
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper mediated C–H amination with oximes: en route to primary anilines†
Lin-Lin
Xu‡
 b,
Xing
Wang‡
a,
Biao
Ma
a,
Ming-Xing
Yin
a,
Hai-Xia
Lin
b,
Hui-Xiong
Dai
*a and
Jin-Quan
Yu
c
b,
Xing
Wang‡
a,
Biao
Ma
a,
Ming-Xing
Yin
a,
Hai-Xia
Lin
b,
Hui-Xiong
Dai
*a and
Jin-Quan
Yu
c
aDepartment of Medicinal Chemistry, Shanghai Institute of Materia Medica, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai, 201203, China. E-mail: hxdai@simm.ac.cn
bDepartment of Chemistry, Innovative Drug Research Center, Shanghai University, 99 Shangda Road, Shanghai, 200444, China
cDepartment of Chemistry, The Scripps Research Institute, 10550N. Torrey Pines Road, La Jolla, California 92037, USA
First published on 14th May 2018
Abstract
Here we report an efficient Cu(I)-mediated C–H amination reaction with oximes as amino donors to introduce NH2 groups directly. Various strongly coordinating heterocycles including quinoline, pyrimidine, pyrazine, pyrazole and triazole were tolerated well. The potential utility was further demonstrated in a late-stage modification of telmisartan (an antagonist for the angiotensin II receptor).
Introduction
Primary anilines are important structural motifs present in many natural and pharmaceutical compounds (Fig. 1).1 For example, retigabine is an anticonvulsant used as an adjunctive treatment for partial epilepsies in treatment-experienced adult patients.2 Anileridine, as a member of the piperidine class of analgesic agents, is used as an analgesic drug.3 Mesalazine is an aminosalicylate anti-inflammatory drug used for treating inflammatory bowel disease,4 and lenalidomide is a tumour necrosis factor (TNF) inhibitor and can be used to treat multiple myeloma.5 The development of efficient and practical approaches to accessing this type of compound is in high demand and remains a challenge in organic synthesis.6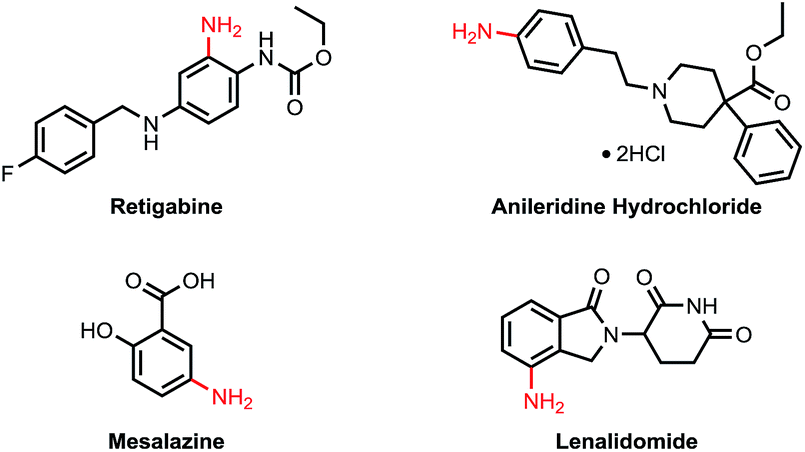 | ||
| Fig. 1 Selected drug molecules containing primary anilines. | ||
Recently, C–H amination via a transition-metal catalyzed C–H activation process has attracted considerable attention and has been proven to be a useful method for preparing amine molecules.7 Among these C–H amination reactions, only a few examples were able to directly introduce an unprotected amino group to form primary anilines.8 These reactions can be classified into two approaches: non-directed and directed (Scheme 1). The former exploits the innate reactivity of the substrates. For example, the non-directed amination of arenes involving photocatalysis was realized by the Tung9a and Nicewicz9b groups independently.
