
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe 3rd degree of biomimetism: associating the cavity effect, ZnII coordination and internal base assistance for guest binding and activation†
A.
Parrot
a,
S.
Collin
a,
G.
Bruylants
 b and
O.
Reinaud
*a
b and
O.
Reinaud
*a
aLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, CNRS UMR8601, Université Paris Descartes, Sorbonne Paris Cité, 45 rue des Saints Pères, 75006 Paris, France. E-mail: olivia.reinaud@parisdescartes.fr
bEngineering of Molecular NanoSystems, Ecole Polytechnique de Bruxelles, Université Libre de Bruxelles (ULB), Avenue F. D. Roosevelt 50, CP165/64, B-1050 Brussels, Belgium
First published on 29th May 2018
Abstract
The synthesis and characterization of a resorcinarene-based tetra(imidazole) ligand is reported. The properties of the corresponding ZnII complex are studied in depth, notably by NMR spectroscopy. In MeCN, acid–base titration reveals that one out of the four imidazole arms is hemi-labile and can be selectively protonated, thereby opening a coordination site in the exo position. Quite remarkably, the 4th imidazole arm promotes binding of an acidic molecule (a carboxylic acid, a β-diketone or acetamide), by acting as an internal base, which allows guest binding as an anion to the metal center in the endo position. Most importantly, the presence of this labile imidazole arm makes the ZnII complex active for the catalyzed hydration of acetonitrile. It is proposed that it acts as a general base for activating a water molecule in the vicinity of the metal center during its nucleophilic attack to the endo-bound MeCN substrate. This system presents a unique degree of biomimetism when considering zinc enzymes: a pocket for guest binding, a similar first coordination sphere, a coordination site available for water activation in the cis position relative to the substrate and finally an internal imidazole residue that plays the role of a general base.
Introduction
Nature has selected proteic structures with remarkable properties for molecular recognition and catalysis. Their structural identification gives key insights into active sites, which is helpful for understanding structure/reactivity relationships. It is also an inexhaustible source of inspiration for chemists interested in the design of smart systems prone to recognition or catalysis, or both. The classical approach to mimic metallo-enzymes consists in the elaboration of a ligand that reproduces the first coordination sphere of the natural system.1–5 Control of the second coordination sphere has recently become a hot topic and has been recognized as a key point for substrate/ligand stabilization or activation.2,3,5–13 We have developed a biomimetic supramolecular approach consisting in the association of a macrocyclic cavity to a biomimetic coordination core in order to control the second coordination sphere (and further) with a macrocyclic structure surrounding the metal ion labile site. This approach also allows controlling the access to the metal ion and thus the selectivity against ligands. The first system we developed uses a calix[6]arene as a funnel to drive the exogenous ligand to the metal ion. In this system, a single guest can bind to the metal ion (Fig. 1).14,15 More recently, we have described systems based on resorcinarenes.16,17 The so-called bowl complexes, presenting three imidazole arms on their large rim,18 were shown, when complexing a zinc cation, to present two labile sites in the cis position relative to each other, which opened new opportunities for recognition and reactivity.19–23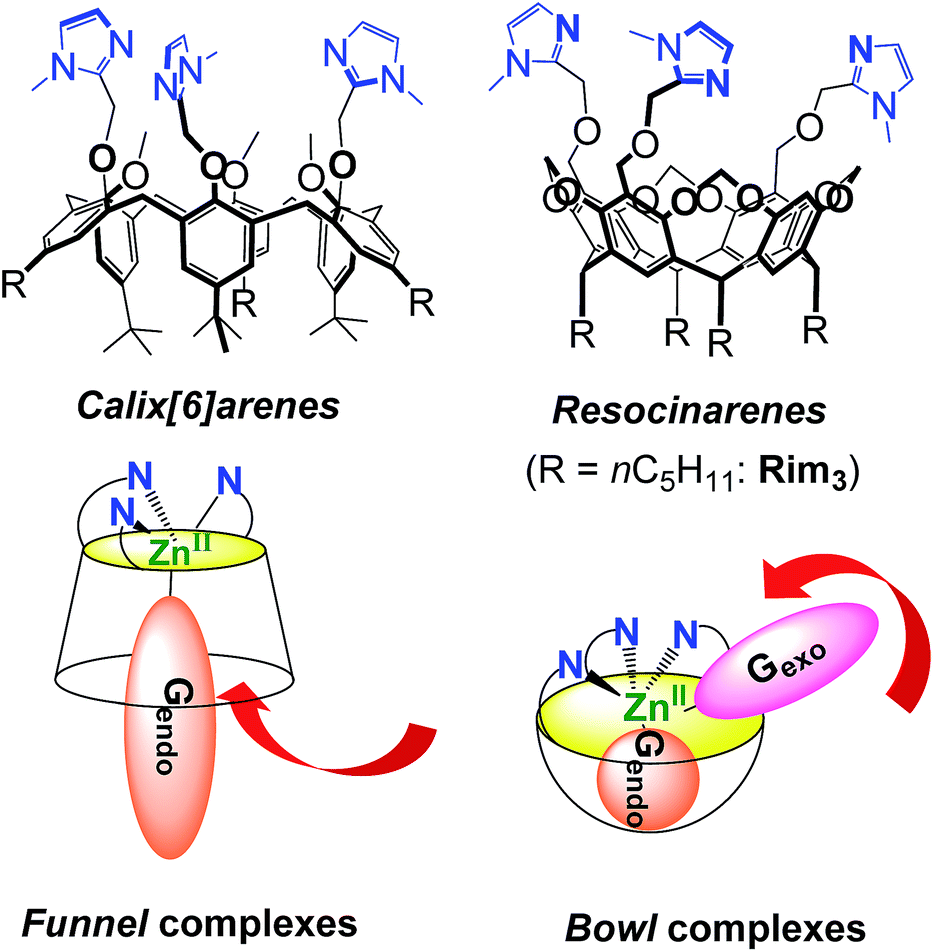 | ||
| Fig. 1 Compared structures and hosting properties of cavity complexes based on calix[6]arene (left) and resorcinarene (right) functionalized with three imidazole moieties that mimic proteic active sites. The red arrows emphasize the different guest binding pathways. G stands for a guest ligand. | ||
In many metallo-enzymes however, the catalytic cycle involves a general base (or acid) carefully positioned vis-à-vis the substrate. Zinc hydrolytic enzymes, in particular, generally display a N2O or N3 coordination core, associated with a general base which is hydrogen-bonded to a water ligand (Fig. 2).24 The latter is often a Glu or Asp residue but in some cases, it is a His residue as for families of histone deacetylases25,26 and metallo-β-lactamases.27 In some enzymes, this general base (an Asp residue) is coordinated to the metal ion, as in adenosine deaminase.28 It has been shown to assist the water nucleophilic attack to the coordinated substrate (e.g. the carbonyl group of a peptidic bond, acetamide moieties or even the pyrimidine group of a nucleic base) as schematized in Fig. 2. Hence, the key features for the enzymatic activity stem from the strong Lewis acidic center, the presence of two labile sites in cis-position relative to each other and from the general base present in the first or second coordination sphere. This pattern allows the simultaneous activation of the electrophilic substrate and the water nucleophile. Upon coordination, both the carbonyl substrate and the water ligand are polarized, whereas the general base activates the water nucleophile to provide the tetrahedral oxyanion intermediate, which is stabilized by coordination to ZnII.
