
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free borylative dearomatization of indoles: exploring the divergent reactivity of aminoborane C–H borylation catalysts†
Arumugam
Jayaraman
 ,
Luis C.
Misal Castro
,
Vincent
Desrosiers
and
Frédéric-Georges
Fontaine
*
,
Luis C.
Misal Castro
,
Vincent
Desrosiers
and
Frédéric-Georges
Fontaine
*
Département de Chimie, Centre de Catalyse et de Chimie Verte (C3V), Université Laval, Québec City, Québec, Canada G1V 0A6. E-mail: frederic.fontaine@chm.ulaval.ca
First published on 7th May 2018
Abstract
While the dearomatization of indoles by carbon–boron bond forming reactions is new and quite promising, they are so far mainly metal-catalyzed. Here, we establish the use of metal-free catalysts in promoting such reactions in an atom-efficient way. The in situ generated ambiphilic aminoborane catalyst (1-Pip-2-BH2-C6H4)2 (Pip = piperidyl) promotes borylative dearomatization of various 1-arylsulfonyl indoles with pinacolborane in a syn addition fashion, with H and Bpin groups added respectively to the 2 and 3 positions of indoles. Catalysis proceeds with good to excellent conversion and essentially with complete regio- and diastereoselectivity. From mechanistic insights and DFT computations, we realized and established that prototypical boranes can also catalyze this borylative dearomatization.
Dearomative functionalization of indoles is of high interest for synthetic chemists and chemical industries to generate the structural motifs of numerous pharmaceuticals, natural products, and materials (Fig. 1).1 While this approach appears more reasonable in terms of the number of synthetic steps, achieving it is usually hard due to the strong aromatic resonance delocalization in heteroarenes,2 which disfavours the kinetics and thermodynamics of dearomatization.2,3 Despite this hurdle, many methods have been developed in recent years to operate these transformations.4 Dearomatization with the introduction of a boron-containing moiety is attractive since organoboron products with a C(sp3)–B bond can readily serve as coupling partners in a variety of metal-catalyzed cross coupling reactions5 and in the emerging metal-free coupling reactions.6 Some examples of stoichiometric and catalytic borylative dearomatization of heteroarenes, mostly pyridine derivatives, have been reported.7 Notably, Rh(I),7f Mg(II)7a and La(III)7c complexes (Scheme 1a) and boron-based species have been used as catalysts.7e In all these stoichiometric and catalytic reactions, dearomatization occurs as a result of the 1,2-addition of H and Bpin groups with boron typically adding to the nitrogen atom of pyridines. In 2015, Ito and co-workers reported the borylative dearomatization of N-protected indoles with bis(pinacolato)diboron using Cu(I) catalysts,8 which forms C–B bonds and affords regio-, diastereo- and enantioselective 3-borylated indolines with the H and Bpin groups added respectively at the 2 and 3 positions in an anti-fashion (Scheme 1a).
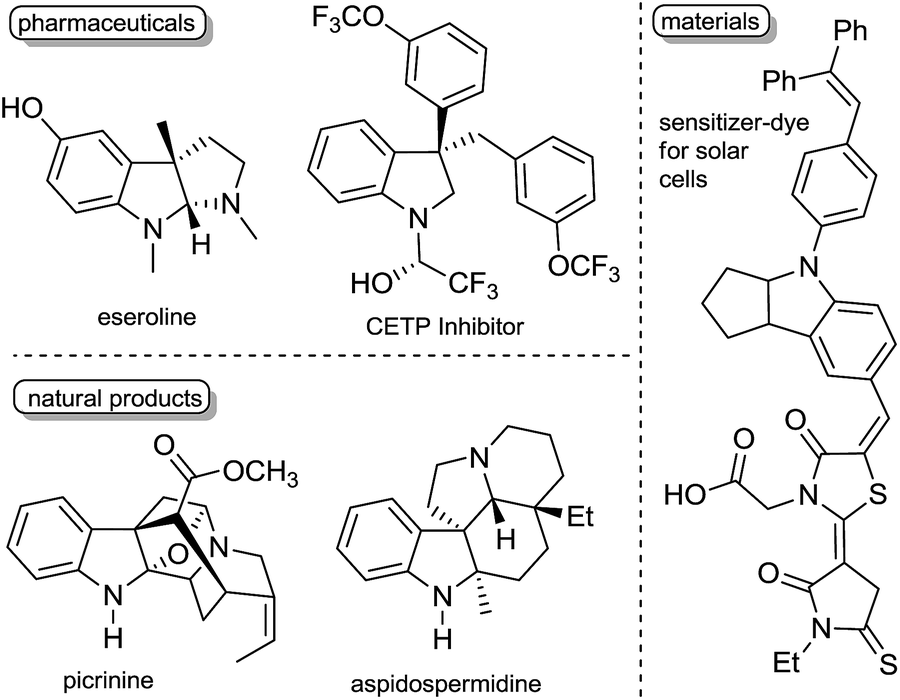 | ||
| Fig. 1 Representative highly valuable compounds with an indoline core. | ||
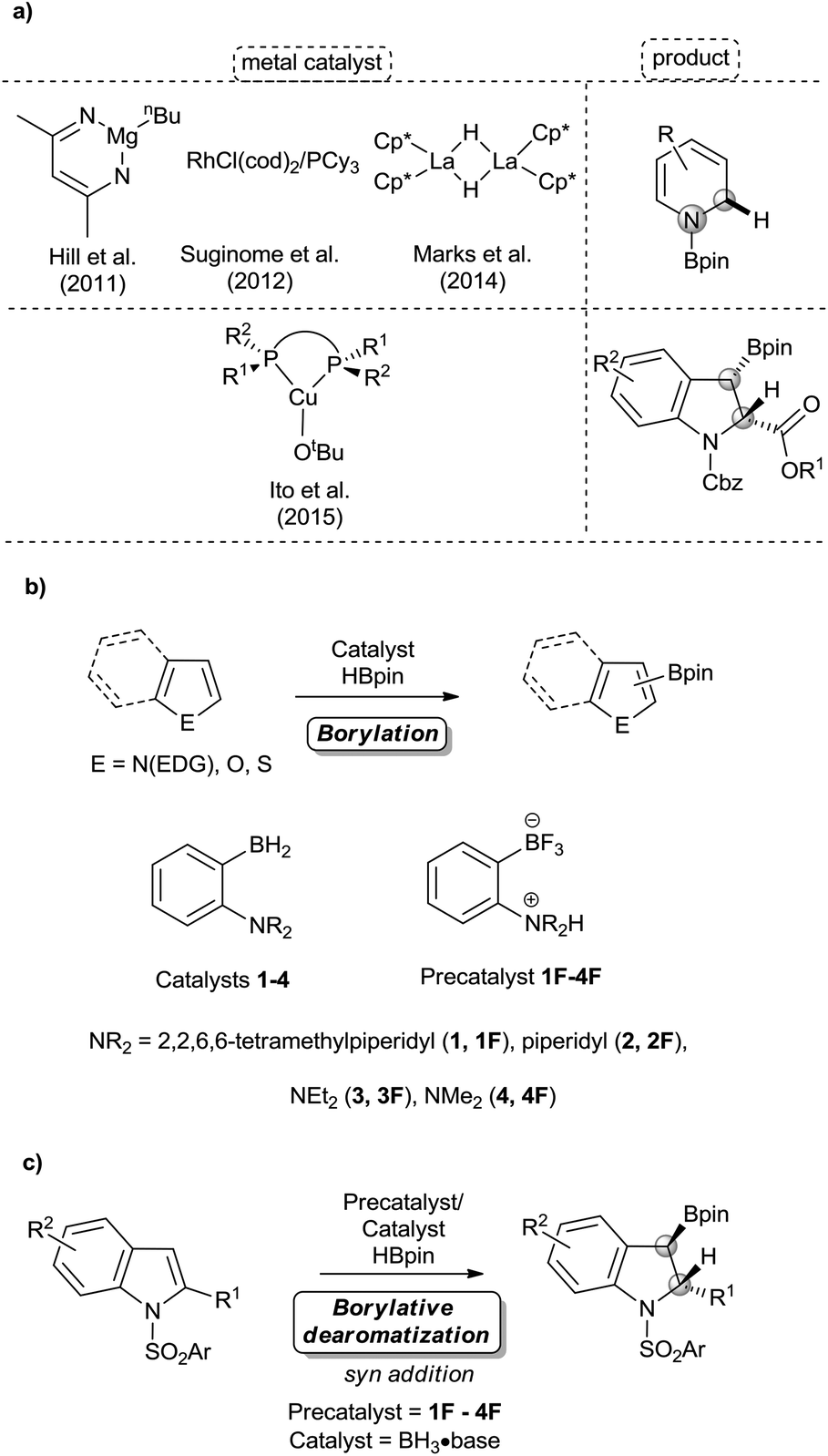 | ||