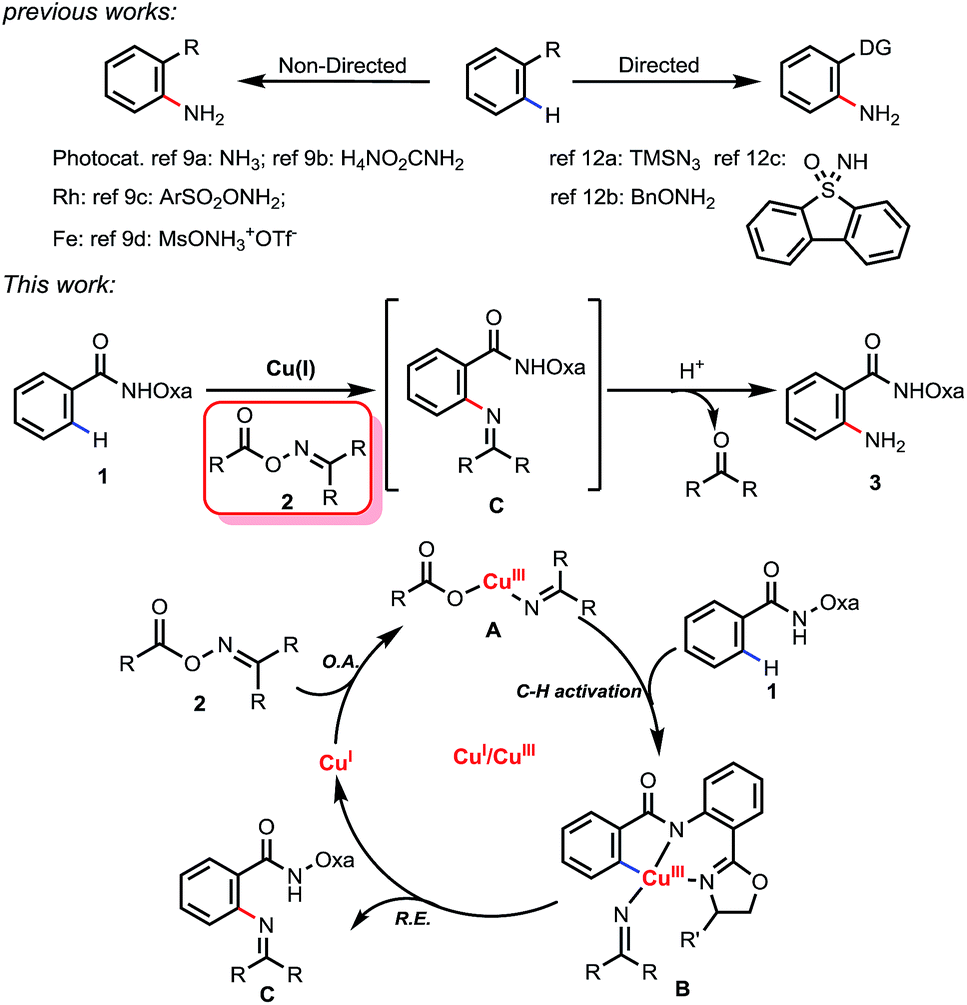 | ||
| Scheme 1 The direct synthesis of primary anilines by C–H amination. | ||
Transition metal-catalyzed C–H aminations of arenes were also reported by the two groups of Falck9c and Morandi.9d However, controlling the positional selectivity effectively remains a great challenge in these reactions. In recent years, the use of directing groups has become a reliable approach to realizing selective C–H functionalization.10 However, few examples of C–H amination for preparing primary anilines are known. The use of chalcogenide amine donors is largely limited to alkyl amines.11 In 2014, the Zhu group reported an example employing TMSN3 as a nitrogen source to form primary anilines.12a Uchiyama and co-workers developed another amination protocol which used BnONH2 as aminating agent and involved deprotonative cupration with a stoichiometric amount of (TMP)2Cu(CN)Li2.12b More recently, Bolm and coworkers have developed a new reagent, dibenzothiophene sulfoximine, as an NH3 surrogate to synthesize primary anilines.12c However, site selective C–H amination in late-stage heterocycle drug discovery is always a thorny problem because heterocycles compete with directing groups for coordination to metal catalysts, which may result in the poisoning of the catalyst or the functionalization of an undesired C–H bond. Herein, we report for the first time a facile Cu(I)-mediated Csp2–H amination using oxime derivatives as an aminating agent, in which the free amine products can be easily obtained upon in situ hydrolysis. The potential use of this method in late-stage diversification for drug discovery was exemplified by the directed amination of telmisartan.
Oxime derivatives as a class of versatile building block have been applied to a number of transition metal-catalyzed C–H functionalizations.13 Due to the N–O σ bond (∼57 kcal mol−1) being weaker than normal C–X (X = C, N, O) σ bonds, oxime derivatives could not only act as amino sources but also as oxidants. Based on our previous work14 and oxime chemistry reported by other groups,13 we propose a Cu(I) to Cu(III) catalytic cycle (Scheme 1): the first step is the oxidative insertion of the Cu(I) species into the N–O bond to give the Cu(III) intermediate A. After coordination of the bidentate directing group oxazoline-amide to the Cu(III) complex A, C–H activation occurs to generate the intermediate B. Subsequent reductive elimination affords imine compound C and regenerates Cu(I). Finally, imine compound C can be hydrolyzed to form primary amine 3 and a ketone.