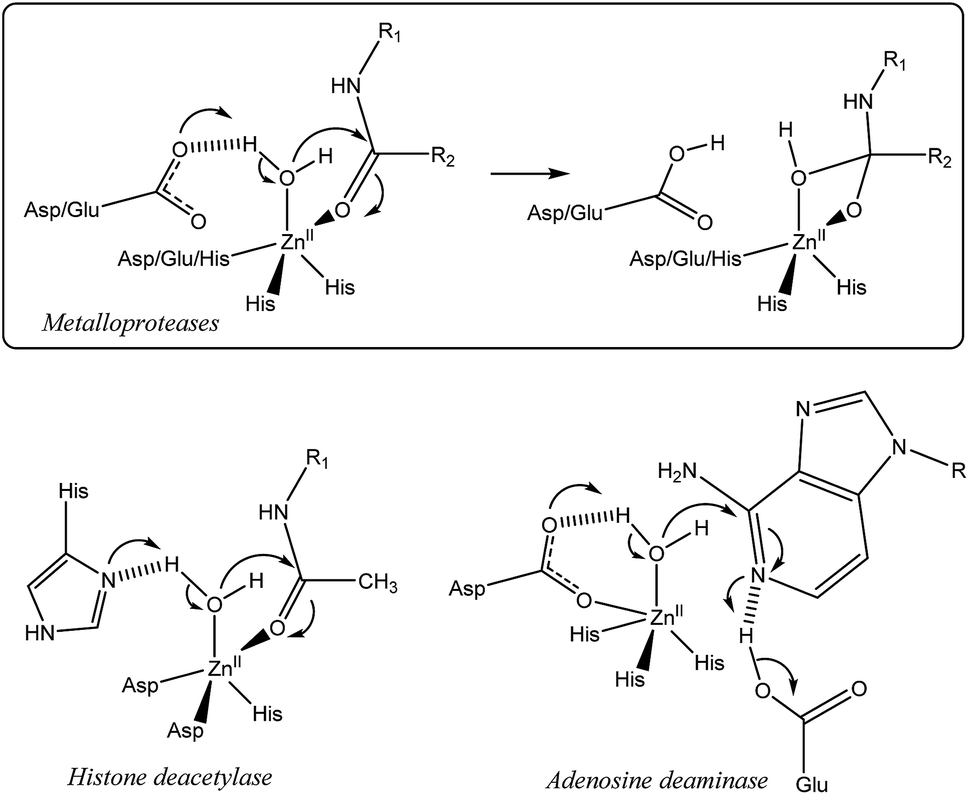 | ||
| Fig. 2 Top: generic active site of ZnII proteases and the key step involving the assistance of an Asp or Glu residue as a general base. Bottom: example of variations of the active site and nucleophilic attack occurring in other zinc hydrolytic enzymes. | ||
Here, we present the new bowl ligand Rim4 (Scheme 1) that displays a fourth imidazole arm. Studies with the corresponding ZnII complex show that, whereas three imidazole groups allow the strong binding of the metal center at the entrance of the cavity, the 4th imidazole arm is hemilabile and can act as an internal base. The consequences on molecular recognition and hydrolytic activity towards the acetonitrile guest are presented and discussed in the context and development of biomimetic catalysts.
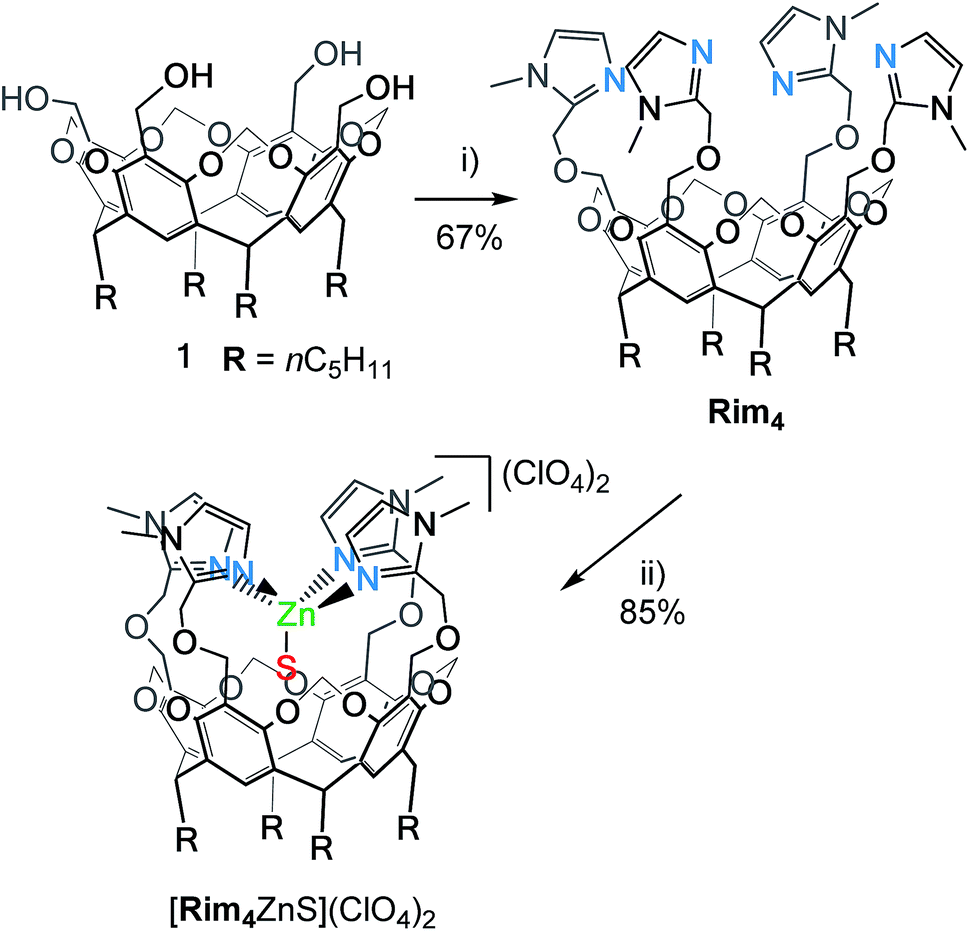 | ||
| Scheme 1 Synthetic route for ligand Rim4 and its corresponding ZnII complex. (i) NaH, 2-chloromethyl-1-methyl-1H-imidazole, and DMF. (ii) Zn(OH2)6(ClO4)2 and EtOH. S stands for solvent (EtOH here) or residual water. | ||
Results and discussion
Synthesis of Rim4 and complexation of ZnII
The resorcin[4]arene tetra(imidazole) ligand Rim4 was synthesized by reacting the tetra(alcohol) precursor 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 with 2-chloromethyl-1-methyl-1H-imidazole in the presence of NaH (Scheme 1).
29 with 2-chloromethyl-1-methyl-1H-imidazole in the presence of NaH (Scheme 1).
The zinc complex was obtained by reacting Rim4 with one equivalent of ZnII perchlorate salt in EtOH. The complex, [Rim4Zn(EtOH)](ClO4)2, spontaneously precipitated out of the solution as an off-white solid. The complex was fully characterized by NMR, IR, and ESI-MS. IR analysis confirmed the presence of ClO4− counterions. Rim4Zn was only soluble in CD3CN, displaying a well-defined signature, corresponding to a complex with an apparent C4v symmetry (Fig. 3), even at 240 K. Only two aromatic signals are assigned to the imidazole, showing that all four imidazoles are equivalent on the NMR chemical shift timescale. NMR experiments in CH3CN with 5% CD3CN showed the coordination of acetonitrile to zinc, with an intracavity peak at −2.20 ppm (see ESI Fig. S32†). The corresponding complexation induced shift (CIS = −4.14 ppm), due to the embedment of the guest methyl group in the bowl cavity, is similar to that measured with Rim3 (δ = −2.14 ppm, CIS = −4.08 ppm). The presence of exactly one equivalent of free ethanol is likely due to its initial hosting in the cavity (in the as-isolated complex) and substitution by the coordinating solvent. DOSY experiments conducted on Rim4 and its ZnII complex in MeCN showed similar diffusion coefficients (7.6 × 10−10 and 7.4 × 10−10 m2 s−1, respectively).‡ ZnII complexation to the bowl ligands Rim3 and Rim4 was also monitored by isothermal calorimetry (ITC, Fig. 3) in dry acetonitrile. In each case, the ZnII complexation, confirmed to be of 1:1 stoichiometry, appeared to be enthalpically driven (ΔH° ≈ −70 kJ mol−1) with a large entropic penalty (Table 1).