| Scheme 1 Catalytic borylation and borylative dearomatization reactions of heteroarenes: (a) previous studies on catalytic borylative dearomatizatization of heteroarenes, (b) our previous work on the B/N FLP catalyzed C–H borylation reaction, and (c) this work on B/N FLP catalyzed borylative dearomatizatization of indoles. EDG = electron donating group; Cp* = pentamethylcyclopentadienyl; Cbz = benzyloxycarbonyl. | ||
In the same year, we described a novel metal-free approach for the borylation of unactivated electron-rich heteroarenes, using pinacolborane (HBpin) as the reagent and ambiphilic aminoborane (1-TMP-2-BH2-C6H4)2 (1, TMP = 2,2,6,6-tetramethylpiperidine, Scheme 1b) as the catalyst.9 The primary mechanistic step of this approach is the concerted C–H activation of heteroarenes by the ambiphilic aminoborane catalyst using the concept of frustrated Lewis pair (FLP) chemistry.10
Although the transition metal-free borylation of heteroarenes using electrophilic boron reagents was already known,11 additional metal-free systems for the borylation reaction appeared in the literature after our report.4e,12 We subsequently showed that the bench-stable fluoroborate salt of 1,1-TMP(H)-2-BF3-C6H4 (1F), can be used as a precatalyst for this reaction.13 Recently, we have expanded this new borylation methodology by designing more active ambiphilic aminoborane catalysts (2–4, Scheme 1b).14 We wish to report here that while covering a wide variety of heteroarenes, some heterocycles undergo borylative dearomatization rather than borylation through C–H activation. Indeed, the syn addition and borylative dearomatization of indoles are catalyzed by in situ generated metal-free ambiphilic aminoborane catalysts (Scheme 1c). From the mechanistic insights rationalizing the preference of the FLP catalysts for either activation modes, we demonstrate that prototypical boranes of the form BH3·base can also catalyze the same borylative dearomatization reaction. Our metal-free approach towards this transformation is atom-efficient and essentially provides regio- and diastereoselective products in good to excellent yields.
Although compounds 1–4 catalyze exclusively the C–H borylation of 1-methyl indole in presence of HBpin, we observed that the substrate 1-tosyl indole rather undergoes a divergent route by performing borylative dearomatization, installing the H and Bpin moieties respectively at the 2- and 3-positions (Scheme 2). Although all four in situ generated ambiphilic aminoboranes catalyze the borylative dearomatization of 1-tosyl indole, we chose to use the piperidine-based precatalyst 2F for further exploration, as its preparation is more convenient. As shown in Table 1, treatment of 1-tosyl indole with 1.3 equiv. of HBpin and 5 mol% of 2F in CDCl3 at 100 °C resulted in the formation of borylative dearomatized product 6a with 67% conversion after 16 h (entry 2). As expected, this reaction does not proceed in the absence of a precatalyst (entry 1). Increasing the precatalyst and HBpin loadings to 10 mol% and 1.5 equiv., respectively, and decreasing the reaction time to 6 h provided again a moderate conversion (entry 3). However, extending the reaction time to 16 h gave a conversion of 99% (entry 4). Although most experiments were carried out in CDCl3, it is possible to change the solvent to THF and 2-Me-THF (entries 5 and 6).
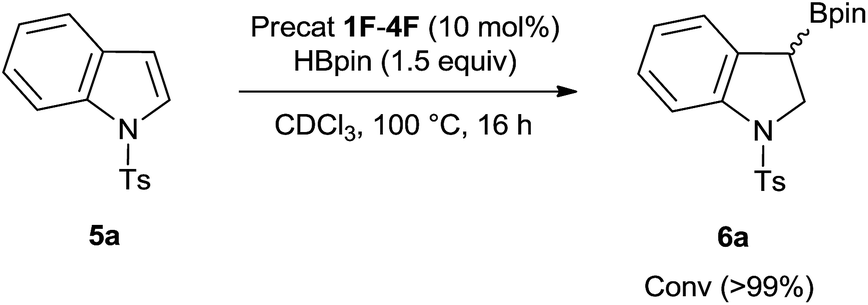 | ||
| Scheme 2 Borylative dearomatization of 1-tosyl indole catalyzed by in situ generated ambiphilic aminoboranes. Ts = SO2(C6H4CH3). | ||
| Entry | 2F (mol%) | HBpin (equiv.) | Conditionsa | Convb (%) |
|---|---|---|---|---|
| a Reaction temperature used is 100 °C. b Percent conversions are 1H NMR conversions determined with an aliquot vs. hexamethylbenzene as internal standard. | ||||
| 1 | 0 | 1.1 | CDCl3, 100 °C, 24 h | 0 |
| 2 | 5 | 1.3 | CDCl3, 100 °C, 16 h | 67 |
| 3 | 10 | 1.5 | CDCl3, 100 °C, 6 h | 62 |
| 4 | 10 | 1.5 | CDCl3, 100 °C, 16 h | 99 |
| 5 | 10 | 1.5 | THF, 100 °C, 16 h | 96 |
| 6 | 10 | 1.5 | 2-MeTHF, 100 °C, 16 h | 95 |
| 7 | 10 | 1.5 | Neat, 100 °C, 12 h | 99 |
Interestingly, this reaction comes to completion under neat conditions in less than 12 hours (entry 7). While the conversion is still taking place, albeit in significantly lower yields, in the presence of benzoic acid, dimethylaniline, and benzonitrile as additives, no product was observed in the presence of phenol, acetophenone and ethyl benzoate additives (see the ESI†).
Next, the scope of this catalytic C–B bond forming borylative dearomatization reaction using different 1-arylsulfonyl indoles was examined (Scheme 3). The 1-tosyl indoles with either electron-donating or electron-withdrawing functionalities at the 4- or 5-position undergo the borylative dearomatization reaction across the C2–C3 aromatic double bond with a high regioselectivity and good-to-excellent conversions (6c, 6e–6h, 6m, and 6o). Likewise, the 1-tosyl indoles possessing the methyl (6d), phenyl (6i), fluoro (6j), chloro (6k), or bromo (6l) substituents at the C6-position were well tolerated. In contrast, the electron-withdrawing nitro substituent at the C6-position of the 1-tosyl indole did not allow borylative dearomatization (6p). While it is possible to catalytically hydroborate 5-nitro-1-tosyl indole (6o) with a moderate conversion, the absence of reaction with 6-nitro-1-tosyl indole suggests that the strong electron withdrawing effect, combined with the negative inductive effect (−I) and negative mesomeric effect (−M), exerted by the nitro group on the para-positioned C3 carbon, is shutting down this process. Some of the formed hydroborated products are unstable under the routine workup procedure or in the presence of silica gel, and thus were subsequently oxidized to the corresponding alcohols using the commercially available household bleach comprised of 6% NaOCl, which is known to oxidize organoboranes stereoretentively.15 In some cases, the oxidized products were further functionalized with silyl protecting groups. The highly regioselective borylative dearomatization of different 1-arylsulfonyl indoles achieved using an ambiphilic aminoborane catalyst tempted us to disclose the diasteroselectivity of this metal-free approach. This was probed using 5-fluoro-2-methyl-1-phenylsulfonyl indole and 2-methyl-1-phenylsulfonyl indole substrates. Catalysis under optimal conditions afforded the syn addition products 6r and 6s, respectively, having the Bpin and methyl substituents positioned trans to each other. A strong support of the syn addition of H and Bpin groups comes from the X-ray crystallographic characterization of product 6r (Fig. 2, left). In the case of 2-methyl-1-phenylsulfonyl indole, the product 6s is less stable; therefore, it was derivatized to 2-methyl 3-siloxy 1-phenylsulfonyl indoline 6s′′ using the stereoretentive oxidant NaOCl (household bleach) and silylating reagent TBDMSCl. The X-ray crystallographic characterization of product 6s′′, as depicted in Fig. 2 (right), clearly indicates the formation of a syn addition product (6s).
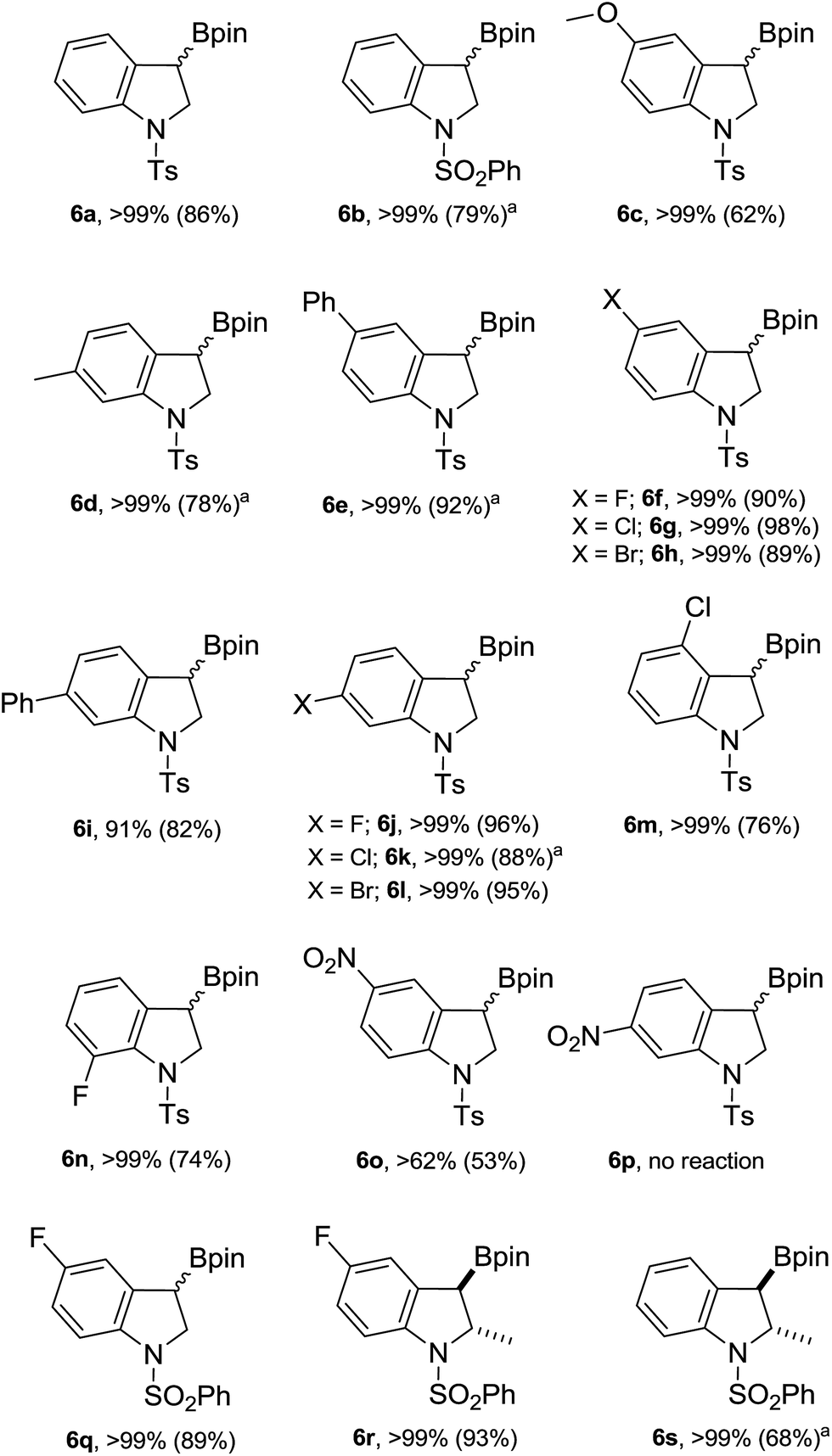 | ||
| Scheme 3 Scope of 1-arylsulfonyl indoles. Reaction conditions: 1-arylsulfonyl indoles (0.5–1.0 mmol), precatalyst 2F (10–20 mol%), HBpin (1.5–2.6 equiv), 100 °C, CDCl3/neat, 16–24 h. The numbers in parentheses are the isolated yield. aIndicates the isolated yield after oxidation. | ||
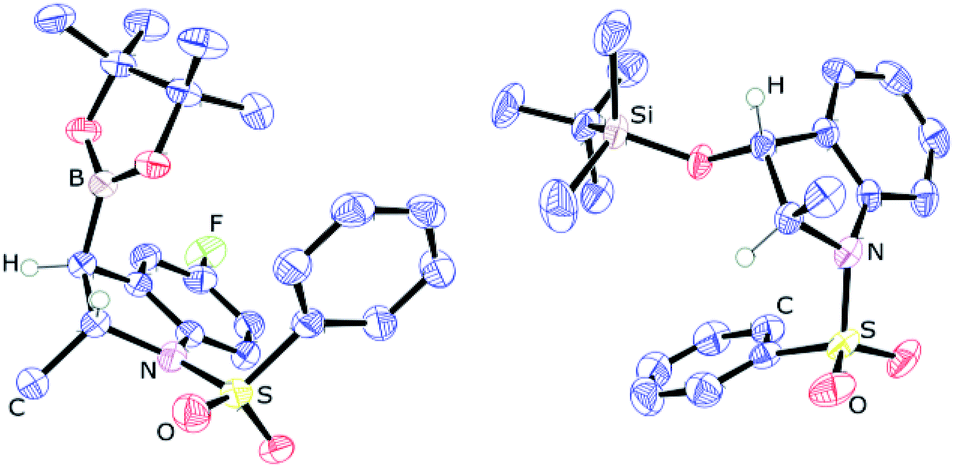 | ||
| Fig. 2 ORTEP diagrams showing the molecular structure of 6r (left) and 6s′′ (right). Thermal ellipsoids are shown at the 50% level, and less informative hydrogens have been omitted for clarity. | ||
Notably, in the framework of transition metal catalysis and organocatalysis, this is the first catalytic method for borylative dearomatization of indoles which ensures addition in a syn fashion. Moreover, this metal-free methodology can be considered complementary to the existing group 10 metal-catalyzed borylative dearomatization of 1-Cbz indoles (Cbz = benzyloxycarbonyl) in which the H and Bpin groups are added in an anti-fashion.8 Our observation and generalization of borylative dearomatization of 1-arylsulfonyl indoles using the same catalyst that can catalyze C–H borylation of electron-rich heteroarenes demonstrates the divergent ability of our catalytic system. The divergent ability of our catalysts also includes the recent report on S–H borylation of thiols.16 Because the synthesis of precatalyst 2F is expedient and the catalytic conversions are quantitative in most cases, we have demonstrated that this metal-free catalysis can be applied to gram-scale synthesis, via preparing 6a in 2 grams from 5a under neat conditions.
A plausible mechanism for the borylative dearomatization reaction is shown in Scheme 4.17 The initial step of this catalytic cycle involves cleavage of the dimeric catalyst. The active monomeric catalyst then undergoes hydroboration with the substrate, which leads to the formation of the 3-borylated intermediate (A1), a possible resting state in the catalytic cycle. The σ-bond metathesis between the C(sp3)–B bond of A1 and the H–B bond of HBpin, as observed in the catalyzed C(sp2)–H borylation of heteroarenes, is unlikely since our computational attempts to optimize the corresponding transition state using catalyst 4 led to the dissociation of HBpin from A1. Thus, we propose that the redistribution of the backbone leads to the product and regeneration of our catalyst. Several precedents of redistribution reactions between RBH2 and R′B(OR′′)2 have been reported (vide infra).18
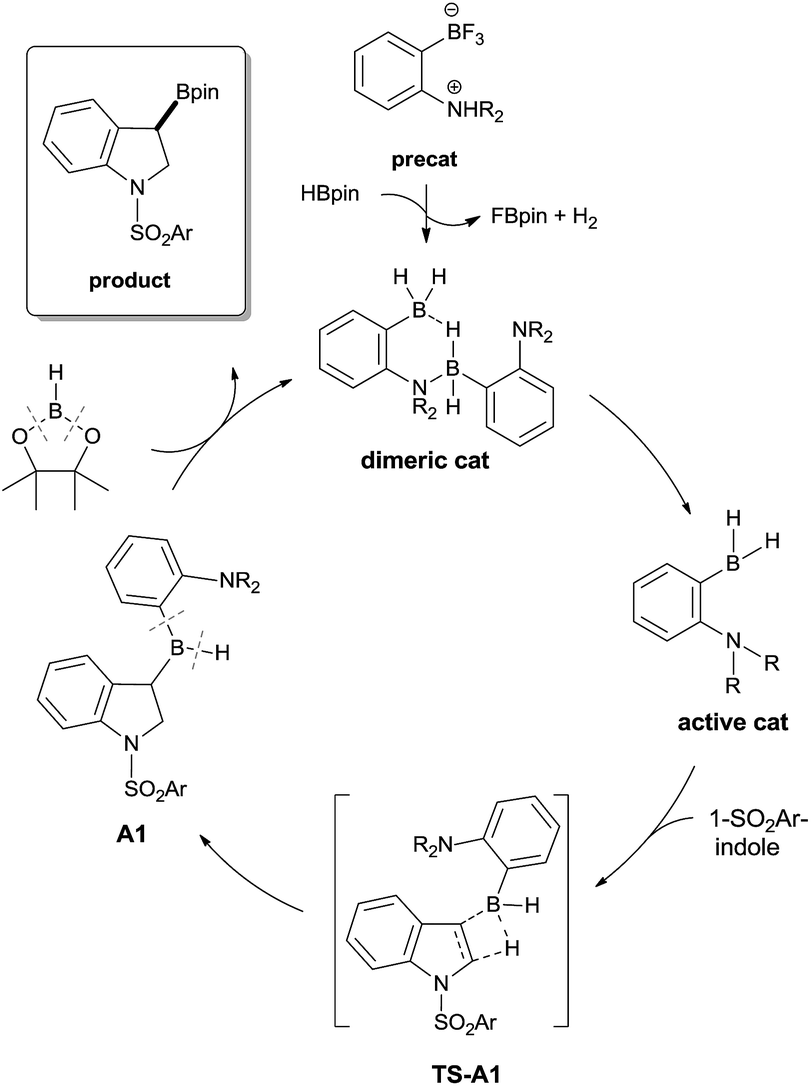 | ||
| Scheme 4 Plausible mechanism for the ambiphilic aminoborane catalyzed borylative dearomatization. | ||
To investigate the proposed initial hydroboration step and to compare it with the possible C–H activation, the activation barriers were computed, using the model chemistry ωB97XD/6-311G+**/PCM(chloroform)//ωB97XD/6-31G** (ref. 19) as implemented in the Gaussian 09 package,20 with the dimethyl catalyst 4. Computations show a barrier of 25.9 kcal mol−1 for the hydroboration of 1-phenylsulfonyl indole while the C–H activation step is higher in energy at 27.8 kcal mol−1, suggesting that the former mechanism is favored by 1.9 kcal mol−1 (Fig. 3, top). In the case of more electron-rich 1-methyl indole, the C–H activation is preferred over hydroboration as the barrier for activation is lower by about 5.2 kcal mol−1 (Fig. 3, bottom), which is in accordance with our experimental observations.9 Since the hydroborated intermediate A1 is 2.8 kcal mol−1 higher in energy compared to the starting materials, this intermediate was not observed experimentally. Indeed, 1H NMR and ESI mass spectrometry of the CDCl3 solution of equimolar catalyst 2 and 1-tosyl- or 1-phenylsulfonyl indole showed only unreacted starting material and no detectable intermediate was seen when the solution was heated at 100 °C for 16 h. These results suggest that the catalytic hydroboration is a competing reaction for the catalytic borylation reaction. Hydroboration dominates when the aromatic π-delocalization in heteroarenes is significantly reduced, and the borylation, through C–H activation, dominates when the π-delocalization is intact or enhanced. This difference in the reactivity pattern explains how our ambiphilic aminoboranes show a divergent catalytic activity according to the nature of the 1-substitution on indoles.9,14
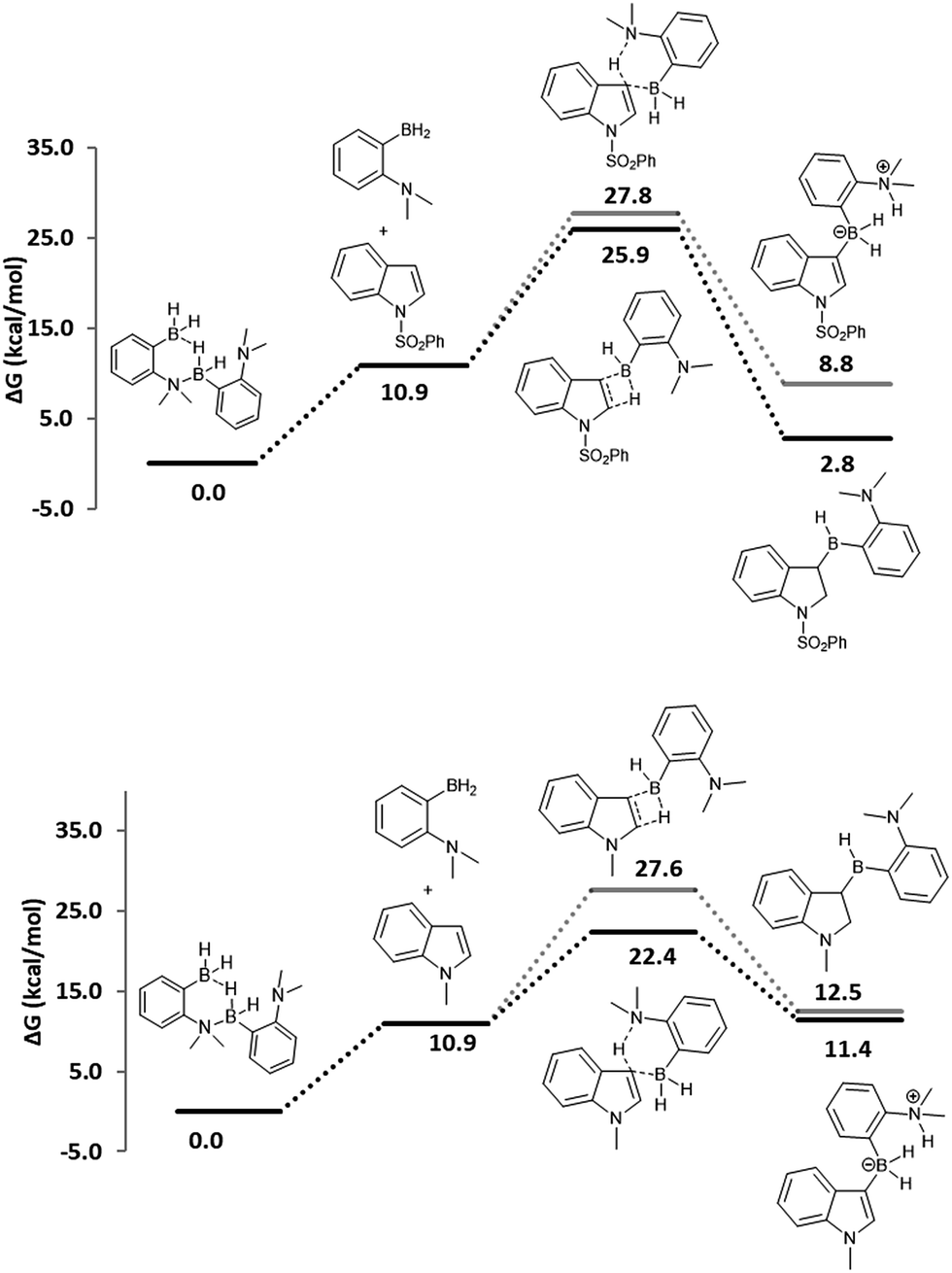 | ||
| Fig. 3 DFT computed free energy profiles for hydroboration vs. C–H activation of 1-phenylsulfonyl indole (top) and 1-methyl indole (bottom) by the ambiphilic aminoborane catalyst 4. | ||
As the computational evidence suggests that the amino group is not required for catalysis, we next proceeded with simple primary and secondary boranes not having an ambiphilic character. Catalysis did not occur when using only catecholborane (HBcat) or the simple phenylborane, generated in situ from tetrabutylammonium trifluoro(phenyl)borate. Surprisingly, the prototypical boranes BH3·base, often reflected as a “trojan horse” in catalytic or stoichiometric hydroboration reactions,18b,21 catalyze the borylative dearomatization quantitatively with no change in the regio- and diastereoselectivity (Scheme 5). Further optimization of this approach using many different BH3 adducts revealed that the BH3·DMS (dimethyl sulfide) and BH3·THF (tetrahydrofuran) adducts are the more efficient catalysts (see Table S2 of the ESI†), as the reaction slowly proceeds even at room temperature. At 100 °C, the reaction comes to completion within 1 h, which is much faster than that observed for all other examined BH3 adducts and aminoborane catalysts. Next, the group of substrates that was successfully transformed using precatalyst 2F was examined with the BH3·DMS catalyst (Table S3 of the ESI†). The same selectivity observed using precatalyst 2F was observed with the exception that BH3 catalyzes the reaction at lower temperature (60 °C) and faster. To demonstrate the viability of this process, product 6q was prepared on a two gram-scale using BH3·DMS as the catalyst. A main advantage of using BH3·base as catalysts is that the purification of the products is much easier as it involves only evaporation of the catalysts and excess reagents in vacuo.
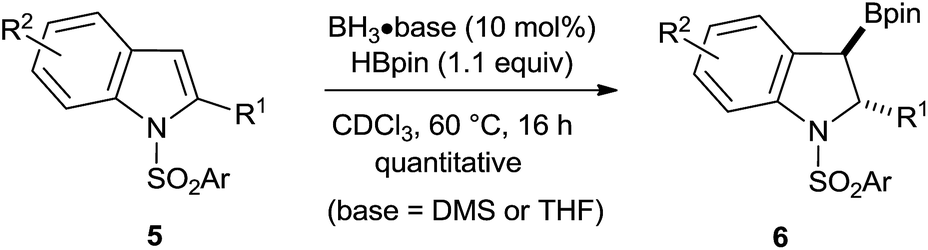 | ||
| Scheme 5 Borylative dearomatization of indoles catalyzed by prototypical boranes. | ||
The mechanism proposed for the BH3·base catalyzed transformation is shown in Fig. 4 and is identical to the one depicted in Scheme 4 for the aminoborane catalysts. Computations show that the initial hydroboration step is exergonic, leading to the formation of hydroborated BH2 dimer, B2 (−8.9 kcal mol−1), which is in equilibrium with the monomeric dimethyl sulfide adduct B2′ (−5.6 kcal mol−1). The unprotected monomeric form B2′′ is much higher in energy (8.1 kcal mol−1). This was demonstrated using NMR spectroscopy, where we observed that the stoichiometric reaction of BH3·DMS with 1-phenylsulfonyl indole at 60 °C for 16 h led to a single hydroboration product in equilibrium between the borane-dimethylsulfide adduct and the diborane dimer. As mentioned above, our inability to observe the similar intermediate from the reaction between 1-phenylsulfonylindole and ambiphilic aminoborane catalysts can be rationalized by thermodynamics of this addition step. The DFT results show that the addition is endergonic (2.8 kcal mol−1 for catalyst 4, Fig. 3) for the aminoborane catalyst, while it is exergonic in the case of BH3 catalysts. In the second step of the mechanism, scrambling of the backbone occurs between this newly formed BH2-intermediate (B2) and HBpin. The scrambling between hydrides and alkoxy groups between primary boranes and secondary boronic esters is well known.18b,22 The first hydride switch leads to the ring-opened intermediate B3 and is endergonic by 12.9 kcal mol−1, with a rate limiting barrier of 23.7 kcal mol−1. The final step involving the second hydride switch is exergonic by −16.9 kcal mol−1 with a barrier of 9.2 kcal mol−1. The direct formation of the final product B4 from B2via a direct σ-bond metathesis between the C–B bond of B2 and the H–B bond of HBpin has a barrier of 34.4 kcal mol−1 (not shown, see the ESI†); thus, this pathway was considered kinetically uncompetitive in comparison to the backbone redistribution process.
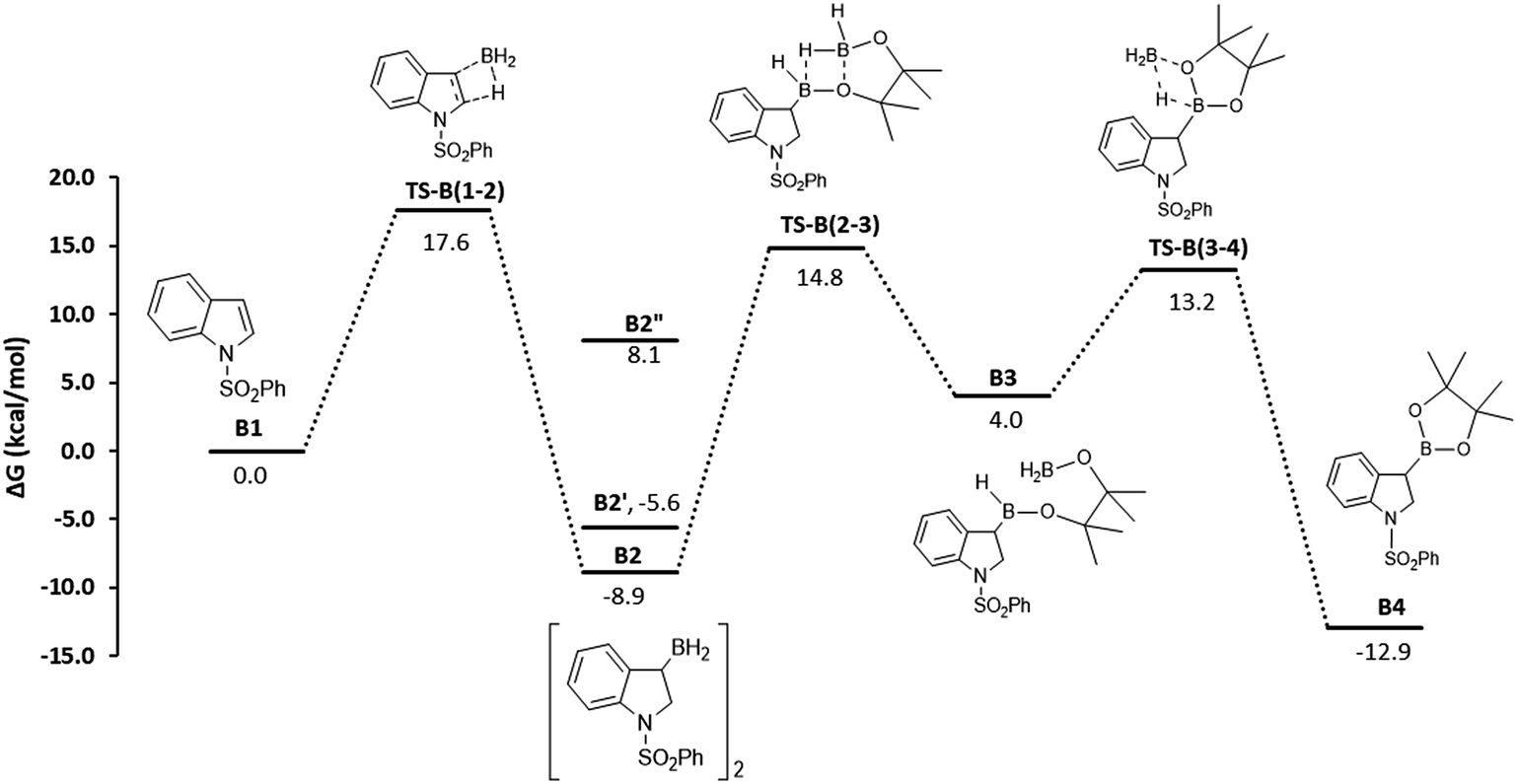 | ||
| Fig. 4 DFT-computed free energies, in kcal mol−1, for the BH3 catalyzed borylative dearomatization of 1-phenylsulfonyl indole. Species B2′ is the dimethyl sulfide adduct, and B2′′ is the monomeric form. | ||
It was recently demonstrated by Thomas and co-workers that a variety of alkenes could undergo hydroboration with HBpin as a reagent and the BH3·base acting as catalysts.23 However, the hydroboration of indoles to generate 3-borylindolines is scarce in the scientific literature even if this transformation is synthetically useful for the construction of complex molecular structures for the pharmaceutical industry.24 While protection of sensitive functionalities is required to apply the borylated products in synthetic applications such as for cross-coupling reactions, many mild and efficient deprotection strategies were well documented for the deprotection of the tosyl group after the execution of a desired transformation.25 We should also note that the borylative dearomatized products can also be readily obtained by the stoichiometric hydroboration of tosyl indoles with BH3·DMS followed by the addition of pinacol, as exemplified here with the 1-tosyl indole substrate in Scheme 6. This route is cheaper relative to the other synthetic routes described herein, but the ability to modify the framework of aminoborane catalysts could potentially lead to asymmetric induction, which prototypical boranes cannot do.
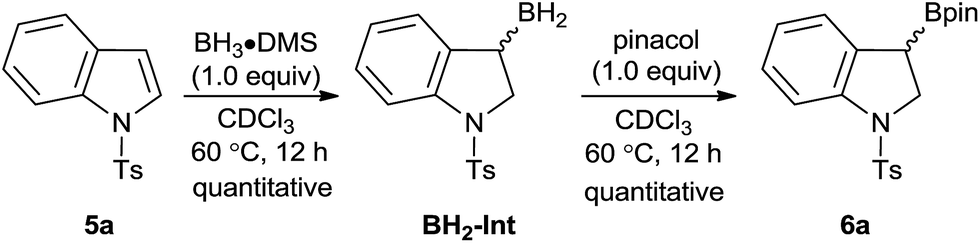 | ||
| Scheme 6 Stoichiometric borylative dearomatization reaction of 1-tosylindole using BH3·DMS and pinacol. Species BH2-Int likely exists in equilibrium between its DMS adduct and the diborane dimer form. | ||
In summary, we have established the catalytic use of a bench-stable precatalyst, 1-Pip(H)-2-BF3-C6H4 (2F), for the C–B bond forming borylative dearomatization of indoles. The in situ generated ambiphilic aminoborane catalyzes borylative dearomatization of various 1-arylsulfonyl indoles with HBpin and proceeded in a syn addition fashion with a high conversion and with complete regio- and diasteroselectivity. From the mechanistic insights, we recognized and demonstrated that prototypical boranes can also catalyze the same reactions with an improvement in the catalytic efficiency. The metal-free catalytic approach we demonstrated can be considered complementary to the existing transition metal-based catalytic system where an anti-addition of H and Bpin is followed exclusively. Eventhough prototypical boranes are being efficient to catalyze borylative dearomatization of N-arylsulfonyl indoles, utilizing ambiphilic aminoboranes as catalysts is still advantageous as they can become chiral catalysts when decorated with chiral substituents and may enable the enantioselective borylative dearomatization reactions. To achieve this, asymmetric ambiphilic borane catalytic systems are currently being developed in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Sciences and Engineering Research Council of Canada (NSERC) and the Centre de Catalyse et Chimie Verte (Quebec) for funding, BASF for the generous supply of pinacolborane, Prof. Jean-François Paquin (Université Laval) and Prof. Alexandre Gagnon (UQAM) for helpful discussion, Mr Jonathan Gauvin-Audet for his assistance on the synthesis of some substrates, Dr Thierry Maris for X-ray data collection and refinement, and Compute Canada (CalculQuebec) for computational resources. V. D. thanks NSERC for the 2017 USRA scholarship. This research was undertaken, in part, thanks to funding from the Canada Research Chairs program.Notes and references
- (a) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications, John Wiley & Sons, 2013 Search PubMed; (b) S. W. Pelletier, Alkaloids: chemical and biological perspective, Springer, 1999, Vol. 13 Search PubMed.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777–2812 CrossRef PubMed.
- (a) T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef PubMed; (b) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef PubMed.
- (a) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef; (b) S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 7720–7738 CrossRef PubMed; (c) K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC; (d) S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2009, 48, 4235–4238 CrossRef PubMed; (e) S. Zhang, Y. Han, J. He and Y. Zhang, J. Org. Chem., 2018, 83, 1377–1386 CrossRef PubMed.
- (a) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082–1096 CrossRef PubMed; (b) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105–5113 RSC; (c) S. C. Matthew, B. W. Glasspoole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828–5831 CrossRef PubMed; (d) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024–5025 CrossRef PubMed; (e) Y. Lou, P. Cao, T. Jia, Y. Zhang, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2015, 54, 12134–12138 CrossRef PubMed; (f) D. L. Sandrock, L. Jean-Gérard, C.-y. Chen, S. D. Dreher and G. A. Molander, J. Am. Chem. Soc., 2010, 132, 17108–17110 CrossRef PubMed; (g) G. A. Molander and S. R. Wisniewski, J. Am. Chem. Soc., 2012, 134, 16856–16868 CrossRef PubMed; (h) J. C. H. Lee, R. McDonald and D. G. Hall, Nat. Chem., 2011, 3, 894–899 CrossRef PubMed; (i) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191–13193 CrossRef PubMed; (j) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738–20741 CrossRef PubMed; (k) S. D. Dreher, P. G. Dormer, D. L. Sandrock and G. A. Molander, J. Am. Chem. Soc., 2008, 130, 9257–9259 CrossRef PubMed; (l) X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989–3992 CrossRef PubMed; (m) L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027–14030 CrossRef PubMed.
- M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef PubMed.
- (a) M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef; (b) H. Braunschweig, M. Colling and C. Hu, Inorg. Chem., 2003, 42, 941–943 CrossRef PubMed; (c) A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100–1107 CrossRef PubMed; (d) C. D. Entwistle, A. S. Batsanov, J. A. K. Howard, M. A. Fox and T. B. Marder, Chem. Commun., 2004, 702–703 RSC; (e) X. Fan, J. Zheng, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2015, 137, 4916–4919 CrossRef PubMed; (f) K. Oshima, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 3699–3702 CrossRef PubMed.
- K. Kubota, K. Hayama, H. Iwamoto and H. Ito, Angew. Chem., Int. Ed., 2015, 54, 8809–8813 CrossRef PubMed.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed.
- (a) F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375 Search PubMed; (b) D. W. Stephan, Science, 2016, 354, 6317 CrossRef PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef PubMed; (d) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef PubMed; (e) G. Erker and D. W. Stephan, Frustrated Lewis Pairs I and II, Springer, New York, 2013 Search PubMed; (f) F.-G. Fontaine, M.-A. Courtemanche, M.-A. Légaré and É. Rochette, Coord. Chem. Rev., 2017, 334, 124–135 CrossRef; (g) F.-G. Fontaine and É. Rochette, Acc. Chem. Res., 2018, 51, 454–464 CrossRef PubMed.
- (a) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef PubMed; (b) A. Del Grosso, J. A. Carrillo and M. J. Ingleson, Chem. Commun., 2015, 51, 2878–2881 RSC; (c) V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef PubMed; (d) A. Prokofjevs, W. J. Kampf and E. Vedejs, Angew. Chem., Int. Ed., 2011, 50, 2098–2101 CrossRef PubMed; (e) A. Del Grosso, P. J. Singleton, C. A. Muryn and M. J. Ingleson, Angew. Chem., Int. Ed., 2011, 50, 2102–2106 CrossRef PubMed.
- (a) A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408–8411 CrossRef PubMed; (b) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef PubMed; (c) Y. L. Liu, G. Kehr, C. G. Daniliuc and G. Erker, Chem.–Eur. J., 2017, 23, 12141–12144 CrossRef PubMed; (d) J. S. McGough, J. Cid and M. J. Ingleson, Chem.–Eur. J., 2017, 23, 8180–8184 CrossRef PubMed; (e) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef; (f) Q. Yin, F. T. Klare Hendrik and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3712–3717 CrossRef PubMed; (g) K. Chernichenko, M. Lindqvist, B. Kótai, M. Nieger, K. Sorochkina, I. Pápai and T. Repo, J. Am. Chem. Soc., 2016, 138, 4860–4868 CrossRef PubMed.
- M.-A. Légaré, E. Rochette, J. Légaré Lavergne, N. Bouchard and F.-G. Fontaine, Chem. Commun., 2016, 52, 5387–5390 RSC.
- J. Légaré-Lavergne, A. Jayaraman, L. C. Misal Castro, É. Rochette and F.-G. Fontaine, J. Am. Chem. Soc., 2017, 139, 14714–14723 CrossRef PubMed.
- S. Radomkit and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 3387–3391 CrossRef PubMed.
- É. Rochette, H. Boutin and F.-G. Fontaine, Organometallics, 2017, 36, 2870–2876 CrossRef.
- Other mechanism involving B-H activation of HBpin by aminoborane catalyst followed by delivering the activated HBpin across the C2-C3 double of indole was proposed initially, but later dimissed on the basis of DFT computed high activation barrier for the delivery step (see ESI†).
- (a) J. V. B. Kanth, M. Periasamy and H. C. Brown, Org. Process Res. Dev., 2000, 4, 550–553 CrossRef; (b) Q. Yin, S. Kemper, H. F. T. Klare and M. Oestreich, Chem.–Eur. J., 2016, 22, 13840–13844 CrossRef PubMed.
- (a) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09 (Rev C.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) C. Villiers and M. Ephritikhine, Tetrahedron Lett., 2003, 44, 8077–8079 CrossRef; (b) S. Harder and J. Spielmann, J. Organomet. Chem., 2012, 698, 7–14 CrossRef.
- Y. Suseela, A. S. B. Prasad and M. Periasamy, Chem. Commun., 1990, 446–447 RSC.
- N. W. J. Ang, C. S. Buettner, S. Docherty, A. Bismuto, J. R. Carney, J. H. Docherty, M. J. Cowley and S. P. Thomas, Synthesis, 2018, 50, 803–808 CrossRef.
- Hydroboration of 3-substituted indoles using 9-BBN has only been postulated see E. M. Ferreira and B. M. Stolz, Tetrahedron Lett., 2006, 47, 8579–8582 CrossRef PubMed.
- P. G. M. Wuts and T. W. Greene, Protection for the Amino Group, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 696–926 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, computational details, energies, cartesian coordinates, and X-ray crystallographic data. CCDC 1584844 and 1819482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01093e |
| This journal is © The Royal Society of Chemistry 2018 |