Results and discussion
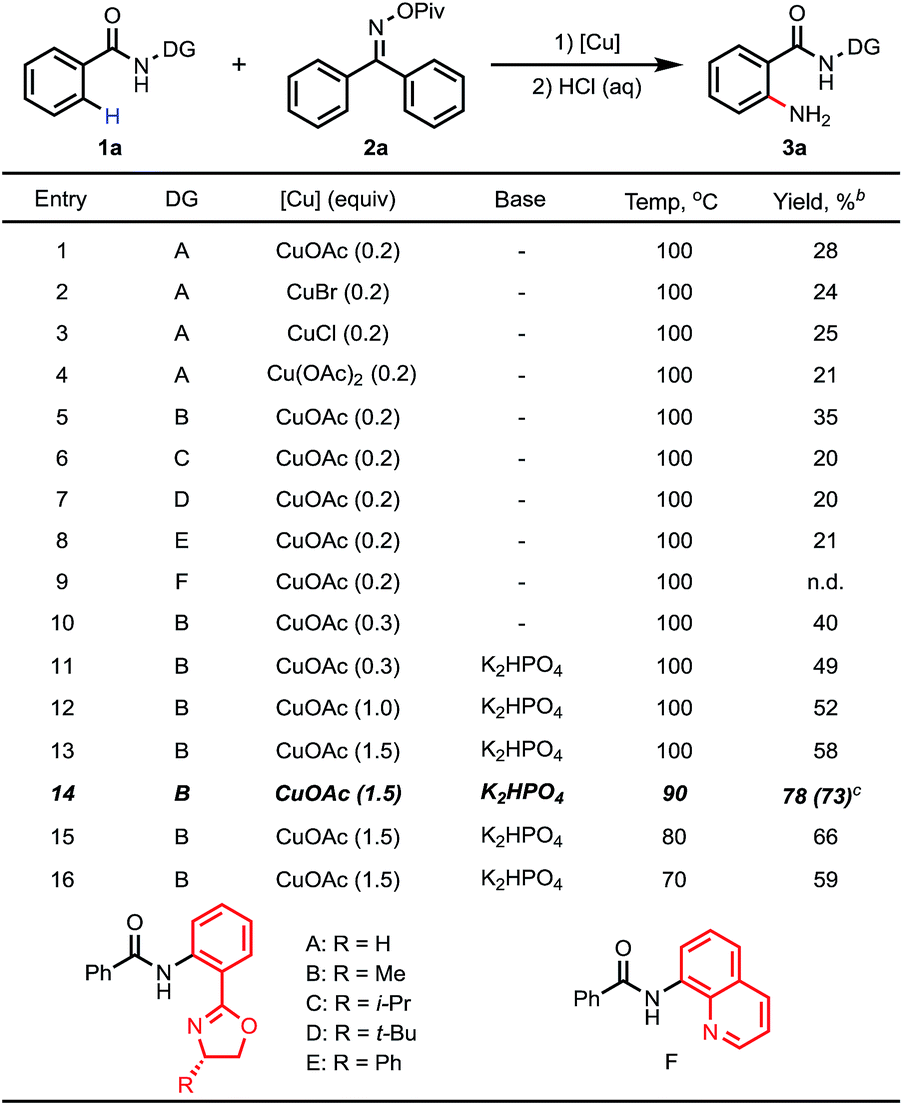
Based on this hypothesis, we initiated our studies by investigating the C–H amination of N-arylbenzamide, substrate 1a, with oxime 2a. Encouragingly, we found that C–H amination of 1a-A with 1.5 equiv. of 2a occurred in the presence of 20 mol% CuOAc in DMSO at 100 °C over 8 h, and the desired aminated product 3a-A was obtained in 28% yield after acid hydrolysis. The structure of the product 3a-A was confirmed by X-ray crystallography (Table 1). Other copper catalysts were also investigated (Table 1, entries 1–4), and CuOAc was still found to be the optimal one. Subsequently, we investigated the influence of the directing groups (DG) (Table 1, entries 5–9) on the efficiency of the amination reaction. We found that the use of substrate 1a-B, containing the methyl-substituted oxazoline DG B, afforded the aminated product 3a-B in 35% yield. Interestingly, the use of Daugulis’s 8-aminoquinoline DG under the same conditions did not provide any desired product (entry 9). This result is consistent with our early observation.14 The yield was improved to 40% by further increasing the amount of CuOAc to 30 mol%. After screening a series of bases (see ESI†), a higher yield of 49% was obtained using 1 equiv. of K2HPO4 as a base. Considering that the product binds to the catalyst better than the starting material, increasing the amount of copper