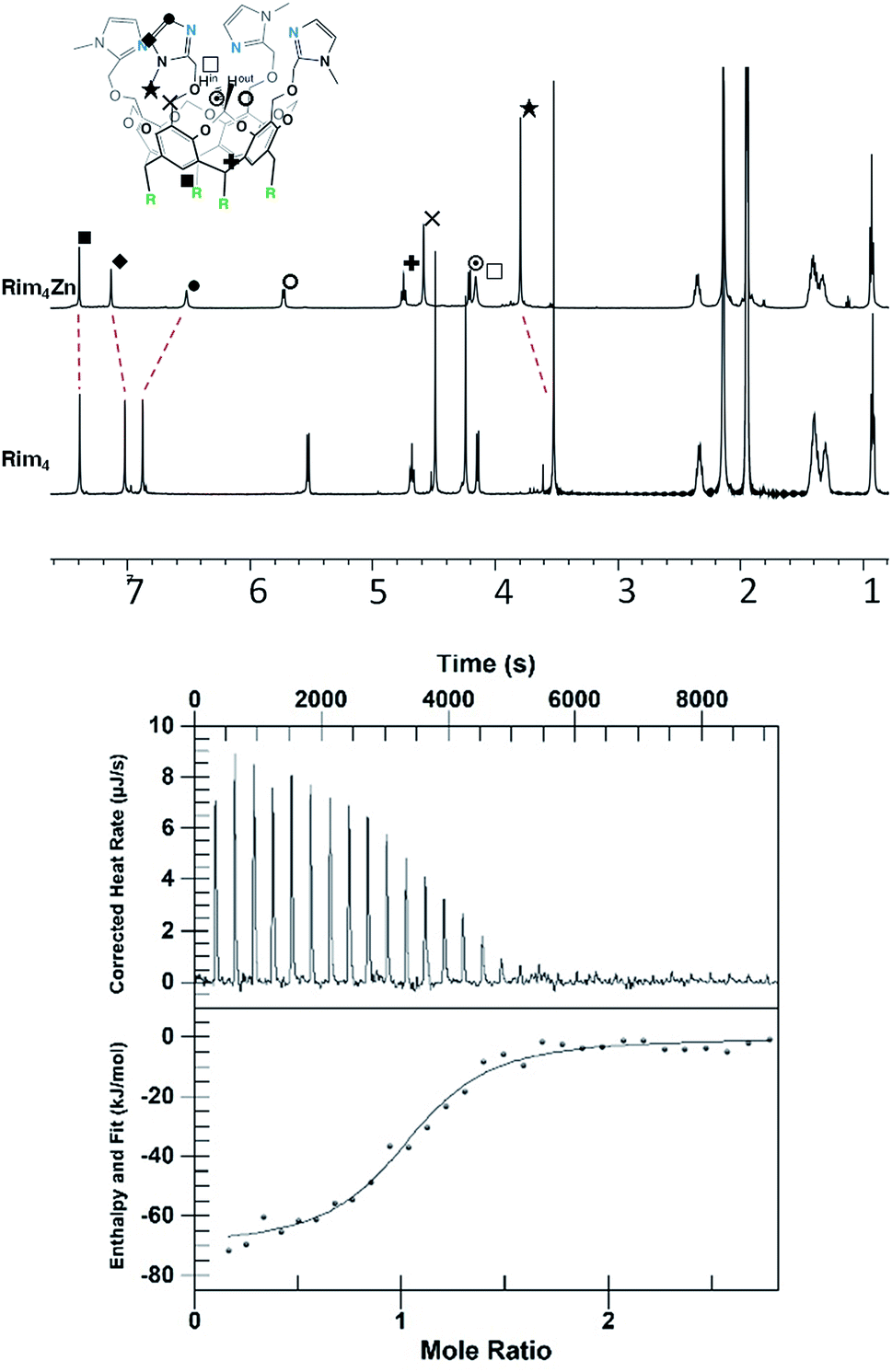 | ||
| Fig. 3 Top: 1H NMR spectra (600 MHz, 300 K, CD3CN) of ligand Rim4 and its corresponding ZnII complex, [Rim4Zn(MeCN)](ClO4)2. Bottom: titration of Rim4 (0.05 mM) with zinc perchlorate (0.5 mM) in dry MeCN. | ||
| Compound | ΔH° (kJ mol−1) | K (M−1) | n | ΔS° (J mol−1 K−1) |
|---|---|---|---|---|
| Rim3 | −67 ± 5 | (6 ± 4) 106 | 0.9 ± 0.2 | −102 ± 26 |
| Rim4 | −69 ± 7 | (7 ± 2) 105 | 1.0 ± 0.1 | −120 ± 23 |
The similarity of the thermodynamic profiles suggests that the fourth imidazole does not play a crucial role in the complexation and is only weakly bound to the metal center.§
Acid–base studies
Acid titration of [Rim4Zn(MeCN)](ClO4)2 was first conducted with a strong, non-coordinating acid, namely picric acid, and monitored by 1H NMR and ITC (Fig. 4). The addition of the first equivalent of acid led to a shift of the imidazole resonances, which is particularly pronounced for the proton in the α-position of the coordinating nitrogen atom (signal at 6.5 ppm). The fact that the other peaks are little affected suggests that the coordination of ZnII to Rim4 is maintained, whereas the imidazole set sees its global environment changed. One explanation is that one imidazole undergoes protonation, whereas the three other imidazoles maintain the ZnII bound to the bowl structure. The fact that only one set of resonances for the imidazole arms is observed is best explained by a fast exchange (vs. the NMR chemical shift time scale) between the protonated and the coordinated ones. Such a “dancing” behavior was observed with the Rim3CuI complex where the CuI ion is 2-coordinate in a non-coordinating solvent.30 Above 4 equivalents of acid, a new sharp signature is obtained, which corresponds to the fully protonated ligand, as attested to by a low-field resonance at ca. 13 ppm accounting for 4 protonated imidazolium arms. Interestingly, between 2 and 4 equivalents of acid, the emergence of new peaks (indicated by arrows) shows the co-existence of the ZnII complex and the tetraprotonated ligand that are in slow exchange relative to the chemical shift time scale. The ITC study, conducted at a much lower concentration (Fig. 4, right), evidenced a clean exothermic event (−26 kJ mol−1) for the mono-protonation, which also appears entropically favorable (ΔS° = 23 J K−1 mol−1). Considering a pKa value of 11 for picric acid in MeCN, the pKa corresponding to the imidazolium arm can be estimated to be 16.7.¶31 Although the pKa of imidazole in MeCN has not been reported (6.95 in water), it can be compared to those of 2-NH2-benzimidazole (15.95 in MeCN vs. 7.51 in water) and 2-NH2-pyridine (ca. 14.5 in MeCN vs. 6.75 in water).32 The similarity of the values in MeCN further confirms the weak coordination of this fourth imidazole arm to the metal ion. Note that the protonation reaction is fully reversible since subsequent addition of a base such as NEt3 led to the quantitative regeneration of the ZnII complex.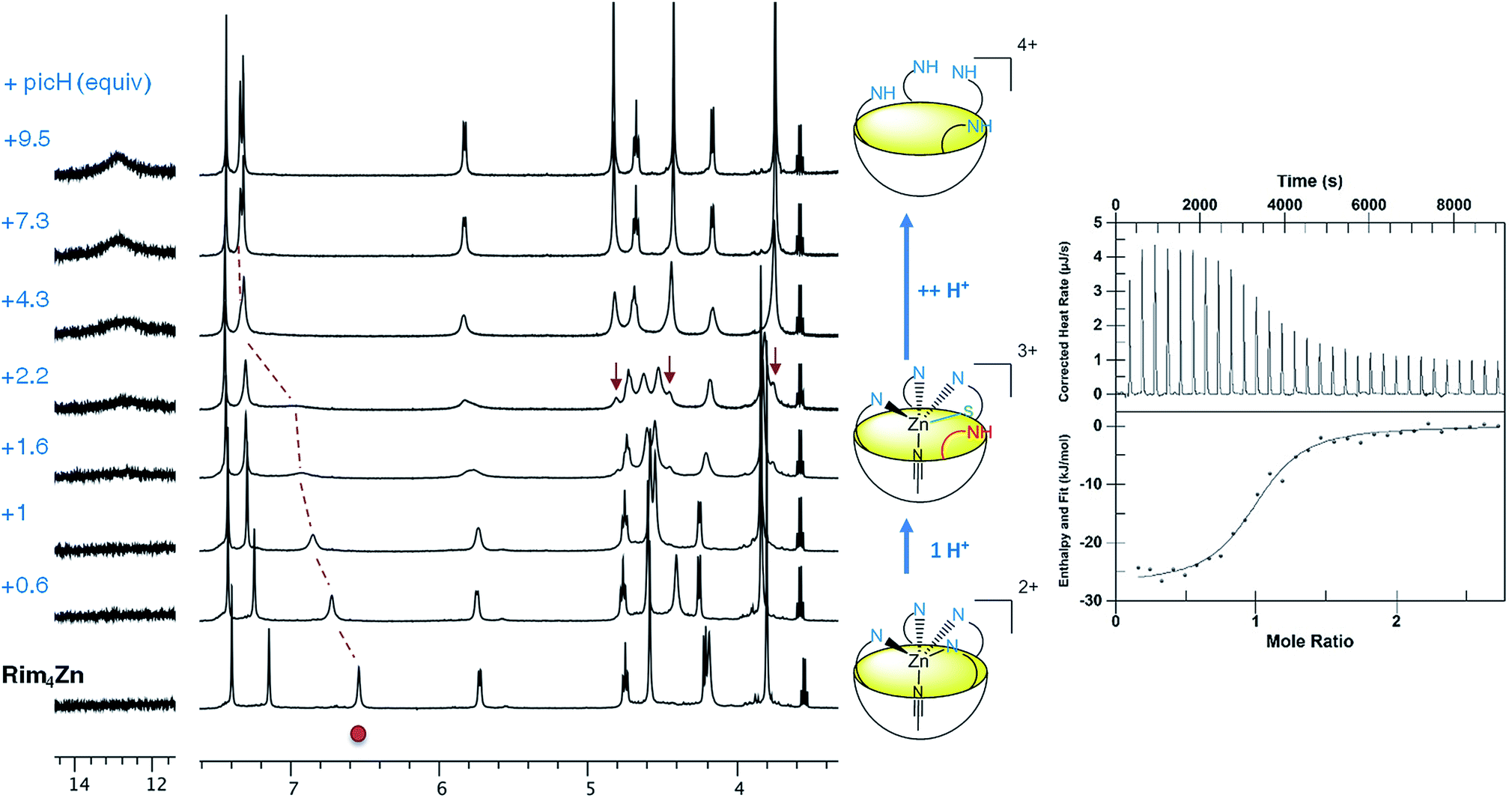 | ||
| Fig. 4 Titration of [Rim4Zn(MeCN)](ClO4)2 with picric acid in CD3CN at 300 K. Left: 1H NMR monitoring (2 mM, 500 MHz). The red dot depicts an imidazole peak; the little red arrows indicate growing peaks assigned to the fully protonated ligand. Right: ITC monitoring of the monoprotonation of the complex (0.05 mM) by picric acid (PicH, 0.5 mM): K = 5.44 × 105 M−1 (n = 0.93), ΔH° = −26 kJ mol−1, ΔS° = 23 J K−1 mol−1; pKa = −5.7 (which gives pKa = 16.7, considering pKa(PicH) = 11.0 in MeCN). | ||
Reaction of the ZnII complex with acetic acid was then studied. Indeed, acetic acid is a weak acid, but a potentially coordinating guest. With Rim3, it was shown that the ZnII center displays very strong affinity for this guest provided a base (triethylamine) was added to the solution, leading to the formation of the monocationic [Rim3Zn(OAc)]+ adduct.33 Having evidenced the lability of the fourth imidazole arm of complex [Rim4Zn(MeCN)]2+ mediated by the addition of a strong acid, we explored the possible concomitant deprotonation/coordination of AcOH by the complex based on Rim4.
Upon stepwise addition of acetic acid to a solution of [Rim4Zn(MeCN)]2+ in acetonitrile, two new resonances appeared in the high-field region [δ(Me) = −2.25 and −2.38 ppm] of the 1H NMR spectra (Fig. 5, bottom), attesting to the inclusion of acetate into the resorcinarene cavity. Concomitantly, new peaks emerged in the aromatic region, which evidenced the formation of three different species. According to integrations of the imidazole peaks relative to endo-bound acetate, one set of imidazole resonances corresponds to the major peak (−2.25) at one equivalent of added acetic acid (identified with large red dots in Fig. 5), whereas the other sets correspond to the minor peak (δ = −2.38 ppm). Lastly, the appearance of a low field resonance at 12.4 ppm (see ESI Fig. S21†) indicated the protonation of one imidazole moiety. All of this information accounts for the quantitative coordination of acetate in the cavity accompanied by the protonation of one imidazole arm. The three associated dicationic acetato complexes actually correspond to different solvated forms. Indeed, we have previously shown that the analogous tris(imidazole) ZnII complex, based on the Rim3 ligand,33 can bind exogenous donors in the exo position, such as solvent (MeCN) or residual water. Here, the protonation of one imidazole arm opens a coordination site in the exo position that can be free (major species under these experimental conditions {[Rim4HZn(OAc)]2+}, or occupied by solvent or residual water {[Rim4HZnS(OAc)]2+}). This was further confirmed by water addition to a solution containing the acetato dicationic complex, which induced growth of the high-field peak at δ = −2.38 ppm, and was thus attributed to the water adduct (S = H2O, see ESI Fig. S22 and S23†).
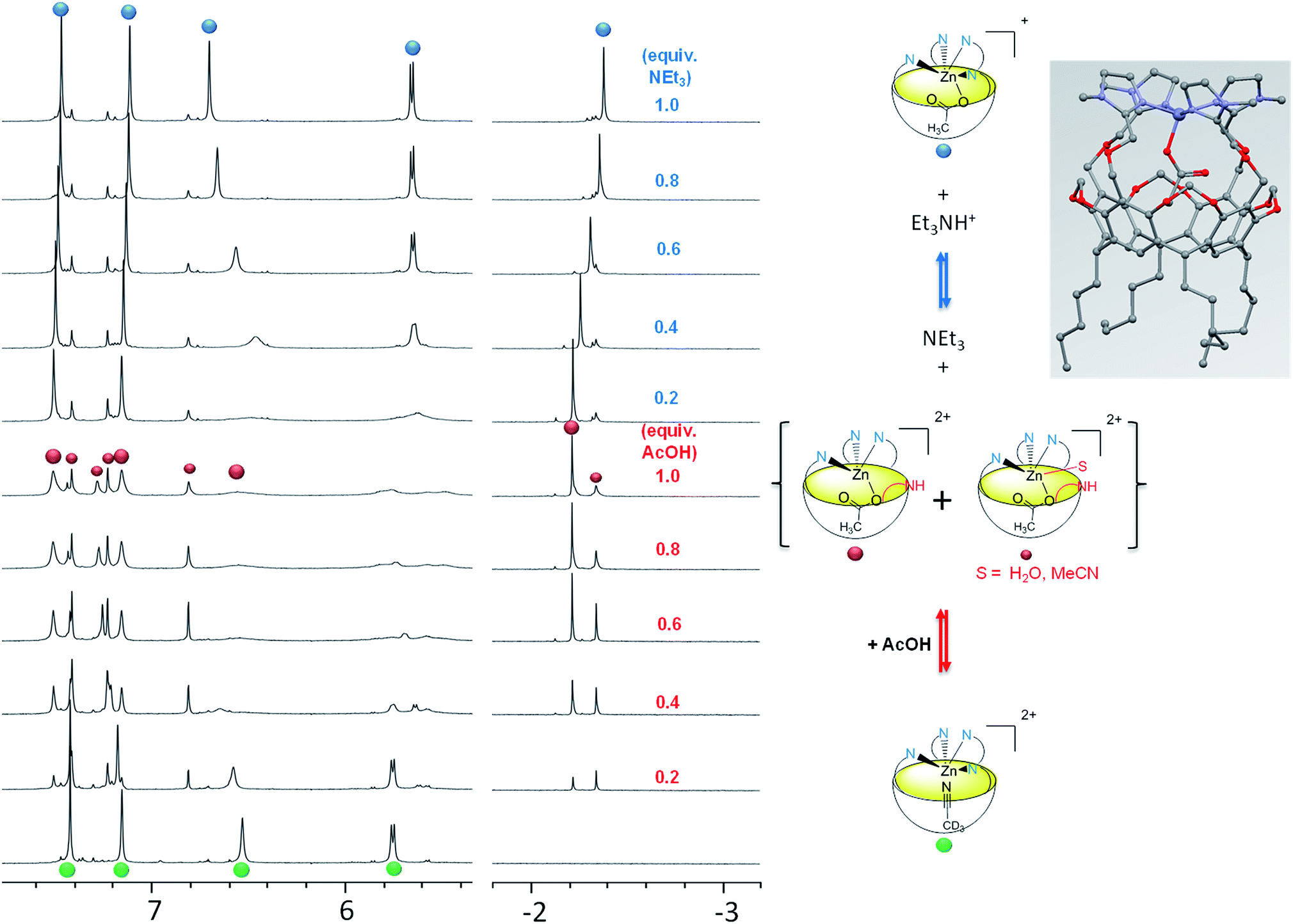 | ||
| Fig. 5 Coordination of acetate by [Rim4Zn(MeCN)](ClO4)2 in CD3CN and evidence of the hemilability of the 4th imidazole arm. Left: 1H NMR titration (500 MHz, 300 K, CD3CN) of [Rim4Zn(MeCN)](ClO4)2 with (from bottom to top) acetic acid, and subsequent titration with Et3N. The small red dots are attributed to the solvated dicationic acetato species (S = H2O or MeCN). Right: various species observed during the titrations. Inset: model of the acetato complex [Rim4Zn(OAc)]+ obtained with software HyperChem.34 | ||
In order to confirm all these observations, subsequent titration of the protonated acetato complex with a base was conducted (Fig. 5, top). Upon progressive addition of Et3N, all resonances converged towards a major set corresponding to the monocationic acetato complex [Rim4Zn(OAc)]+. Indeed, a similar signature was obtained upon addition of AcONa to the nitrilo complex (see ESI Fig. S9†). The resonance at 12.4 ppm corresponding to the protonated imidazolium arm vanished, whereas those corresponding to Et3N (at 3.1 ppm) attested to its quantitative protonation (see ESI Fig. S21†). The solvated species almost disappeared, in agreement with the deprotonation of the fourth imidazole arm that favors its coordination to the detriment of exogenous donors.
