can conclude that the boron–phosphorus distance must be larger than 600 pm. Thus, all the experimental data are consistent with well-separated phosphonium and borate ions.
 | ||
| Fig. 3 1H/31P (left) and 1H/11B (right) HETCOR spectra of a HB(C6F5)3−/HPCy3+ mixture 4a after the solid state reaction with H2. Cross-peaks denote the B–H and P–H correlations. The additional 1H/11B crosspeak seen in the right part of the figure at (1.3/−5.2) ppm arises from the substitution product 3a. | ||
The reaction of the phosphane PPhCy2 (1b) with the Lewis acid B(C6F5)3 (2) in the solid state proceeds similarly. The reaction was carried out analogously as the one described above (r.t., 50 bar H2, 3 days). Our analysis of a product sample dissolved in CD2Cl2 revealed almost exclusive formation of the dihydrogen FLP splitting product 4b [Scheme 1b (H2)]. It shows a characteristic 1H NMR [P]H doublet at δ 6.13, 1JPH = 459.3 Hz (31P: δ 30.6) and a broad 1:1:1:1 intensity [B]H quartet at δ 3.64 (11B: δ −25.3, d, 1JBH ∼94 Hz).
Keeping the PPhCy2/B(C6F5)3 mixture (1b/2) in CD2Cl2 solution for 12 hours under H2 (50 bar) gave a different result. The NMR analysis showed the formation of a ca. 1:6 mixture of the salt 4b with the substitution product 3b. [Scheme 1a (H2)]. The zwitterionic phosphonium/fluoroborate product 3b was isolated from a separate experiment as a white solid in 82% yield. It shows a 11B NMR doublet (1JBF ∼70 Hz) at δ −0.6 and a sharp 31P NMR signal at δ 33.6. The 19F NMR spectrum shows the [B]F resonance at δ −192.9, two signals of the bridging C6F4 group and three resonances (o, p, m) of the remaining B(C6F5)2 unit with a Δδ19Fm,p shift difference of 4.9 ppm, which is typical for a borate type structure (for details and the depicted NMR spectra see the ESI†).
In this case the nucleophilic aromatic substitution by the markedly less nucleophilic phosphane PPhCy2 (1b) relative to PCy3 (1a) seems to allow the FLP reaction to compete as a minor pathway in solution. This becomes continued in the reaction of the much less nucleophilic phosphane PPh2(tBu) (1c) which in solution together with the B(C6F5)3 Lewis acid gave a ratio of the SNAr and FLP products of 3c:4c ∼ 2:1 under our typical reaction conditions (r.t., CH2Cl2 solution, 12 hours, 50 bar H2). [Scheme 1a (H2)]. Without H2 only the substitution product 3c was formed in solution (isolated in 76% yield, see the ESI† for its characterization). On the contrary, the reaction of the PPh2(tBu)/B(C6F5)3 Lewis base/Lewis acid mixture in the solid state (r.t., 50 bar H2, 3 days) gave almost pure H2-splitting product 4c, [Scheme 1b (H2)] [31P NMR: δ 31.4 (1JPH ∼474 Hz), 11B: δ −25.2 (1JBH ∼91 Hz), 19F: Δδ19Fm,p = 3.0 ppm] which we isolated from the workup procedure involving recrystallization from CH2Cl2/pentane in 60% yield (details of the characterization of the compounds 3c and 4c see the ESI†).
Quantum chemical simulations were used to investigate the mechanistic details of the different FLP reactivities in all three states of matter, i.e. gas, liquid, and solid phase. The FLP/H2 thermochemistry is for the first time investigated here in the solid state using relatively high-level periodic quantum chemistry methods. We employed a hierarchy of theoretical methods, ranging from semi-empirical tight-binding Hamiltonians to accurate London dispersion corrected hybrid density functionals.38–45 More discussion of methodological points and computational details can be found in our previous benchmark study46 and in the ESI.† The main representative results of the PCy3/B(C6F5)3 FLP are shown in Fig. 4.