may be beneficial to the reaction. Indeed, we found that when 1.5 equiv. of CuOAc was used, the yield could be increased to 58% with full conversion of substrate 1a (Table 1, entry 13), which implied that the substrate partially decomposed at this temperature. Finally, we screened the temperature (Table 1, entries 14–16), and found that a satisfactory yield of 78% could be obtained at 90 °C (entry 14).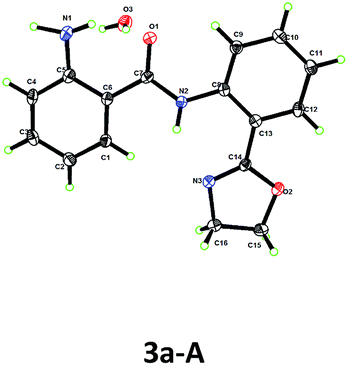
After the optimal reaction conditions were found, we proceeded to examine the substrate scope. As shown in Table 2, a wide variety of substituted benzamides were reactive. 2-Methyl substituted substrate 1b gave the aminated product in a moderate yield of 54%. Various electron-donating and -withdrawing substituents including methyl 1c, t-butyl 1d, methoxyl 1e, fluoro 1f, chloro 1g, bromo 1h, acetyl 1i, trifluoromethyl 1j, phenyl 1k and vinyl 1l at position 4 of the benzamides were compatible in the reaction, affording the desired products in moderate to good yields. When position 3 of the benzamides was substituted with methyl, methoxyl or fluoro groups, C–H activation took place at positions 2 and 6 to give two regioisomeric products, with the less hindered position 6-aminated product as the major one. In the case of the benzamide bearing 3-CF3 (3p), only the position 6-aminated product was obtained. Gratifyingly, this copper-mediated C–H amination protocol was compatible with a wide variety of heterocycles, including pyridines, quinoline, and benzothiphene-derived substrates (3q–t). Notably, 0.3 equiv. of CuOAc was sufficient to enable the amination reaction (the yields are also shown in parentheses), and the yields were even higher than those obtained in the reactions using 1.5 equiv. of CuOAc in some cases (3g, 3h and 3t).
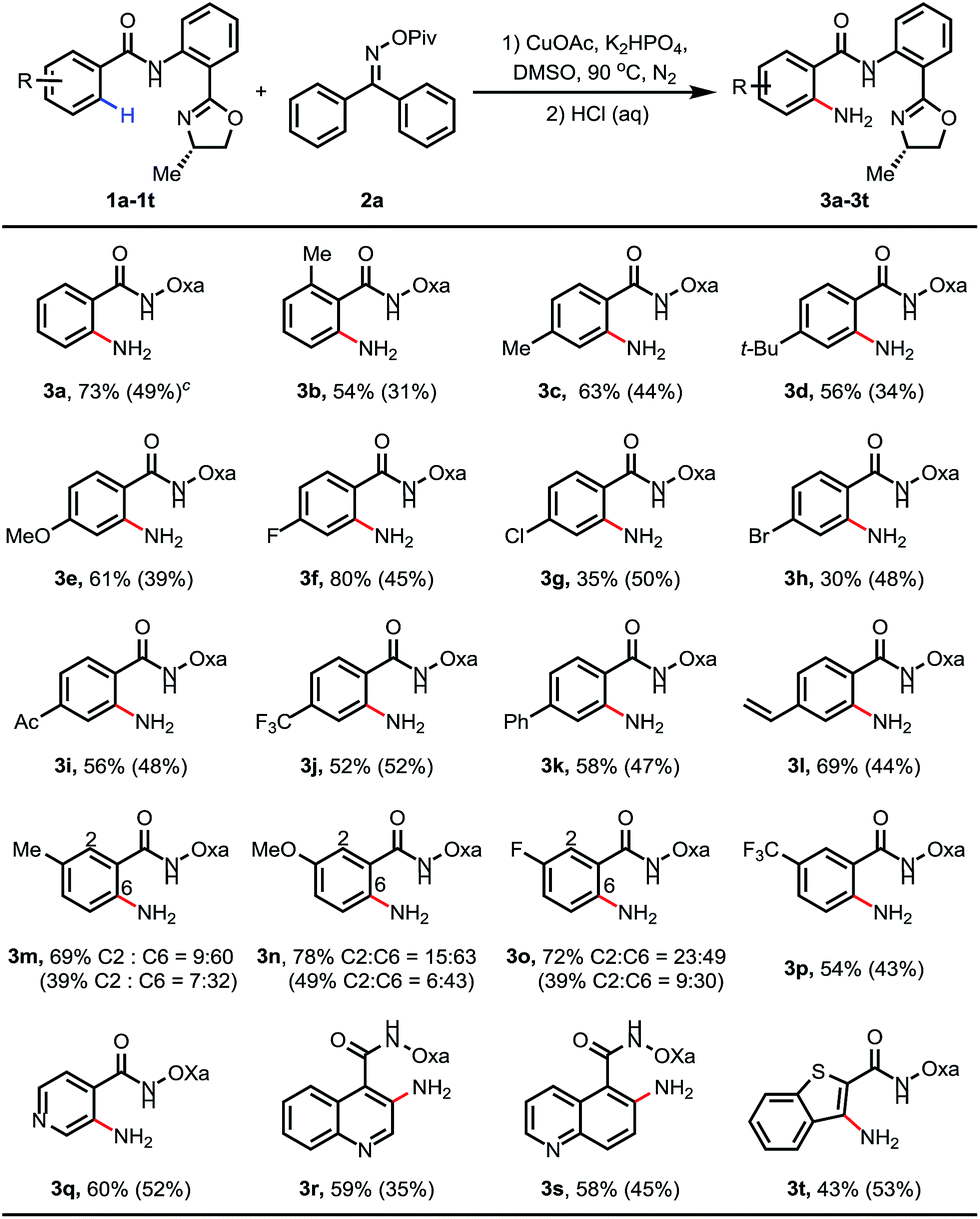
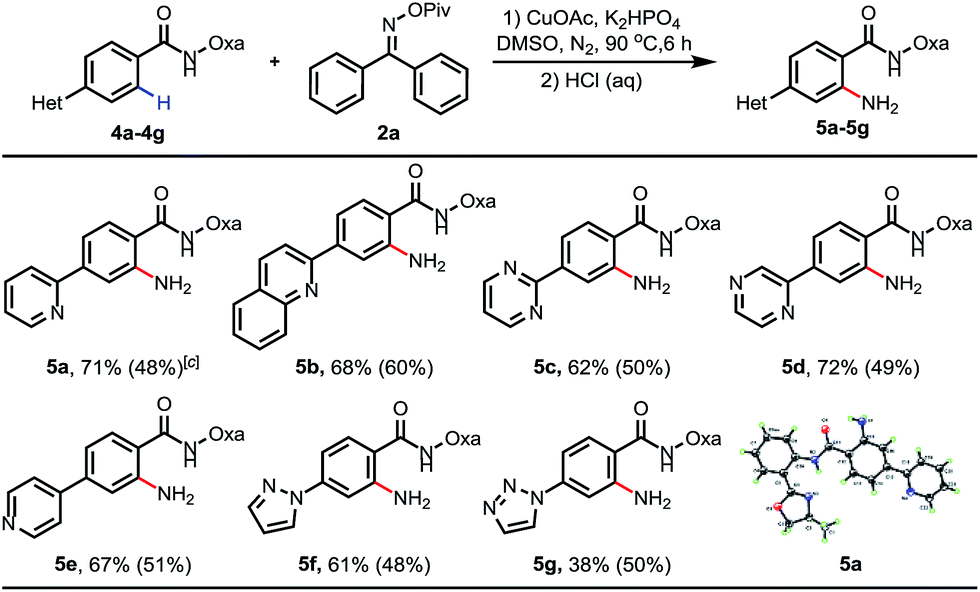
Inspired by previous examples overcoming the positional selectivity with substrates containing strongly coordinating heterocycles,15 we subsequently examined whether the C–H amination system could override the heterocycle-directed cyclometalation (Table 3). We chose para-(2-pyridyl)benzamide as a test substrate. Under the standard conditions, C–H amination proceeded exclusively at the position ortho to the amide group, providing the desired product in 71% isolated yield. The structure of 5a was confirmed using X-ray crystallography. Furthermore, when pyridine was replaced with other medicinally important heterocycles, including quinoline, pyrimidine, pyrazine, pyrazole and triazole, the amide group was still an effective directing group, and the desired aminated products were formed in moderate to good yields (38–72%). Furthermore, reactions using 0.3 equiv. of CuOAc as the catalyst were also examined (the yields are also shown in parentheses).
To probe the reaction mechanism, we measured intra- and intermolecular kinetic isotope effects. Significant isotope effects were observed in both cases (Scheme 2). In addition, the addition of the radical scavenger 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) did not affect the reaction efficiency under optimized conditions, which indicates that a radical pathway is unlikely (Scheme 3). These combined data support a mechanism which involves a copper-mediated C–H cleavage step rather than an electrophilic aromatic substitution (SEAr) or radical pathway. Therefore, we propose a Cu(I) to Cu(III) mechanism as shown in Scheme 1.
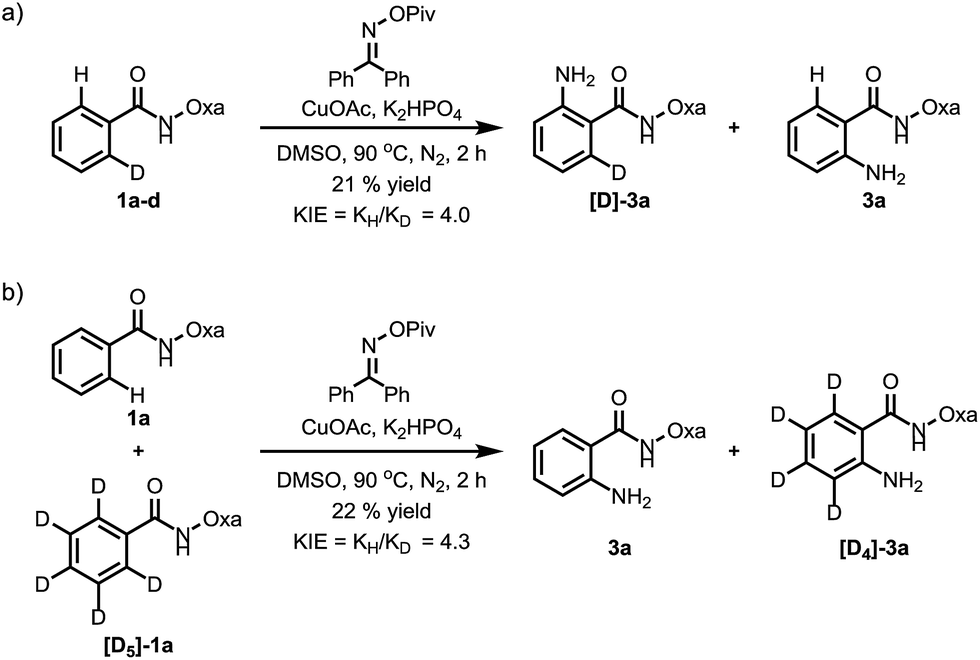 | ||
| Scheme 2 (a) Intramolecular kinetic isotope effect (KIE); (b) intermolecular KIE. | ||
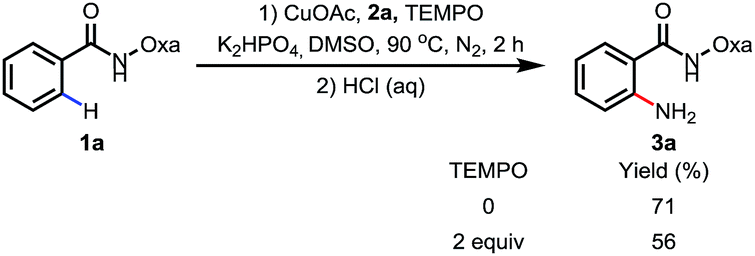 | ||
| Scheme 3 The effect of TEMPO. | ||
We also conducted the reaction on a gram scale with 0.3 and 1.5 equiv. of CuOAc and 3a was obtained in 45% and 62% yield respectively, and most of the benzophenone was recovered (see ESI†). Removal of this amide-oxazoline DG was demonstrated by treating product 3a with 2 N KOH/EtOH at 80 °C to release the corresponding carboxylic acid and oxazolyamide DG in good yields (Scheme 4). Interestingly, when the aminated products were treated with concentrated HCl at 190 °C, a series of different substituted primary anilines were obtained with complete removal of the amide-oxazoline part (Table 4).16 In particular, primary anilines containing heterocycles are accessible via this approach, making it a good complement to early reported nondirected C–H aminations.9
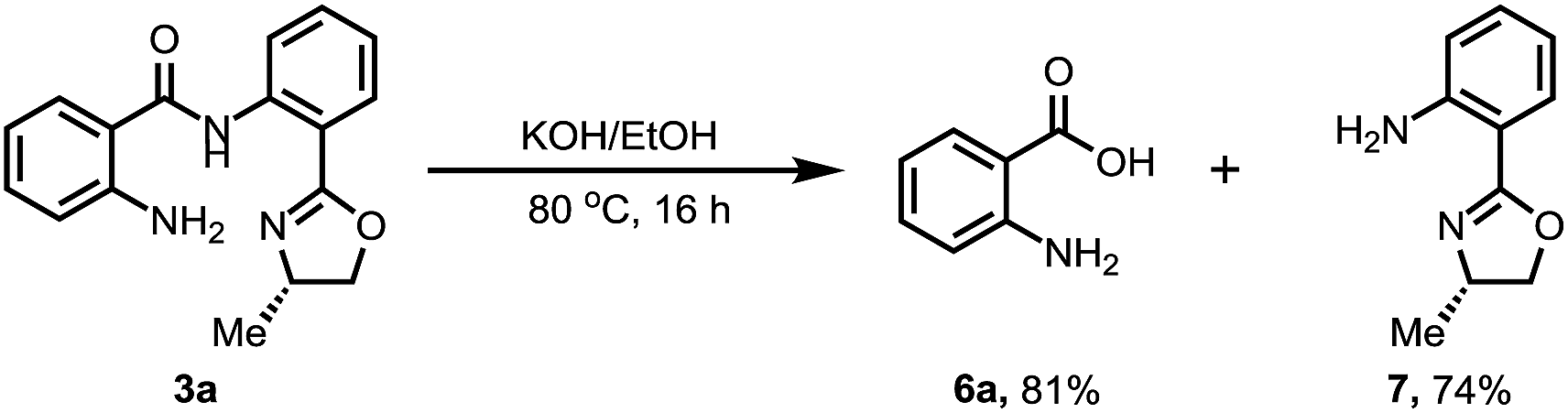 | ||
| Scheme 4 Removal of the directing group. | ||
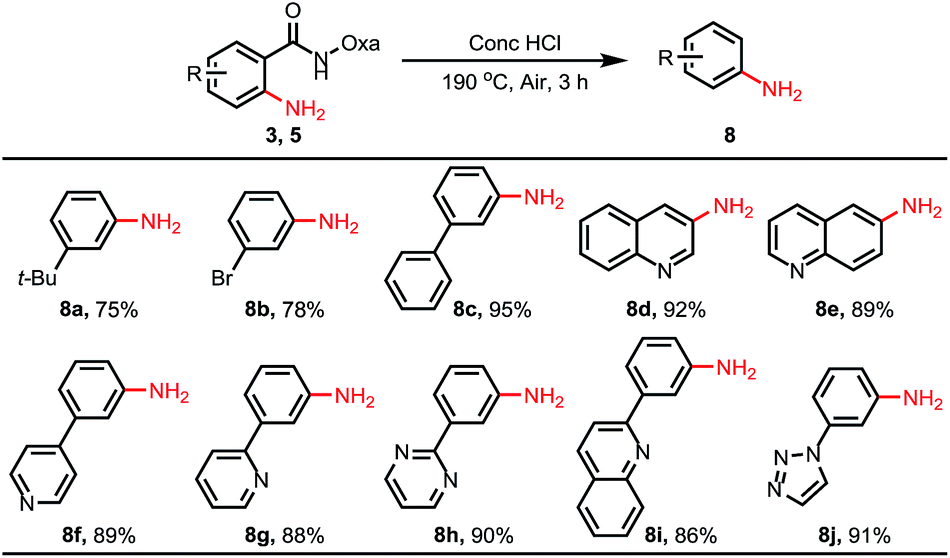
To further demonstrate the viability of the C–H amination reactions for late-stage diversification in drug discovery, we chose telmisartan, an antagonist for the angiotensin II receptor, for functionalization (Scheme 5). As we mentioned earlier,15f there are multiple reactive C–H bonds in this molecule, which significantly enhance the difficulty of selective functionalization of the target C–H bond. By installing the directing group amide oxazoline into the molecule, we were pleased to find that C–H amination occurred exclusively at the amide-directed position, while other C–H functionalization products were not observed. Furthermore, the resulting aminated product was treated under acidic or alkaline conditions to afford two derivatives 8k and 6b, respectively.
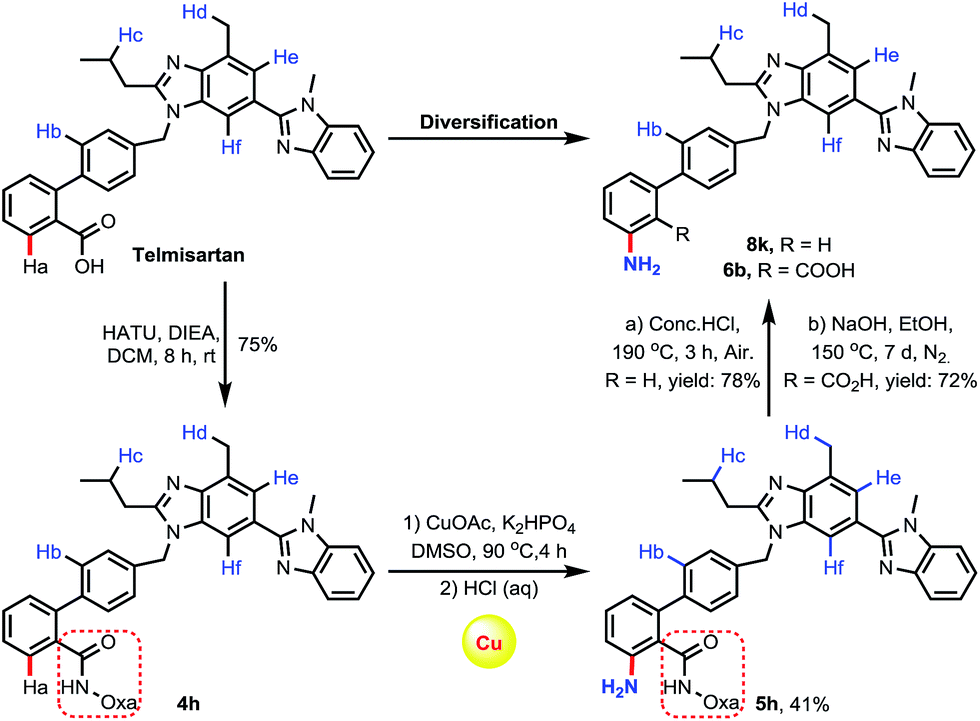 | ||
| Scheme 5 Late-stage diversification of telmisartan. | ||
Conclusions
In summary, we have developed a Cu(I)-mediated C–H amination reaction for the synthesis of primary anilines. A catalytic cycle involving Cu(I) to Cu(III) was proposed, and the oxime derivatives not only act as amino sources but also as oxidants. This practical reaction is compatible with various functional groups as well as diverse strongly coordinating heterocycles. The potential utility of this method was demonstrated by the late-stage diversification of the drug molecule telmisartan.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, NSFC-21472211, NSFC-21772211, the Youth Innovation Promotion Association CAS (No. 2014229), the Institutes for Drug Discovery and Development, Chinese Academy of Sciences (No. CASIMM0120163006) and the Science and Technology Commission of Shanghai Municipality (17JC1405000) for their financial support. We gratefully acknowledge NSF under the CCI Center for Selective C–H Functionalization, CHE-1205646 for their financial support.References
-
(a)
Amines: Synthesis Properties and Applications, ed. S. A. Lawrence, Cambridge University Press, Cambridge, 2004 Search PubMed
; (b) The Chemistry of Anilines, Parts 1 and 2, ed. Z. Rappoport, John Wiley & Sons, New York, 2007 Search PubMed
- R. J. Porter, V. Nohria and C. Rundfeldt, Neurotherapeutics, 2007, 4, 149 CrossRef PubMed
- J. Weijlard, P. D. Orahoyats, A. P. Sullivan Jr, G. Purdue, F. K. Heath and K. Pfister, J. Am. Chem. Soc., 1956, 78, 2342 CrossRef
- L. P. Yang and P. L. Mccormack, Drugs, 2011, 71, 221 CrossRef PubMed
- J. B. Bartlett, K. Dredge and A. G. Dalgleish, Nat. Rev. Cancer, 2004, 4, 314 CrossRef PubMed
-
(a) Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028 CrossRef PubMed
-
(a) P. Subramanian, G. C. Rudolf and K. P. Kaliappan, Chem.–Asian J., 2016, 11, 168 CrossRef PubMed
- L. Legnani, B. N. Bhawal and B. Morandi, Synthesis, 2017, 49, 776 Search PubMed
-
(a) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080 CrossRef PubMed
-
(a) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef
-
(a) Y.-H. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef PubMed
-
(a) J. Peng, M. Chen, Z. Xie, S. Luo and Q. Zhu, Org. Chem. Front., 2014, 1, 777 RSC
-
(a) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268 CrossRef PubMed
-
(a) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef PubMed
-
(a) H. A. Malik, B. L. Taylor, J. R. Kerrigan, J. E. Grob, K. Houk, J. Du Bois, L. G. Hamann and A. W. Patterson, Chem. Sci., 2014, 5, 2352 RSC
-
(a) J. Sinnreich, Synthesis, 1980, 578 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 1821862 and 1821863. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01256c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |