
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β C–H di-halogenation via iterative hydrogen atom transfer†
Ethan A.
Wappes‡
,
Avassaya
Vanitcha‡
and
David A.
Nagib
 *
*
The Ohio State University, Department of Chemistry and Biochemistry, Columbus, OH 43210, USA. E-mail: nagib.1@osu.edu
First published on 30th April 2018
Abstract
A radical relay strategy for mono- and di-halogenation (iodination, bromination, and chlorination) of sp3 C–H bonds has been developed. This first example of β C–H di-halogenation is achieved through sequential C–H abstraction by iterative, hydrogen atom transfer (HAT). A double C–H functionalization is enabled by in situ generated imidate radicals, which facilitate selective N˙ to C˙ radical translocation and tunable C–X termination. The versatile, geminal di-iodide products are further elaborated to β ketones and vinyl iodides. Mechanistic experiments explain the unique di-functionalization selectivity of this iterative HAT pathway, wherein the second C–H iodination is twice as fast as the first.
Introduction
The halogenation of an sp3 C–H bond1 enables direct conversion of an inert motif into a versatile synthetic handle that permits broad reactivity via cross-coupling and substitution.2 Generally, C–H halogenation occurs by radical-mediated3 or organometallic4 mechanisms. Each approach exhibits complementary reactivity and selectivity – especially for incorporation of the most versatile halide: an iodide (Fig. 1a). In the realm of metal-mediated sp3 C–H iodination, there are just a few methods that can install this reactive handle; they are stoichiometric5 or catalytic6 in Pd. In the latter cases, only Yu and Rao have reported directed sp3 C–H iodination – employing oxazolines, amides, or oximes as directing groups (Fig. 1b).6 These Pd-catalyzed methods exclusively effect primary C–H conversion to a terminal mono-iodide, which is deactivated to further reactivity. In this mechanism, a second iodination at a distal, primary C–H affords a 1,3-di-iodide.7 | ||
| Fig. 1 Directed, mono- and di-iodination of sp3 C–H bonds. | ||
Alternatively, radical mechanisms can promote efficient iodination of various types of sp3 C–H bonds via hydrogen atom transfer (HAT).8 Moreover, intramolecular HAT provides unique, δ selective C–H functionalizations.9 Yet, non-directed methods10 surpass the few, pioneering examples of δ (or γ) C–H halogenation.11 Notably, a directed C–H iodination has yet to be developed, despite the key intermediacy of a distal iodide in several δ C–H aminations (or etherification) mediated by 1,5-HAT.12 Due to the penchant for iodide displacement, intercepting this alkyl iodide intermediate is challenging. As an alternate strategy, we proposed a cascade mechanism – involving abstraction of the adjacent, α-iodo C–H – might enable geminal C–H di-iodination (Fig. 1c).
We noted that Suárez observed a minor di-iodide byproduct upon intramolecular δ amination of 8-membered lactams.13 Benzylic tri-iodination mechanisms have also been proposed,14 but no method yet exists to isolate them.
Given the limited synthetic accessibility (and potential pharmacological value15) of gem-di-iodides – an important, versatile motif (previously only accessible from hydrazones or vinyl iodides)16 – we sought to design a strategy to harness a directed, iterative HAT mechanism to introduce geminal di-halides at remote carbons. Notably, this new type of double C–H iodination at a single carbon atom is complementary to Pd-catalyzed methods and uniquely possible via a radical mechanism (Fig. 1).
To develop a versatile β C–H di-iodination via iterative, intramolecular HAT and sequential iodination, we chose to employ imidates as readily accessible, radical relay precursors (Fig. 1c). In our proposed di-iodination mechanism, we envisioned that in situ formation of a weak imidate sp2 N–I bond would enable its rapid homolysis by visible light. Selective translocation of the ensuing N-centered radical to a β C˙ can occur via thermodynamically favored 1,5-HAT. Finally, either radical recombination with I˙ (derived from the initial N–I homolysis), or homolytic substitution by I2 (or N–I), can afford a reactive β iodide. However, we were cognizant of two major challenges (Fig. 1d) for trapping the δ iodide intermediate of HAT mechanisms, including its reactivity: (1) as a leaving group, and (2) towards further oxidative decomposition.