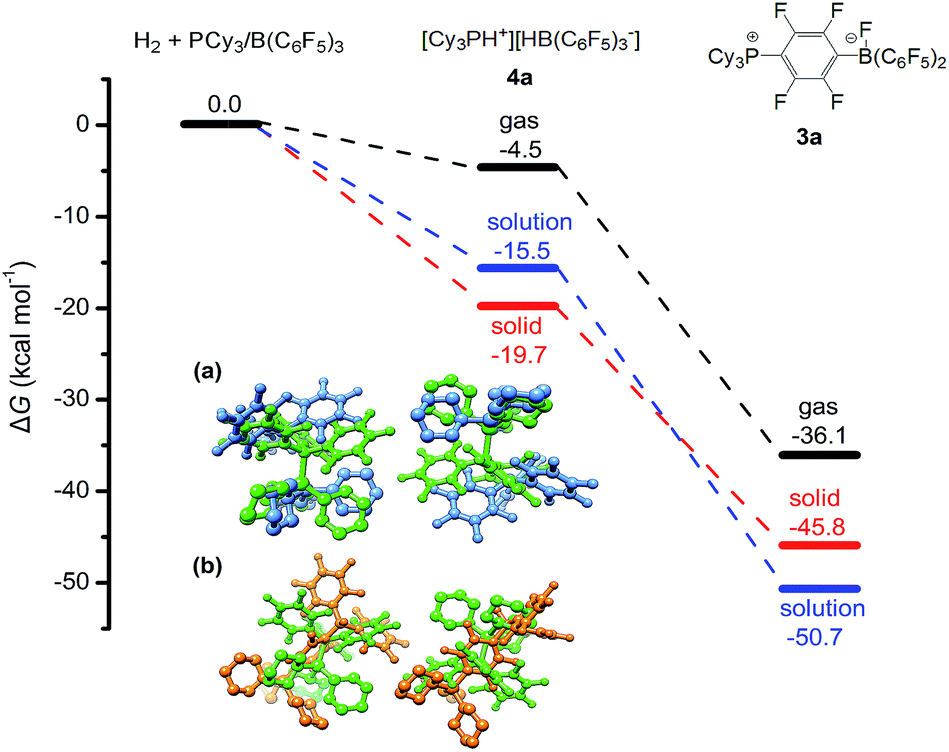 | ||
| Fig. 4 Calculated Gibbs free energies for the reaction of PCy3/B(C6F5)3 with H2 in the solution (toluene) and in the solid state (see the ESI† for computational details). All energy values are given in kcal mol−1. Inserted figures are the overlays of the HF-3c calculated crystal structures of PCy3/B(C6F5)3 (green) and [HPCy3+][HB(C6F5)3−] (blue) (a), and crystal structures of PCy3/B(C6F5)3 (green) and the SN2Ar product 3a (orange) (b). Hydrogen atoms except P–H and B–H are omitted for clarity. | ||
All shown reactions are exergonic, and both the solvent and the crystal phase stabilize the products 4a and 3a by 10 to 15 kcal mol−1 compared to the gas phase. In this regard the FLP reaction to 4a is in fact thermodynamically feasible. However, the competing product 3a is in solution significantly preferred over 4a (−50.7 kcal mol−1vs. −15.5 kcal mol−1) and due to the expected high mobility it can readily react and prevent the desired FLP reaction. In contrast, the crystal field provides much more pronounced energy barriers, which kinetically stabilizes the reactant and enables the targeted reaction to product 4a. These higher energy barriers can be rationalized by a simple geometric comparison (inset of Fig. 4). Apparently, the solid state reaction to 4a requires substantially less rearrangements of the crystal compared to 3a. Thus, we can identify two key roles that drive the reaction in the solid phase: (1) the crystal environment can adopt the solvent role in enhancing the FLP reactivity, typically explained by both the electrostatic screening and undirectional London dispersion stabilization of the FLP products. (2) The static crystal field can selectively suppress certain undesired reaction routes, which is not possible in a liquid or gas environment with high molecular mobility.
While point (1) makes the heterogeneous formulation of typical FLP reactions possible, (2) goes beyond it and opens possibilities for new FLP systems as compellingly demonstrated for the here discussed compound. An additional important prerequisite for the discussed reaction is the possible diffusion of H2 gas through the reactant crystal. Our molecular dynamics (MD) simulation (for details see the ESI†) confirm that H2 can actually move more or less freely through the channels of the crystal thereby generating the correct conditions for the H2 activation to take place. Moreover, we have conducted MD simulations for a model of the interface between the Lewis acid and base as it may occur experimentally in a mixture of the solid particles. According to these results which are shown in Fig. 5, at the interface the components (in particular the Lewis base) are spatially not constrained, possibly due to a mismatch of the molecular surfaces (a kind of interface strain). This leads partially to a “liquid phase” behavior of the FLPs in the solid state. It is seen that the Lewis acid and base components could move rather freely at the contact surface and adopt molecular FLP conformations enabling hydrogen activation as in solution (for details see the ESI†).
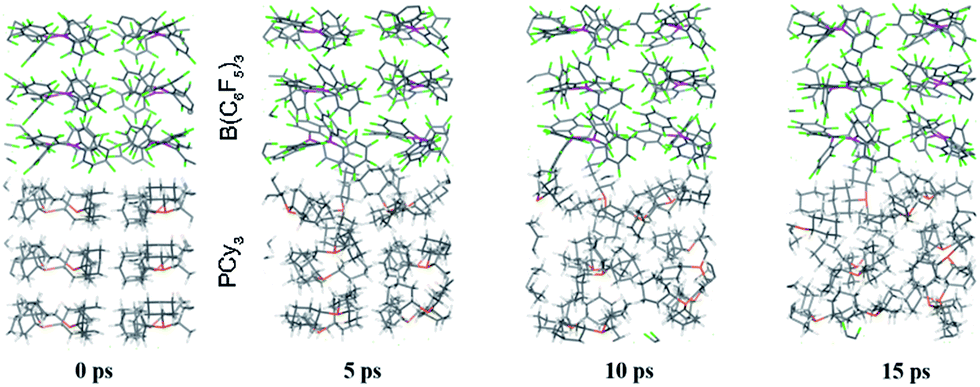 | ||
| Fig. 5 Snapshots of the periodic BO-MD simulation at the DFTB-D3 level of theory for the PCy3 + B(C6F5)3 FLP (1a/2). Color legend: P yellow, B pink, C black, F green and H white. | ||
The new solid state phosphane/borane FLP reactions are not limited to the splitting of dihydrogen. We exposed the 1:1 mixture of PCy3 (1a) and B(C6F5)3 (2) for 4 hours at r.t. in the solid state to SO2 gas (1.5 bar).47 A sample was dissolved in CD2Cl2 and its NMR spectra revealed the almost quantitative formation of the P/B FLP SO2 addition product 5. [Scheme 1b (SO2)] the product was also directly identified from the solid obtained by solid state NMR spectroscopy (see the ESI†), indicating essentially quantitative conversion. The distorted four-coordinate boron environment in 5 is characterized by δiso = −0.6 ppm, and a nearly axially symmetric electric field gradient, with CQ = 1.54 MHz and ηQ = 0.15. The 31P MAS-NMR spectrum shows a single sharp signal at 51.5 ppm. In this case, 11B{31P} REDOR and 31P{11B} REAPDOR experiments consistently point towards a B⋯P internuclear distance of 450 pm, which is in good agreement with the distance of 434 pm from the crystal structure.
We performed the reaction on a preparative scale and isolated the product 5 as a white solid after recrystallization from CH2Cl2/pentane in 84% yield. The product shows the typical 11B (δ −0.3) and 19F NMR features (three resonances, Δδ19Fm,p = 6.4 ppm) of the borate section of the molecule and a phosphonium 31P NMR signal at δ 50.0 (for further details see the ESI†).
Compound 5 was characterized by X-ray diffraction (Fig. 6). The X-ray crystal structure analysis shows the newly formed P1–S1 and O2–B1 bonds. Both the phosphorus and the boron atoms show pseudo-tetrahedral coordination geometries. The sulfur coordination geometry is distorted trigonal-pyramidal. We also exposed the PCy3/B(C6F5)3 pair to SO2 in solution but only observed the formation of the substitution product 3a [Scheme 1a (SO2); see the ESI† for details].
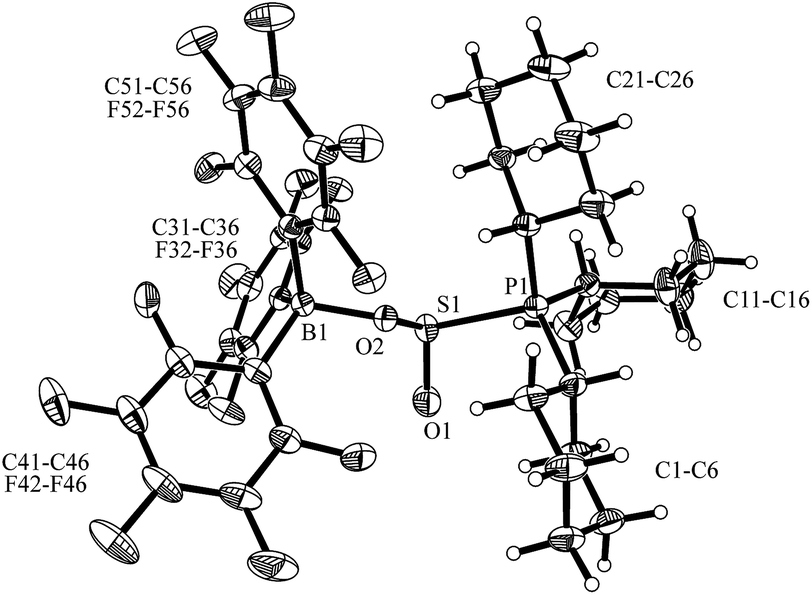 | ||
| Fig. 6 Molecular structure of the solid state P/B FLP SO2 addition product 5. Selected bond lengths (Å): S1–O1 1.445(2), S1–O2 1.564(2), P1–S1 2.268(1), B1–O2 1.545(3) and bond angles (°): O1–S1–O2 110.8(1), O1–S1–P1 106.3(1), O2–S1–P1 93.4(1), ΣB1CCC 334.0, ΣP1CCC 334.1. | ||
Our study has shown so far that an agitated mixture of particles of the phosphanes 1a–c with particles of the B(C6F5)3 Lewis acid 2 did very effectively evade the deactivating SNAr reaction that these pairs rapidly undergo in solution. Instead, they retained their frustrated Lewis pair character and, consequently, showed the ability to split dihydrogen heterolytically. While this observation is probably of a far-reaching principal interest, the rather harsh conditions of the solid state FLP H2-splitting (50 bars of dihydrogen, 3 to 10 d reaction time) made this far from a conveniently applicable procedure.
Fluorous liquids show some extraordinary properties.48–55 They do not mix with a variety of common organic solvents; they show an enhanced solubility of many gases in them, among them dihydrogen.48,55–57 Moreover, many organic and element-organic compounds, among them the phosphanes 1a–c and B(C6F5)3 (2) are insoluble in them. Therefore, we decided to carry out the solid state FLP dihydrogen splitting reaction in perfluoromethylcyclohexane (F11C6–CF3). In a typical experiment (see the ESI† for details) we suspended an equimolar mixture of PCy3 (1a) and B(C6F5)3 in perfluoromethylcyclohexane and stirred the suspension for 10 h in a dihydrogen atmosphere at near to ambient conditions (r.t., 1.5 bar H2). Workup was simply done by evaporation of the volatiles. A sample of the obtained white powdery solid was then subjected to NMR analysis in D2-dichloromethane solution. It showed that a ca. 60% conversion to the hydrogen splitting product HPCy3+/HB(C6F5)3− (4a) had been achieved. The remaining starting material had become converted to the SNAr reaction product 3a under the conditions of the NMR analysis in solution. The solid state NMR spectra of the products obtained after the suspension reaction showed the formation of 4a.
The reaction of the PPhCy2 (1b)/B(C6F5)3 pair with dihydrogen proceeded at least equally well in this fluorous liquid. Under analogous conditions a ca. 95% conversion to the H2-splitting product 4b was achieved within the 10 h reaction time. The PPh2tBu (1c)/B(C6F5)3 system even slightly surpassed this result. We obtained a near to quantitative conversion to the HPPh2tBu+/HB(C6F5)3− salt within 10 h at near to ambient conditions in the inert perfluoromethylcyclohexane liquid (Table 1, see the ESI† for details).
| No. | solutionc | Dry solid | In F11C6–CF3b | |
|---|---|---|---|---|
| a 10 days, r.t., 50 bar H2. b 10 hours, r.t., 1.5 bar H2. c D 2-Dichloromethane, 12 hours, r.t., 50 bar H2. d 3 days, r.t., 50 bar H2. | ||||
| PCy3 | 1a | Only 3a | >90% conv. to 4aa | 60% conv. to 4a |
| PPhCy2 | 1b |
3b:4b ∼ 6:1 |
ca. 95% conv. to 4bd | ca. 95% conv. to 4b |
| PPh2tBu | 1c |
3c:4c ∼ 2:1 |
ca. 95% conv. to 4cd | ca. 98% conv. to 4c |
Conclusions
Our study has shown that frustrated Lewis pair behavior can be achieved by other means than the usual electronic3,19 or steric modifications4–9 of Lewis acids and bases. In the cases reported here we have deviated from the ubiquitous method of using sterically bulky substituents at the phosphorus Lewis base in order to prevent neutralizing adduct formation or other deactivating reactions with the strong B(C6F5)3 boron Lewis acid. On the contrary, the phosphanes used in this study are rather nucleophilic and in solution they undergo rapid nucleophilic aromatic substitution at one C6F5 ring of the Lewis acid replacing fluoride which consequently then becomes bonded to the boron atom31 with annihilation of its Lewis acidic features. In all the cases looked at in our study this FLP deactivating reaction is very efficiently suppressed by localizing the individual Lewis acidic and Lewis basic molecules inside their solid state lattices. This prevents them effectively from undergoing the bimolecular deactivation reaction.From the solid state NMR and the DFT analysis in conjunction with general principles about molecular diffusion in the solid state, we assume that the individual Lewis acid and base components do not easily mix on a molecular level in our experiments, but that initially we are dealing with separate solid state phosphane and borane particles. This lets us assume that the FLP dihydrogen splitting reaction must take place at the surface, respectively the interface between phosphane and borane solid state entities; the ensuing reaction is, however, probably facilitated by the easy permeability of the respective crystal lattices by dihydrogen (and other gases). The dihydrogen splitting reaction may then have formed the phosphonium/hydridoborate salt initially at the surface, but it may be assumed that accumulation of that species creates a local situation resembling an ionic liquid, which might facilitate diffusion and mixing since eventually we obtained homogeneous solid samples of the respective dihydrogen splitting products. The MD simulations which support this view are currently due to the large system size too approximate (short) to draw any quantitative conclusions nor are we able to simulate further parts of the solid state reaction dynamically. Nevertheless, the MD and static theoretical results clearly support the above described picture of partially “molten” material at the interface with sufficient molecular flexibility for activation of almost freely diffusing molecular hydrogen.
Although the mechanistic aspects of our solid state FLP reactions must remain somewhat speculative at this time, we have greatly improved its practical applicability by using the fluorous liquid effect.55–57 This has made dihydrogen splitting reactions readily available from frustrated Lewis pair combinations which cannot be kept active in other ways.33 We have further found first indications that the resulting [P]H+/[B]H− product 4a can be usefully employed in imine reduction and we have shown that the solid state FLP reactions are not confined to the dihydrogen splitting reactions but can be developed beyond. The solid state approach can make FLPs available for small molecule activation beyond using the conventional methods leading to Lewis pair “frustration”. It needs to be explored if this will open new pathways of extending FLP chemistry beyond its existing scope, for example by opening FLP routes to the large field of heterogeneous catalysis, here to be performed without the aid of metals.27–30
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (G. E.) and the Deutsche Forschungsgemeinschaft (Leibniz award (S. G.)) is gratefully acknowledged. We thank Prof. Beat Meier and the ETH Zürich for supporting the high field solid state NMR measurements. L. W. did the synthetic work, C. G. D. did the X-ray crystal structure analyses, G. K. supervised the work and checked the experimental data, G. E. initiated and supervised the study, M. B., T. W., A.-L. W. and H. E. did the solid state NMR studies, L. L, J. G. B. and S. G. did the theoretical study.Notes and references
- F. H. Stephens, V. Pons and R. Tom Baker, Dalton Trans., 2007, 2613–2626 RSC.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- A. Stute, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2013, 42, 4487–4499 RSC.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Frohlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 1–350 CrossRef CAS PubMed.
- Y. Hasegawa, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 12168–12171 CrossRef CAS PubMed.
- M. Sajid, L.-M. Elmer, C. Rosorius, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 2243–2246 CrossRef CAS PubMed.
- M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118–1121 CrossRef CAS PubMed.
- M. Sajid, A. Lawzer, W. Dong, C. Rosorius, W. Sander, B. Schirmer, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 18567–18574 CrossRef CAS PubMed.
- A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7567–7571 CrossRef CAS PubMed.
- M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156–10168 CrossRef CAS PubMed.
- R. Liedtke, F. Scheidt, J. Ren, B. Schirmer, A. J. P. Cardenas, C. G. Daniliuc, H. Eckert, T. H. Warren, S. Grimme, G. Kehr and G. Erker, J. Am. Chem. Soc., 2014, 136, 9014–9027 CrossRef CAS PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS PubMed.
- T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302–4305 CrossRef CAS PubMed.
- E. Theuergarten, J. Schlosser, D. Schluns, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2012, 41, 9101–9110 RSC.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2012, 18, 16938–16946 CrossRef PubMed.
- B. Inés, D. Palomas, S. Holle, S. Steinberg, J. A. Nicasio and M. Alcarazo, Angew. Chem., Int. Ed., 2012, 51, 12367–12369 CrossRef PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206–1214 CrossRef CAS PubMed.
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Phys. Chem. Chem. Phys., 2015, 17, 14623–14635 RSC.
- (a) S. Zhang, Z.-Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C.-R. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS PubMed, and references cited therein; (b) Z.-Q. Huang, L.-P. Liu, S. Qi, S. Zhang, Y. Qu and C.-R. Chang, ACS Catal., 2018, 8, 546–554 CrossRef CAS; (c) Review: N. J. Coville and L. Cheng, J. Organomet. Chem., 1998, 571, 149–169 CrossRef CAS.
- (a) J.-Y. Xing, J.-C. Buffet, N. H. Rees, P. Norby and D. O'Hare, Chem. Commun., 2016, 52, 10478–10481 RSC; see also: (b) Y.-J. Wanglee, J. Hu, R. E. White, M.-Y. Lee, S. M. Stewart, P. Perrotin and S. L. Scott, J. Am. Chem. Soc., 2012, 134, 355–366 CrossRef CAS PubMed; (c) K. C. Szeto, W. Sahyoun, N. Merle, J. L. Castlebou, N. Popoff, F. Lefebvre, J. Raynaud, C. Godard, C. Claver, L. Delevoye, R. M. Gauvin and M. Taoufik, Catal. Sci. Technol., 2016, 6, 882–889 RSC.
- S. Döring, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Organometallics, 1998, 17, 2183–2187 CrossRef.
- M. Ullrich, A. J. Lough and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 52–53 CrossRef CAS PubMed.
- Y. Jiang, B. Schirmer, O. Blacque, T. Fox, S. Grimme and H. Berke, J. Am. Chem. Soc., 2013, 135, 4088–4102 CrossRef CAS PubMed.
- T. Wiegand, H. Eckert, O. Ekkert, R. Fröhlich, G. Kehr, G. Erker and S. Grimme, J. Am. Chem. Soc., 2012, 134, 4236–4249 CrossRef CAS PubMed.
- C. Jaeger and F. Hemmann, Solid State Nucl. Magn. Reson., 2014, 57–58, 22–28 CrossRef CAS PubMed.
- A. Böckmann, M. Ernst and B. H. Meier, J. Magn. Reson., 2015, 253, 71–79 CrossRef PubMed.
- T. Wiegand, M. Siedow, H. Eckert, G. Kehr and G. Erker, Isr. J. Chem., 2015, 55, 150–178 CrossRef CAS.
- R. Sure and S. Grimme, J. Comput. Chem., 2013, 34, 1672–1685 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- J. G. Brandenburg and S. Grimme, J. Phys. Chem. Lett., 2014, 5, 1785–1789 CrossRef CAS PubMed.
- L. Liu, J. G. Brandenburg and S. Grimme, Philos. Trans. R. Soc., A, 2017, 375, 20170006 CrossRef PubMed.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- J. A. Gladysz, D. P. Curran and I. T. Horvath, Handbook of Fluorous Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- I. Ryu, H. Matsubara, C. Emnet, J. A. Gladysz, S. Takeuchi, Y. Nakamura and D. P. Curran, in Green Reaction Media in Organic Synthesis, Blackwell Publishing Ltd, 2007, pp. 59–124 Search PubMed.
- H. Nakamura, T. Usui, H. Kuroda, I. Ryu, H. Matsubara, S. Yasuda and D. P. Curran, Org. Lett., 2003, 5, 1167–1169 CrossRef CAS PubMed.
- D. P. Curran, Aldrichimica Acta, 2006, 39, 3–9 CAS.
- D. P. Curran, in Stimulating Concepts in Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 25–37 Search PubMed.
- A. Studer, S. Hadida, R. Ferritto, S.-Y. Kim, P. Jeger, P. Wipf and D. P. Curran, Science, 1997, 275, 823–826 CrossRef CAS PubMed.
- A. P. Dobbs and M. R. Kimberley, J. Fluorine Chem., 2002, 118, 3–17 CrossRef CAS.
- E. de Wolf, G. van Koten and B.-J. Deelman, Chem. Soc. Rev., 1999, 28, 37–41 RSC.
- J. A. Gladysz and M. Jurisch, Top. Curr. Chem., 2012, 308, 1–24 CAS.
- G. Serratrice, J. J. Delpuech and R. Diguet, Nouv. J. Chim., 1982, 6, 489 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, further spectral and crystallographic data, additional data from the solid state NMR and theoretical studies. CCDC 1515080–1515082. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01089g |
| This journal is © The Royal Society of Chemistry 2018 |