Probing metal access and basic assistance
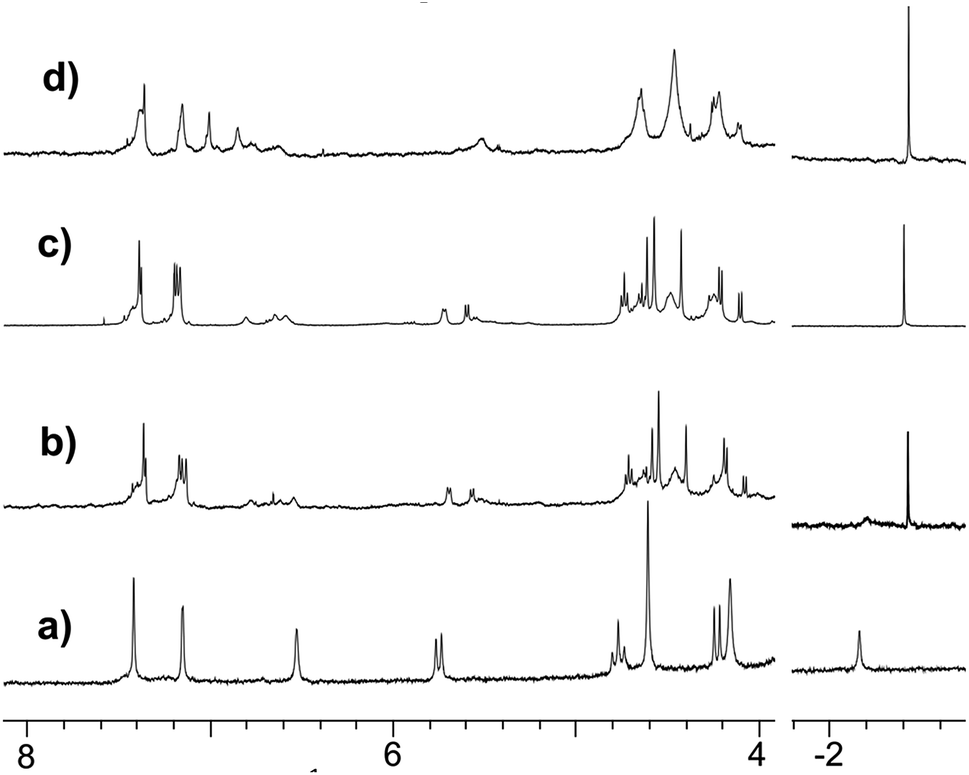 | ||
| Fig. 6 Comparative 1H NMR signatures (250 MHz, 300 K) of the nitrilo and acetamido Rim4ZnII complexes in acetonitrile: (a) the initial spectrum of the freshly dissolved nitrilo complex in CH3CN + 5% CD3CN; (b) spectrum of the Rim4ZnII complex (4.7 mM) in CH3CN + 5% CD3CN after two weeks at RT; (c) a fresh solution of the Rim4Zn complex dissolved in CD3CN to which 1.5 equiv. of CH3CONH2 was added; and (d) the same sample to which 1 equiv. of Et3N was added. | ||
As in the case of the Rim3-based acetamido complex, several sets of peaks corresponding to the associated Rim4 core in the δ = 4–8 ppm region denote the presence of different species attributed to different coordination modes of the acetamido guest. Addition of Et3N affected the Rim4 signature but not the guest resonance (Fig. 6d), thus confirming its coordination as an anion.**
Acetyl- and benzoylacetone, which were shown to readily bind to the Rim3ZnII complex in a bidentate fashion in the presence of Et3N,33 were tested with the Rim4ZnII complex. As for carboxylic acids and acetamide, direct titrations showed up-field methyl peaks attesting to the coordination of both β-diketones, and concomitant broadening and down-field shifts of the imidazole resonances. Upon subsequent addition of Et3N, all peaks sharpened, and the corresponding NMR signatures attested to the formation of the monocationic host–guest adducts (Scheme 2). For acetylacetone, a variable temperature study evidenced concomitant broadening (as T increases) of the peaks at δ = −2.56 ppm and 1.78 ppm, attributed to the methyl groups of the guest in endo and exo positions, respectively. This indicates that these methyl groups are in dynamic exchange. As previously described with Rim3, the acetylacetonate guest undergoes an intramolecular exchange, with its endo and exo methyl groups switching positions.34 For benzoylacetone, due to the cavity size, the phenyl group sits in the exo-position, as indicated by the corresponding low CIS values (−0.09 ppm for phenyl and −0.33 for CH), while the methyl group is selectively bound in the endo-position (CIS = −4.05 ppm). Finally, the apparent C4v signature attests to the fast exchange of the imidazole groups in the vicinity of the metal ion, even at low T (240 K).††
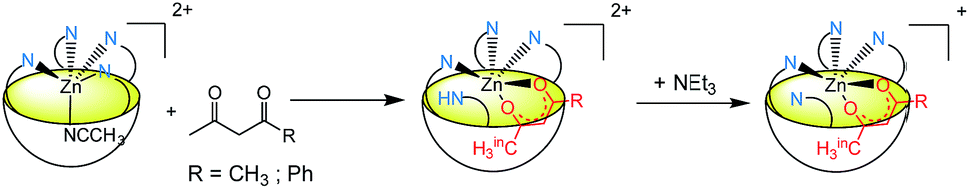 | ||
| Scheme 2 Coordination of diketones to [Rim4Zn(MeCN)](ClO4)2 in MeCN. | ||
In summary, in MeCN and in the absence of a specific guest, the Rim4ZnII complex presents the following characteristics:
– Three imidazole arms maintain firmly the metal center above the cavity, at its entrance.
– A molecule of solvent (MeCN) is bound in the endo position and is exchangeable.
– The fourth imidazole is labile and reacts readily as a base. It can promote the coordination of poorly acidic guests and their binding as an anion. It is interesting to consider the corresponding host–guest process from the acid–base point of view: the 1H NMR studies showed quantitative guest binding upon addition of ca. 2 equiv. of acidic guest GH at 2 mM in MeCN (Fig. 7). This indicates that the corresponding equilibrium constant is greater than 104 M−1. For comparison, the proton exchange between acetamide (GH) and simple imidazole in a non-protic solvent (DMSO) is ca. 10−19. Hence, the association of Lewis acid coordination, cavity-hosting and internal base of the Rim4ZnII system allows displacing the acid–base equilibrium by a factor higher than 10 (ref. 23)!
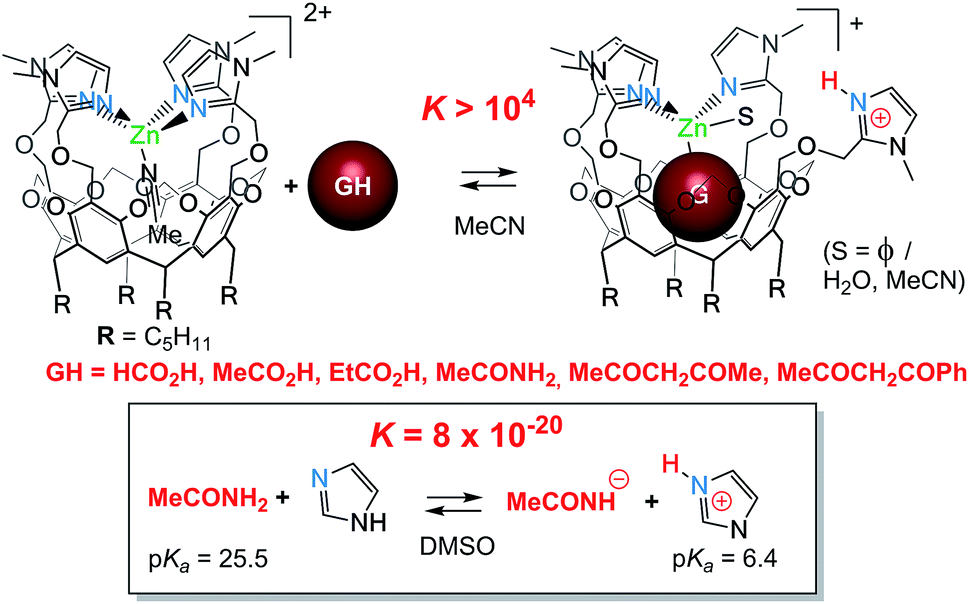 | ||
| Fig. 7 Basic assistance for acidic guest binding and comparison with a simple acid/base equilibrium.|| | ||
– Lastly, the two coordination sites open for guest binding are in the cis position relative to each other. One site is in the endo-position under the steric control of the bowl cavity, and the other one is in the exo position, exposed to the solvent.
All these characteristics are highly reminiscent of the active site of hydrolytic enzymes, which drove us to explore its reactivity.
Acetonitrile hydration
The reactivity of the nitrilo [Rim4Zn(MeCN)]2+ complex towards water was first scrutinized by 1H NMR spectroscopy. After two weeks at room temperature, the NMR signature of the nitrilo complex [in CH3CN containing 5% CD3CN and residual water (around 0.1%)] was replaced by that of the acetamido complex (Fig. 6a and b). GC-MS analyses of the solution (following a literature procedure)35 confirmed the formation of acetamide.The hydration reaction was then studied at 70 °C by 1H NMR spectroscopy with various contents of water. With Rim4ZnII (3.8 mM), in CH3CN/H2O (8:2 v/v) and after 6 days at 70 °C, new broad signals at 6.15 and 5.59 ppm were observed and a sharp peak at 1.84 ppm appeared. This indicated the presence of free acetamide, the singlet at 1.84 ppm corresponding to the methyl group and the broad signals to the NH2 moiety, which was confirmed by IR, HMBC, HSQC and 13C experiments (see ESI Fig. S32–37†). Integration of the peaks indicated the formation of 7 equivalents of acetamide after 14 days at 70 °C. Under the same conditions, no reaction was observed using 3 mM Zn(ClO4)2 or 3 mM Rim3ZnII instead of Rim4ZnII, even in the presence of an additional base (N-methyl imidazole). Kinetic studies were conducted by 1H NMR spectroscopy with DMF as an internal reference for acetamide quantification. The recorded data revealed a 2-phase kinetics (Fig. 8): a burst phase from 0 to 10 minutes corresponding to the formation of the first equivalent of acetamide (t1/2 = 4 min), which is endo-bound. After 10 minutes, the reaction slowed down significantly. This suggests that the endo-bound acetamide partially inhibits the reaction due to cavity filling. Increasing the catalyst concentration by a factor of two doubled the reaction rate in agreement with first-order kinetics with respect to the complex. This two-phase kinetics is well described by a classical model for reaction inhibition due to product displacement (see the ESI†),36 and the associated reaction rates for each phase are reported in Table 2. Whereas the water content seems to have little impact on the burst phase, it enhanced the reaction rate in the second phase, reaching a TOF of 0.22 h−1 at 35% H2O (highest water percentage for solubility reasons).
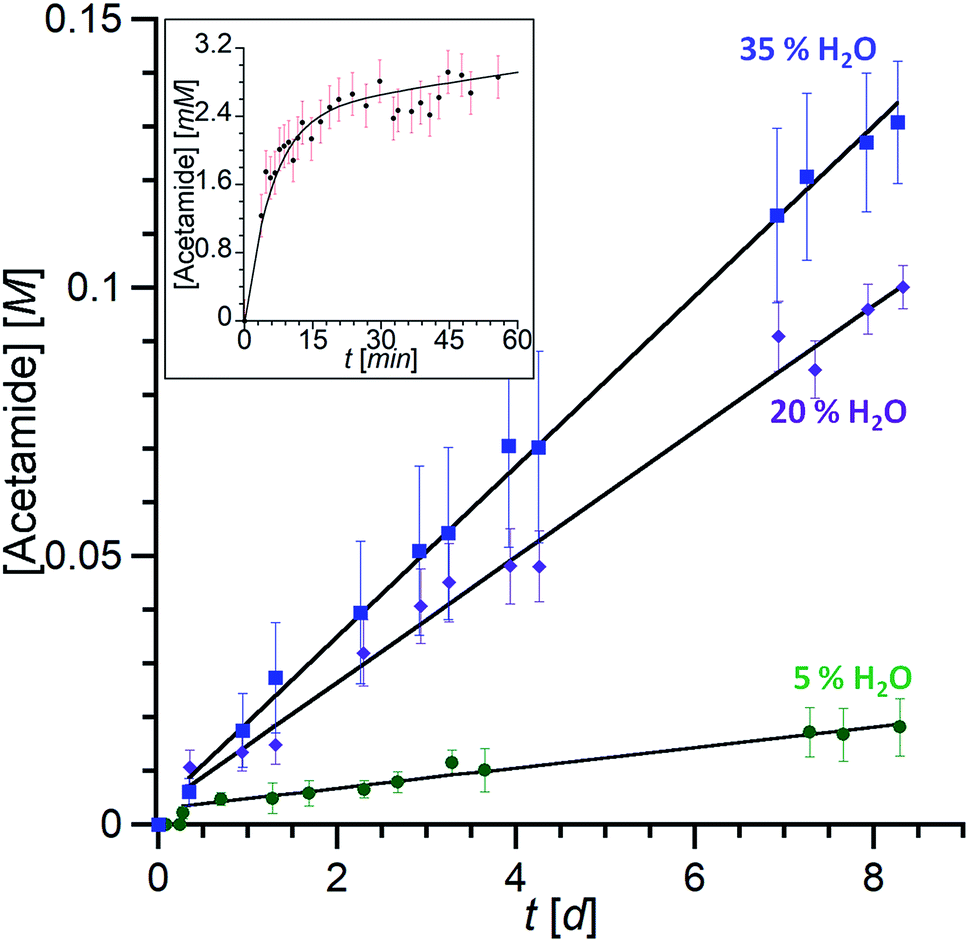 | ||
| Fig. 8 Kinetic traces of acetamide formation. [Rim4Zn] = 3 mM, acetonitrile, 70 °C. Total formation of acetamide (free and endo-bound) over several days with different water contents (measured by 1H NMR (CH3CN/CD3CN = 4:1)). Inset: burst phase with the formation of the acetamido endo complex followed by integration of the 1H NMR trace at δ = −2.4 ppm (20% H2O). The data are fitted according to the equation corresponding to the classical model for reaction inhibition due to product displacement (see the ESI†).36 | ||
| [Rim4Zn] (mM) | % H2O | T (°C) | v 1 (10−3 M min−1) | v 2 (10−6 M min−1) |
|---|---|---|---|---|
| a nd = not determined. | ||||
| 3 | 5 | 70 | 0.4 ± 0.1 | 2 ± 1 |
| 3 | 20 | 70 | 0.5 ± 0.1 | 8 ± 2 |
| 6 | 20 | 70 | 1.1 ± 0.2 | nd |
| 3 | 35 | 70 | 0.5 ± 0.1 | 11 ± 2 |
When conducted in D2O, no change in the reaction rates was detected, which means no (or very little) isotopic effect for the hydration reaction.
Based on all of this information, a mechanism is proposed in Scheme 3.
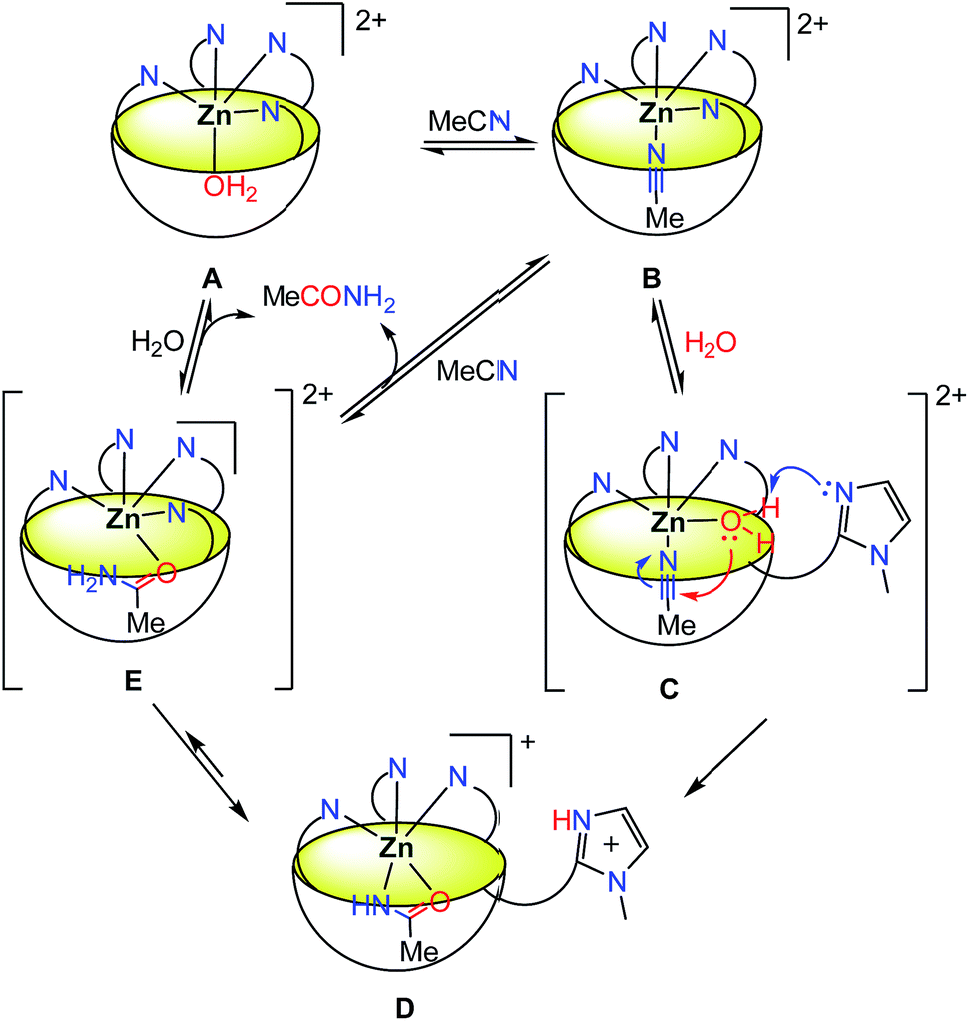 | ||
| Scheme 3 Proposed mechanism for the hydration of MeCN catalyzed by Rim4Zn. | ||
The addition of water leads to a decrease of MeCN occupancy: with 5% water content, only 50% of the resorcinarene sites are occupied by MeCN, and with 20% water content, the major species is the aqua complex A. This complex is in fast equilibrium with the nitrilo complex B (as evidenced by NMR spectroscopy, and in comparison to the rate of hydration). Knowing that one imidazole arm of complex B is hemilabile, it is likely that a fast equilibrium also exists between species B and C where the 4th imidazole arm is substituted by a water molecule.‡‡ The corresponding complex C presents in the cis position an acetonitrile guest substrate and a water ligand, the latter becoming acidic due its coordination to ZnII. The free imidazole arm can then act as a base for the activation of the water ligand37,38 and its nucleophilic addition to the polarized nitrile that sits in close proximity. The resulting complex D is the complex observed during the reaction course, in the stationary state. The equilibrium between species D and E is the most probable pathway for the substitution of acetamide (or the corresponding iminol, a neutral and thus labile ligand in E) by the solvent. This allows the regeneration of the initial form of the catalyst.
The kinetic study highlighted two different phases. At first, during the burst phase, A and B being in fast equilibrium, the limiting step is within B → C → D. The absence of noticeable KIE with D2O does not allow the identification of the rate determining step. In the second (slow) phase, a turn-over of 0.22 h−1 was observed at 70 °C and 35% water content. Under these conditions, it reached a TON of 43 after 198 hours with a 3 mM catalyst concentration. The slowness of the catalysis (the reaction rate v2 is two orders of magnitude lower than v1) is proposed to be due to the relatively strong binding of acetamide as an anion [K = (1.2 ± 0.5) × 103 M−1 in a 95:5 v/v MeCN/D2O mixture, see ESI Fig. S31†] that must be displaced through a sequence involving protonation and exchange for acetonitrile.
Nitrile hydration is a challenging process since the nitrilo function is a poor electrophile (especially acetonitrile) and water a poor nucleophile. Nature provides nitrile hydratases, which are metallo-enzymes that combine the activation of the nitrilo substrate by coordination to FeIII or CoIII to the attack of a coordinated sulfenate (derived from a cysteine residue) on either the water molecule (thus acting as an internal base)37,38 or directly onto the substrate.39 Although the biomimetic approach has led to the synthesis of a great variety of metal complexes, a relatively small number has been reported as active catalysts for the hydration of acetonitrile.40–42 With mononuclear CoIII complexes based on tetraaza ligands, simultaneous coordination of the nitrile and water molecules in the cis-position has been proposed to explain the efficiency of the catalyst.43,44 This route was also suggested by theoretical studies on ZnII-exchanged zeolites.45
Most efficient catalysts are based on metals not following a biomimetic approach, with the drawback of poor selectivity (vs. substrate as there is no substrate recognition step, and vs. products), as often hydrolysis follows the hydration step.46–51 Relatively recently however, the so-called bifunctional catalysts have been reported for the hydration of nitriles.52–56 These catalysts either employ a heteroatom in the ligand backbone to direct the water attack to the nitrile carbon atom, or use a hemilabile basic group for water activation. They are based mostly on precious metals such as Ru, Rh or Pt, although a very recent report describes a NiII catalyst displaying hemilability-driven water activation for the hydration of a coordinated nitrile.57 Examples of biomimetic ZnII catalysts are very rare.6–13,43,44 One of the most efficient is a ketoxime complex, which is proposed to act as a nucleophilic catalyst.58 Finally, it is worth noting that none of these catalysts involves molecular recognition through cavity binding. Breslow reported a CD functionalized by an oximate–nickel complex that is active for the catalytic hydrolysis of activated esters via transient nucleophilic attack of the cavity-hosted substrate by the oximate ligand, but not for nitrile hydration.59
Conclusions
Here, we have described the synthesis of a new ligand based on a resorcinarene macrocycle rigidified into a bowl structure by methylene bridges between the resorcinol units. It presents a tetra(imidazole) coordination core at the large rim of the bowl structure. Such an environment allows the formation of mononuclear ZnII complexes with a well-defined coordination sphere displaying an endo site available for the inclusion of a guest ligand. In acetonitrile, it is occupied by a molecule of solvent that can be readily displaced by anionic guests. The most important new features, when compared to the Rim3-based ZnII complex, stem from the presence of the 4th imidazole arm that is hemilabile. This latter can be selectively protonated without disruption of the ZnII environment, keeping its “bowl-complex” structure and endo-hosting properties. Such a property finds a direct application through the reaction of the dicationic nitrilo complex with acidic guests. The combination of the coordination of the ligand to the Lewis acidic ZnII center with its deprotonation by the 4th imidazole arm allows direct binding of carboxylic acids, β-diketones and even acetamide, under the control of the endo-binding site. An important consequence of such acido-basic/hemilabile features relates to the reactivity of the metal center as illustrated here with the hydration of acetonitrile. Indeed, the Rim4ZnII complex has been shown to catalyze the formation of acetamide when dissolved in MeCN in the presence of various amounts of water. Preliminary kinetic studies evidence biphasic kinetics. An initial burst phase corresponds to the fast appearance of endo-bound acetamide (t1/2 = 4 min at 70 °C), which is followed by a slow phase attributed to slow product release. A general base assistance behavior is proposed to explain the efficiency of the initial burst phase.In spite of its moderate activity relative to precious metal-based catalysts,52–57 this complex displays highly significant and unique biomimetic features relative to the water activation process in hydrolytic Zn enzymes. Indeed, this is the first time that the following criteria are put forward:
(i) A biomimetic 1st coordination sphere (three imidazole groups),
(ii) cis-coordination availability for two exogenous molecules,
(iii) basic assistance by an imidazole group acting as an internal base, and
(iv) the cavity effect for substrate and product binding.
This leads to spectacular assistance for acidic guest binding (e.g. MeCONH2) and acetonitrile hydration (a rare example of a ZnII based catalyst).
We are now further exploring the mechanism through in-depth kinetic studies and theoretical modeling as well as reactivity with other metal ions and other substrates with which cavity binding and general base assistance may be key features.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the CNRS (Institut de Chimie) and the Ministère de l’Enseignement Supérieur et de la Recherche. The COST Action CM1005 “Supramolecular Chemistry in Water” is kindly acknowledged for supporting scientific discussions and short-term scientific missions. G. B. acknowledges the Belgian FNRS (FRFC 2010: 2.4592.10F) and the van Buuren Foundation for the funding of the ITC equipment. The authors thank Dr Lionel Marcelis for fruitful discussions about kinetics and NMR analyses.Notes and references
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef PubMed.
- D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360 RSC.
- K. E. Dalle and F. Meyer, Eur. J. Inorg. Chem., 2015, 3391–3405 CrossRef.
- T. Joshi, B. Graham and L. Spiccia, Acc. Chem. Res., 2015, 48, 2366–2379 CrossRef PubMed.
- H. Aït-Haddou, J. Sumaoka, S. L. Wiskur, J. F. Folmer-Andersen and E. V. Anslyn, Angew. Chem., 2002, 114, 4185–4188 CrossRef.
- G. Feng, J. C. Mareque-Rivas, R. Torres Martín de Rosales and N. H. Williams, J. Am. Chem. Soc., 2005, 127, 13470–13471 CrossRef PubMed.
- R. A. Allred, K. Doyle, A. M. Arif and L. M. Berreau, Inorg. Chem., 2006, 45, 4097–4108 CrossRef PubMed.
- M. Livieri, F. Mancin, U. Tonellato and J. Chin, Chem. Commun., 2004, 2862 RSC.
- E. Y. Tirel, Z. Bellamy, H. Adams, V. Lebrun, F. Duarte and N. H. Williams, Angew. Chem., Int. Ed., 2014, 53, 8246–8250 CrossRef PubMed.
- E. Y. Tirel and N. H. Williams, Chem.–Eur. J., 2015, 21, 7053–7056 CrossRef PubMed.
- G. Feng, J. C. Mareque-Rivas and N. H. Williams, Chem. Commun., 2006, 1845 RSC.
- M. Livieri, F. Mancin, G. Saielli, J. Chin and U. Tonellato, Chem.–Eur. J., 2007, 13, 2246–2256 CrossRef PubMed.
- D. Coquière, S. Le Gac, U. Darbost, O. Sénèque, I. Jabin and O. Reinaud, Org. Biomol. Chem., 2009, 7, 2485 Search PubMed.
- N. Le Poul, Y. Le Mest, I. Jabin and O. Reinaud, Acc. Chem. Res., 2015, 48, 2097–2106 CrossRef PubMed.
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Chem. Soc. Rev., 2015, 44, 467–489 RSC.
- J.-N. Rebilly and O. Reinaud, Supramol. Chem., 2014, 26, 454–479 CrossRef.
- A. Višnjevac, J. Gout, N. Ingert, O. Bistri and O. Reinaud, Org. Lett., 2010, 12, 2044–2047 CrossRef PubMed.
- N. Natarajan, E. Brenner, D. Sémeril, D. Matt and J. Harrowfield, Eur. J. Org. Chem., 2017, 6100–6113 CrossRef.
- D. S. Auld, BioMetals, 2001, 14, 271–313 CrossRef PubMed.
- A. Messerschmidt, W. Bode and M. Cygler, Handbook of Metalloproteins, Wiley, 2004, vol. 3 Search PubMed.
- D. W. Christianson and W. N. Lipscomb, Acc. Chem. Res., 1989, 22, 62–69 CrossRef.
- W. N. Lipscomb and N. Sträter, Chem. Rev., 1996, 96, 2375–2434 CrossRef PubMed.
- N. Cerdà-Costa and F. Xavier Gomis-Rüth, Protein Sci., 2014, 23, 123–144 CrossRef.
- M. Hernick and C. A. Fierke, Arch. Biochem. Biophys., 2005, 433, 71–84 CrossRef PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 184–188 CrossRef PubMed.
- F. Fonseca, E. H. C. Bromley, M. J. Saavedra, A. Correia and J. Spencer, J. Mol. Biol., 2011, 411, 951–959 CrossRef PubMed.
- D. K. Wilson, F. B. Rudolph and F. A. Quiocho, Science, 1991, 252, 1278–1284 Search PubMed.
- H. E. Moll, D. Sémeril, D. Matt, M.-T. Youinou and L. Toupet, Org. Biomol. Chem., 2009, 7, 495–501 Search PubMed.
- J. Gout, A. Višnjevac, S. Rat, O. Bistri, N. Le Poul, Y. Le Mest and O. Reinaud, Eur. J. Inorg. Chem., 2013, 5171–5180 CrossRef.
- A. Kütt, I. Leito, I. Kaljurand, L. Sooväli, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829–2838 CrossRef PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef PubMed.
- J. Gout, S. Rat, O. Bistri and O. Reinaud, Eur. J. Inorg. Chem., 2014, 2819–2828 CrossRef.
- HyperChem Professional Release 7, HyperCube Inc., 1115 NW 4th St, Gainesville, FL 32601, USA Search PubMed.
- Y.-H. Cho and H.-S. Shin, Anal. Chim. Acta, 2013, 787, 111–117 CrossRef PubMed.
- C. Frieden, J. Biol. Chem., 1970, 245, 5788–5799 Search PubMed.
- K. Hashimoto, H. Suzuki, K. Taniguchi, T. Noguchi, M. Yohda and M. Odaka, J. Biol. Chem., 2008, 283, 36617–36623 CrossRef PubMed.
- S. Mitra and R. C. Holz, J. Biol. Chem., 2007, 282, 7397–7404 CrossRef PubMed.
- M. T. Nelp, Y. Song, V. H. Wysocki and V. Bandarian, J. Biol. Chem., 2016, 291, 7822–7829 CrossRef PubMed.
- F. Meyer, E. Kaifer, P. Kircher, K. Heinze and H. Pritkow, Chem.–Eur. J., 1999, 5, 1617–1630 CrossRef.
- P. J. Zinn, T. N. Sorrell, D. R. Powell, V. W. Day and A. S. Borovik, Inorg. Chem., 2007, 46, 10120–10132 CrossRef PubMed.
- L. M. Berreau and W. B. Tolman, Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, Wiley VCH, 2006 Search PubMed.
- J. H. Kim, J. Britten and J. Chin, J. Am. Chem. Soc., 1993, 115, 3618–3622 CrossRef.
- J. Chin, Acc. Chem. Res., 1991, 24, 145–152 CrossRef.
- L. A. Barbosa and R. A. van Santen, J. Mol. Catal. Chem., 2001, 166, 101–121 CrossRef.
- V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef PubMed.
- V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta., 2005, 358, 1–21 CrossRef.
- T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef.
- R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC.
- R. García-Álvarez, J. Francos, E. Tomás-Mendivil, P. Crochet and V. Cadierno, J. Organomet. Chem., 2014, 771, 93–104 CrossRef.
- R. González-Fernández, P. Crochet, V. Cadierno, M. I. Menéndez and R. López, Chem.–Eur. J., 2017, 23, 15210–15221 CrossRef PubMed.
- P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790–3797 CrossRef.
- T. Šmejkal and B. Breit, Organometallics, 2007, 26, 2461–2464 CrossRef.
- M. Muranaka, I. Hyodo, W. Okumura and T. Oshiki, Catal. Today, 2011, 164, 552–555 CrossRef.
- D. B. Grotjahn, Chem.–Eur. J., 2005, 11, 7146–7153 CrossRef PubMed.
- T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287–6290 CrossRef.
- K. Singh, A. Sarbajna, I. Dutta, P. Pandey and J. K. Bera, Chem.–Eur. J., 2017, 23, 7761–7771 CrossRef PubMed.
- M. N. Kopylovich, V. Y. Kukushkin, M. Haukka, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Inorg. Chem., 2002, 41, 4798–4804 CrossRef PubMed.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2012 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full NMR spectra, DOSY, IR, and ITC titrations. See DOI: 10.1039/c8sc01129j |
| ‡ Likewise, the diffusion coefficients obtained for Rim3 and Rim3Zn are very similar (8.2 × 10−10 and 8.0 × 10−10 m2 s−1, respectively). |
| § The reverse titration with Rim3, monitored via ITC, gave the same results as the direct one, which was not the case for Rim4. Due to the fourth imidazole, an excess of zinc may have an effect on the protonation form of the ligand, which results in a phenomenon different from complexation. |
| ¶ The pKa value was calculated considering the following equilibrium: [Rim4Zn(S)]2+ + PicH ⇔ [Rim4Zn(S)H]3+ + Pic−, which results from the reaction of two acid/base couples [pKa(PicH) in MeCN = 11]. The associated constant K in acetonitrile can thus be written as K = Ka,PicH/Pic−/Ka,[Rim4ZnH]3+/[Rim4Zn]2+, from which Ka,[Rim4ZnH]3+/[Rim4Zn]2+ is deduced. |
| || Compared pKa values in DMSO: imidazolium: 6.4; AcOH: 12.3; acetylacetone: 13.5; AcNH2: 25.5 (values from the Bordwell pKa table). The corresponding values in MeCN have not been reported. |
| ** With all acidic guests (GH = RCO2H, MeCONH2, and MeCOCH2COMe), the δ shift of added Et3N showed its quantitative protonation. A detailed structural analysis of the acetamido complex is currently under investigation, as well as the kinetics of its binding, which is slow under anhydrous conditions, and fast in the presence of water. These studies will be the subject of another article. |
| †† The mono-cationic complexes obtained with β-diketones and Et3N present most probably a pending imidazole arm that is in rapid exchange with the three-coordinated one. |
| ‡‡ This was substantiated by an NMR experiment where the gradual addition of water to a solution of [Rim4Zn(OAc)](ClO4) in CD3CN led to the appearance of new peaks in the low- and high-field regions, thereby attesting to the formation of a solvated (by water) species. See ESI Fig. S22.† |
| This journal is © The Royal Society of Chemistry 2018 |