Whereas, we previously observed weak C–H bonds (e.g. benzyl, allyl) provide activated iodides that are rapidly displaced (in a formal C–H amination),17 secondary (2°) C–H bonds yield complete decomposition. Given our knowledge that I3− efficiently mediates HAT of 2° C–H bonds,18 we hypothesized a β iodide intermediate is formed, yet is prone to further I-oxidation. In this case, decomposition may ensue from the resulting sp3 hypervalent iodide, which is an excellent nucleofuge for elimination or cyclization.19 Instead, to enable access to gem-di-iodides, we proposed an alternate N-selective oxidation may promote a second HAT of the slightly weaker β C–H (103 vs. 105 kcal mol−1).20 Importantly, however, this iterative HAT mechanism for directed, di-functionalization is only possible if N-oxidation is more rapid than the previously observed, I-oxidation pathway.
Results and discussion
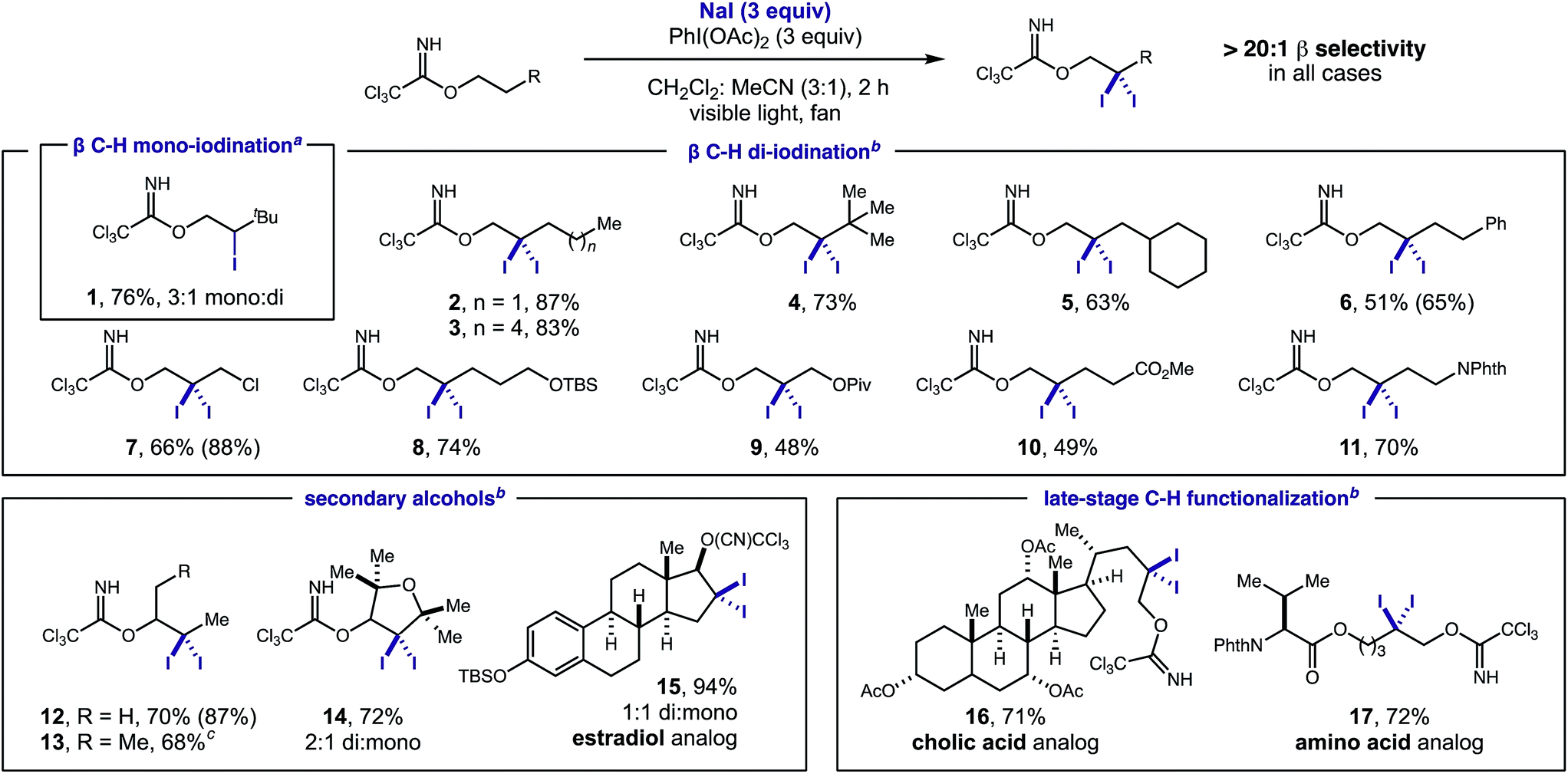
To our delight, adaptation of our radical relay strategy allowed us to intercept the 2° β iodide intermediate for the first time to access both mono- and di-β C–H iodides. The key factors that enabled discovery of these new reactions included judicious choice of oxidant, increased reaction concentration, and shorter reaction duration – all essential to limit product decomposition. Notably, NIS oxidant was found to favor β mono-iodide 1 formation, while a combination of NaI and PhI(OAc)2 provides desired β di-iodide 2–17. For the latter, a strong solvent effect was also observed, wherein greater solubility of NaI (in HFIP or CH2Cl2) affords less product (3, <30%), while more polar, but less solubilizing MeCN affords a higher yield of β di-iodide 3 (58%). Ultimately, a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH2Cl2:MeCN was found to provide the gem-di-iodide most efficiently (3, 88%, 83% isolated yield) (see ESI† for full details of optimization).
:1 mixture of CH2Cl2:MeCN was found to provide the gem-di-iodide most efficiently (3, 88%, 83% isolated yield) (see ESI† for full details of optimization).
Having developed the first method for β C–H di-iodination, we next investigated the generality of this radical-mediated transformation with a variety of imidates – derived from base-induced addition of alcohols into Cl3C–CN. In all cases, we observed efficient formation of β di-iodides with greater than 20:1 regioselectivity (Table 1).
|
a Conditions: C–H mono-iodination: NIS (1 equiv.), MeCN, visible light (26 W CFL).
b Conditions: C–H di-iodination: NaI (3 equiv.), PhI(OAc)2 (3 equiv.), 3:1 CH2Cl2:MeCN, visible light (26 W CFL).
c Conditions: 2 equiv. NaI and PhI(OAc)2; <10% distal di-iodide. Isolated yields. 1H NMR yields in parenthesis.
|
|---|
|
Except for the NIS-based conditions that afford mono-iodide 1, di-iodide is always the major product, typically isolated in high yields (2–3). Interestingly, this reaction is tolerant of steric congestion (4–5) and remains β selective even in the presence of weaker C–H bonds adjacent to arenes, halides, ethers, esters, and amides at the γ or δ positions (6–11). Secondary alcohols are also amenable to this di-iodination with selectivity observed for secondary over primary C–H bonds (12) – in contrast to Pd-mediated pathways.6 While acyclic 2° alcohols efficiently yield di-iodide (13), cyclic alcohols afford a 2:1 mixture of di- and mono-iodide (14) – illustrating conformational constraints for the HAT mechanism. Similarly, an estradiol-derived imidate affords a 1:1 mixture of mono- and di-iodide (15). Imidates derived from cholic acid and amino acid, valine, yield gem-di-iodides (16–17) efficiently.
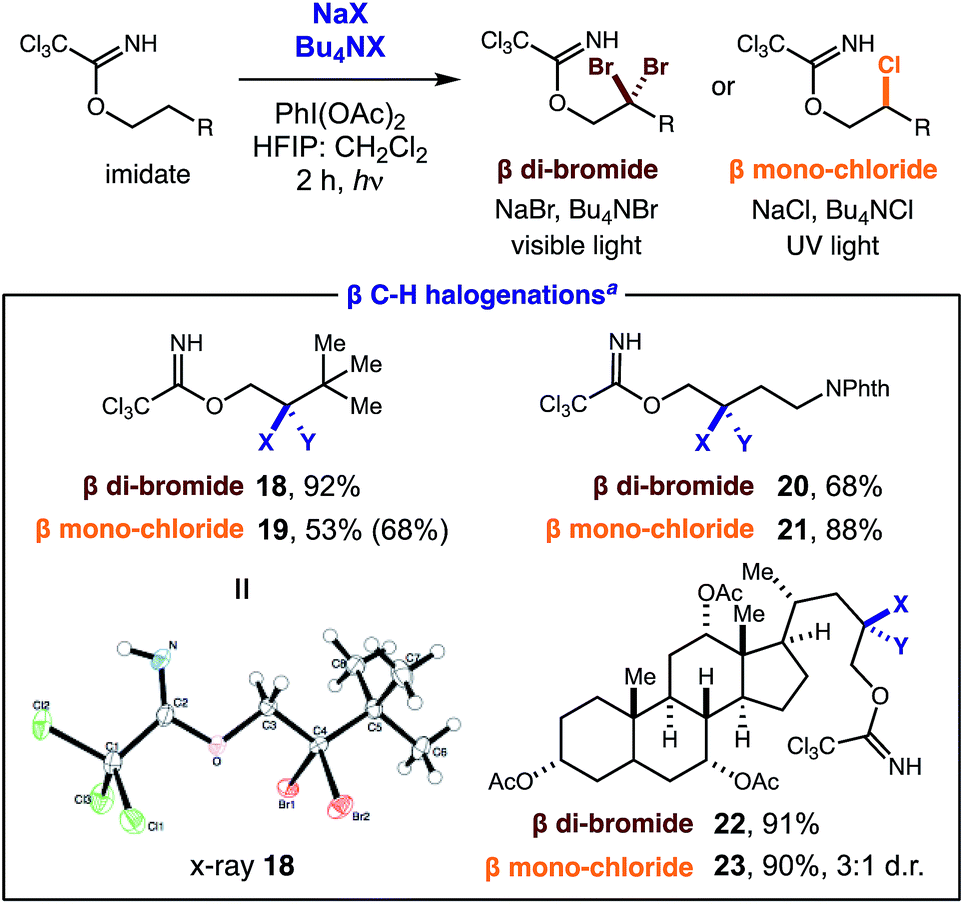
Cognizant of the synthetic utility of gem-di-halides, we sought to extend this unique di-iodination mechanism to other halides. To this end, we found that the use of NaBr or NaCl (instead of NaI) affords analogous β halogenation (Table 2). These new transformations require slight deviation from standard reaction conditions since NaBr and NaCl are less soluble. In these cases, increased halide concentration via phase transfer catalysts (Bu4N+X−) and a more solubilizing solvent mixture (3:1 HFIP:CH2Cl2) are the key factors that enable these new reactions.
|
a C–H di-bromination: NaBr (3 equiv.), Bu4NBr (1 equiv.), PhI(OAc)2 (3 equiv.), 3:1 HFIP:CH2Cl2, visible light (26 W CFL). C–H mono-chlorination: NaCl (3 equiv.), Bu4NCl (1 equiv.), PhI(OAc)2 (3 equiv.), 3:1 HFIP:CH2Cl2, UV light (300 nm). Isolated yields. 1H NMR yields in parenthesis.
|
|---|
|
Notably, a stronger N–Cl intermediate requires UV light (300 nm) for initiation of the radical relay. It is also noteworthy that C–H chlorination ceases after the first halogenation despite a relative similarity in the α-Cl and α-Br C–H bond strengths (±1 kcal).21 The scope is as general as the iodination, with three representative examples shown for each halide (18–23). X-ray crystallographic analysis of di-bromide 18 confirms the structure of these distal geminal halides.
Interested in further understanding this exceptionally efficient sequential di-iodination (which provides orthogonal reactivity and selectivity to Pd catalysis), we sought to explore our hypothesis that the weaker α-iodo C–H bond enables this transformation. First, a kinetic study by 1H NMR illustrates a rapid conversion of the mono-iodide intermediate to the di-iodide product (Fig. 2). After an initial induction period (ca. 10 min), mono-iodide 24 is formed in ∼30% yield, before rapid conversion to di-iodide 2.
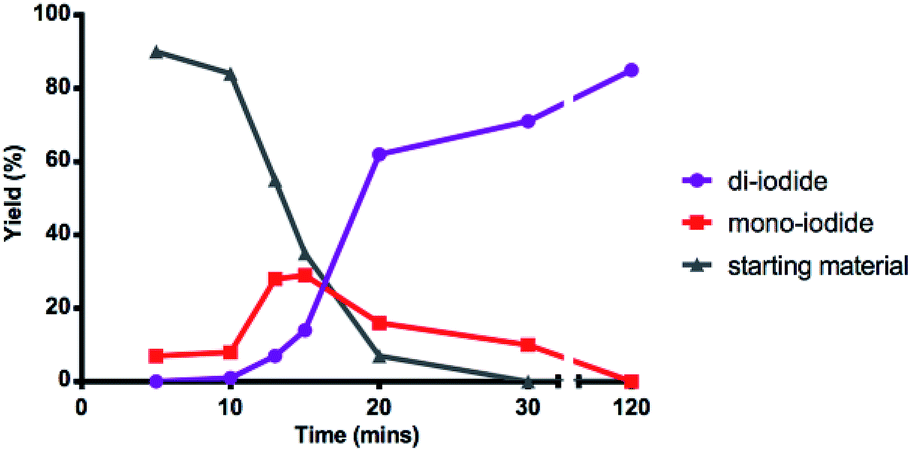 | ||
| Fig. 2 Kinetics of mono and di C–H iodination. | ||
In separate experiments, initial rates of formation of mono-iodide 24 and di-iodide 2 were independently measured from their respective starting materials (Fig. 3a), using 1 equiv. of oxidant, for more accurate measurements. A relative rate of 2.2 was observed in the second iodination, supporting the expectation it is more rapid than the first due to a weaker C–H bond. In the course of our studies, we were also interested in comparing the relative rates of reactivity among the various halides. To this end, we performed competition experiments between NaI & NaBr/NaCl (Fig. 3b). In the I/Br competition, a statistical mixture of products is formed (1:1:2 di-iodide 4:di-bromide 18:mixed 25) – suggesting both reaction rates are comparable. On the other hand, an I/Cl competition provides greater selectivity. Only mono- and di-iodide products (4) are observed with visible light irradiation (since chlorination requires UV light); yet UV irradiation (which unproductively consumes iodinated species) exclusively affords chlorination (19). Lastly, we exploited the difference in halide reactivity to enable a synthetically useful, iterative C–H halogenation (Fig. 3c). In the sequence, mono C–H chlorination (26) and subsequent C–H iodination affords β geminal halide 27 that contains two different halides (Cl, I).
 | ||
| Fig. 3 Mechanistic experiments: (a) initial rates of mono vs. di C–H iodination; (b) competitive and (c) iterative C–H halogenation. | ||
Equipped with the first method to access β gem-di-halides via C–H functionalization, we sought to elucidate the synthetic utility of these versatile handles. Fig. 4 illustrates five post-synthetic transformations we investigated to further elaborate the β di-iodide imidates. First, aminolysis with NH3 affords β di-iodo-alcohol 28. Alternatively, reduction of one of the iodides by Zn in AcOH affords vinyl iodide 29via imidate elimination. Otherwise, imidate hydrolysis to ester 30 occurs under acidic conditions (HBF4·H2O), leaving the di-iodide intact. From the β di-iodo-ester, hydrolysis to α-oxy ketone 31 is possible (AgBF4, Na2HPO4·H2O); or conversion to allyl alcohol 32, bearing a vinyl iodide, is realized via addition of AgOTf and K2HPO4.
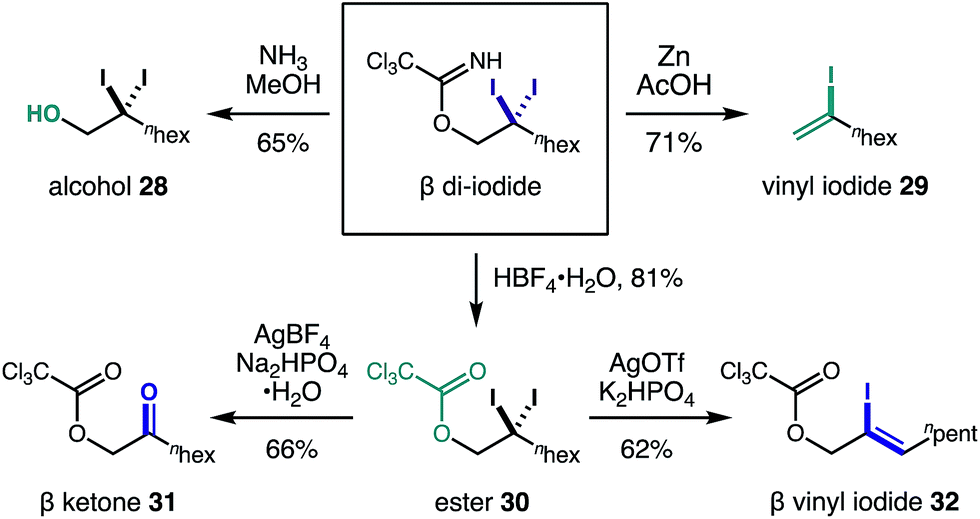 | ||
| Fig. 4 Synthetic versatility of the geminal β di-iodides. | ||
Conclusions
In summary, a radical relay strategy has enabled the one-step conversion of imidates to mono- or di-halides via iterative β C–H halogenation. In particular, synthetic access to the versatile, geminal di-halides is uniquely facilitated by an imidate radical-based 1,5-HAT mechanism. By developing a new strategy to bypass oxidative decomposition pathways, reactive alkyl halide intermediates of a radical relay reaction mechanism were intercepted. Along with new methods for mono- and di-C–H halogenation (X = I, Br, Cl), competitive rates and kinetic profiles have also been investigated. Finally, the versatility of the β di-iodides is showcased in the synthesis of functionally rich molecules – uniquely enabled by an HAT-based β C–H functionalization mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank The Ohio State University, National Institutes of Health (NIH R35 GM119812), National Science Foundation (NSF CAREER 1654656), and American Chemical Society Petroleum Research Fund for financial support.Notes and references
- C–H halogenation reviews: (a) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed; (b) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed; (c) F. Lied, T. Patra and F. Glorius, Isr. J. Chem., 2017, 57, 945 CrossRef CAS.
- The Chemistry of Halides Pseudo-Halides and Azides, ed. S. Patai and Z. Rappoport, John Wiley & Sons, Ltd., Chichester, UK, 2010 Search PubMed.
- HAT-mediated C–H functionalization reviews: (a) R. Breslow, Acc. Chem. Res., 1980, 13, 170 CrossRef CAS; (b) A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (d) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef CAS.
- Pd-catalyzed C–H halogenation reviews: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- K. Carr and J. K. Sutherland, J. Chem. Soc., Chem. Commun., 1984, 1227 RSC.
- (a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed; (b) X. Yang, Y. Sun, T. Sun and Y. Rao, Chem. Commun., 2016, 52, 6423 RSC; (c) R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724 CrossRef CAS PubMed; (d) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 12394 CrossRef CAS PubMed.
- R. Giri, M. Wasa, S. P. Breazzano and J.-Q. Yu, Org. Lett., 2006, 8, 5685 CrossRef CAS PubMed.
- Non-directed C–H iodination via HAT: (a) D. D. Tanner and G. C. Gidley, J. Am. Chem. Soc., 1968, 90, 808 CrossRef CAS; (b) J. Barluenga, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2002, 41, 2556 CrossRef CAS; (c) R. Montoro and T. Wirth, Org. Lett., 2003, 5, 4729 CrossRef CAS PubMed; (d) A. Artaryan, A. Mardyukov, K. Kulbitski, I. Avigdori, G. A. Nisnevich, P. R. Schreiner and M. Gandelman, J. Org. Chem., 2017, 82, 7093 CrossRef CAS PubMed.
- Recent examples of directed C–H functionalization via 1,5-HAT: (a) Y.-F. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980 CrossRef CAS PubMed; (b) T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871 CrossRef CAS PubMed; (c) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340 CrossRef CAS PubMed; (d) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS PubMed; (e) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef PubMed; (f) J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872 CrossRef CAS PubMed; (g) C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495 CrossRef CAS PubMed; (h) W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881 CrossRef CAS PubMed; (i) P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004 CrossRef CAS PubMed; (j) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857 CrossRef CAS PubMed; (k) D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897 CrossRef CAS PubMed; (l) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692 CrossRef CAS PubMed; (m) M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712 CrossRef CAS PubMed; (n) X.-Q. Mou, X.-Y. Chen, G. Chen and G. He, Chem. Commun., 2018, 54, 515 RSC.
- Recent, non-directed C–H halogenations via HAT: (a) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847 CrossRef CAS PubMed; (b) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed; (c) V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389 CrossRef CAS PubMed; (d) R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696 CrossRef CAS PubMed; (e) Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679 RSC.
- Directed C–H halogenation (X = Br, Cl, F) via intramolecular HAT: (a) C. Walling and A. Padwa, J. Am. Chem. Soc., 1963, 85, 1597 CrossRef CAS; (b) R. Breslow, R. J. Corcoran, B. B. Snider, R. J. Doll, P. L. Khanna and R. Kaleya, J. Am. Chem. Soc., 1977, 99, 905 CrossRef CAS PubMed; (c) G. I. Nikishin, E. I. Troyansky and M. I. Lazareva, Tetrahedron Lett., 1985, 26, 3743 CrossRef CAS; (d) L. R. Reddy, B. V. S. Reddy and E. J. Corey, Org. Lett., 2006, 8, 2819 CrossRef CAS PubMed; (e) R. Kundu and Z. T. Ball, Org. Lett., 2010, 12, 2460 CrossRef CAS PubMed; (f) K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed; (g) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894 CrossRef CAS PubMed; (h) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771 CrossRef CAS PubMed; (i) T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306 CrossRef CAS PubMed; (j) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296 CrossRef CAS PubMed; (k) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef CAS PubMed; (l) S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100 RSC.
- G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095 CrossRef CAS.
- R. L. Dorta, C. G. Francisco and E. Suárez, J. Chem. Soc., Chem. Commun., 1989, 1168 RSC.
- (a) M. Katohgi, H. Togo, K. Yamaguchi and M. Yokoyama, Tetrahedron, 1999, 55, 14885 CrossRef CAS; (b) N. R. Paz, D. Rodríguez-Sosa, H. Valdés, R. Marticorena, D. Melián, M. B. Copano, C. C. González and A. J. Herrera, Org. Lett., 2015, 17, 2370 CrossRef CAS PubMed.
- M. J. Tozer and T. F. Herpin, Tetrahedron, 1996, 52, 8619 CrossRef CAS.
- Synthesis of gem-di-iodides: (a) D. H. R. Barton, R. E. O'Brien and S. Sternhell, J. Chem. Soc., 1962, 470 RSC; (b) M. Shimizu, T. Toyoda and T. Baba, Synlett, 2005, 16, 2516 CrossRef; (c) A. Spaggiari, D. Vaccari, P. Davoli, G. Torre and F. Prati, J. Org. Chem., 2007, 72, 2216 CrossRef CAS PubMed; (d) H. Lu, Q. Chen and C. Li, J. Org. Chem., 2007, 72, 2564 CrossRef CAS PubMed.
- E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204 CrossRef CAS PubMed.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef CAS PubMed.
- (a) C. Martínez and K. Muńiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed; (b) L. Bering and A. P. Antonchick, Chem. Sci., 2017, 8, 452 RSC; (c) Ref. 8b ; (d) Alkenes were not observed, under these oxidative conditions..
- BDE calculations (see ESI† for full details) are supported by literature values: 105 (CH4) vs. 103 (CH3I) kcal mol−1 in: Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, Taylor & Francis, Boca Raton, FL, 2010 Search PubMed.
- The increased electrophilicity of Cl (vs. I or Br) may polarize the C–H to such an extent that it is no longer polarity-matched with the imidate radical. For example, see: B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC.
- CCDC 1581032 contains the supplementary crystallographic data for this paper.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1581032. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01214h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |