
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt-catalysed alkene hydrogenation: a metallacycle can explain the hydroxyl activating effect and the diastereoselectivity†
Glenn R.
Morello
a,
Hongyu
Zhong
 b,
Paul J.
Chirik
b and
Kathrin H.
Hopmann
*a
b,
Paul J.
Chirik
b and
Kathrin H.
Hopmann
*a
aHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Tromsø – The Arctic University of Norway, N-9037 Tromsø, Norway. E-mail: kathrin.hopmann@uit.no
bDepartment of Chemistry, Princeton University, New Jersey 08544, USA
First published on 4th May 2018
Abstract
Bis(phosphine)cobalt dialkyl complexes have been reported to be highly active in the hydrogenation of tri-substituted alkenes bearing hydroxyl substituents. Alkene substrates containing ether, ester, or ketone substituents show minimal reactivity, indicating an activating effect of the hydroxyl group. The mechanistic details of bis(phosphine)cobalt-catalysed hydrogenation were recently evaluated computationally (X. Ma, M. Lei, J. Org. Chem. 2017, 82, 2703–2712) and a Co(0)–Co(II) redox mechanism was proposed. However, the activating effect of the hydroxyl substituent and the accompanying high diastereoselectivity were not studied. Here we report a computational study rationalizing the role of the hydroxyl group through a key metallacycle species. The metallacycle is part of a non-redox catalytic pathway proceeding through Co(II) intermediates throughout. The preference for alcohol over ether substrates and the high diastereoselectivity of terpinen-4-ol hydrogenation are correctly predicted in computations adopting the new pathway, whereas the alternative redox mechanism predicts ethers rather than alcohols to be more reactive substrates. Additional experimental evidence supports the role of the hydroxyl group in the metallacycle mechanism. Our work highlights the importance of employing known substrate preferences and stereoselectivities to test the validity of computationally proposed reaction pathways.
Introduction
The majority of homogeneous hydrogenation catalysts employed today are based on precious metals such as rhodium, ruthenium, or iridium. During the last two decades, there has been an increasing focus on developing hydrogenation catalysts involving more earth abundant transition metals. Hydrogenation of alkenes or carbonyl compounds with transition metal complexes based on iron, cobalt, nickel, or manganese has been achieved by several groups, including those of Budzelaar,1 Chirik,2 Casey,3 Milstein,4 Hanson,5 Peters,6 Kempe,7 Morris,8 Fout,9 and Beller.10 Despite these promising results, more efforts are required to develop non-precious catalysts for diastereo- and enantioselective hydrogenation reactions. To date only a limited number of non-precious metal complexes are able to catalyse enantioselective alkene hydrogenations.2d,11,12,13Recently, Chirik and co-workers reported bis(phosphine)cobalt dialkyl complexes for the hydrogenation of alkenes under mild conditions (Fig. 1).2f A significant activating effect by hydroxyl groups was observed for the cobalt catalysts. Ether, ester, or ketone groups did not provide such an effect.2f The reported cobalt dialkyl catalyst dppeCo(CH2SiMe3)2 (C1, dppe = 1,2-bis(diphenylphosphino)ethane) catalyses hydrogenation of terpinen-4-ol providing 99% conversion (entry 2, Fig. 1), whereas the corresponding methyl ether displays <5% conversion despite higher catalyst loading and longer reaction time (entry 3, Fig. 1).2f Interestingly, hydrogenation of terpinen-4-ol gives a high diastereoselectivity with a diastereomeric ratio (d.r.) of 99.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 (entry 2, Fig. 1). Compared to tri-substituted alkenes, di-substituted terminal alkenes could be hydrogenated without a hydroxyl group present (entry 4, Fig. 1).
:0.2 (entry 2, Fig. 1). Compared to tri-substituted alkenes, di-substituted terminal alkenes could be hydrogenated without a hydroxyl group present (entry 4, Fig. 1).
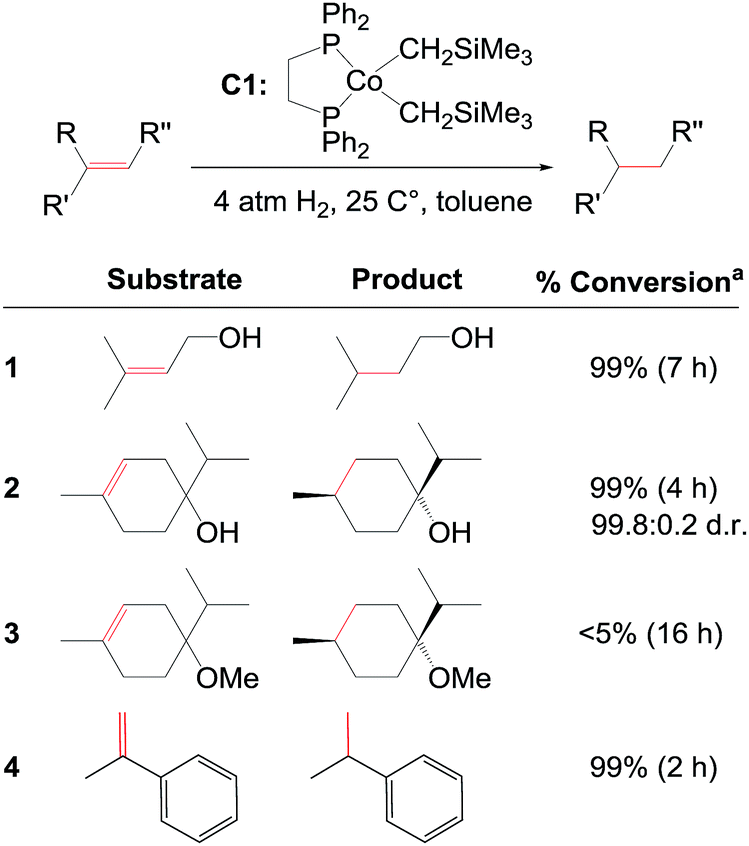 | ||
| Fig. 1 C1-catalysed alkene hydrogenation (data from ref. 2f). a5% C1 for entry 1 and 3, 1% C1 for entry 2 and 4. | ||
Recent computational work by Ma and Lei on C1 suggests that hydrogenation of hydroxylated tri-substituted alkenes proceeds through a Co(0)–Co(II) redox mechanism (Fig. 2).14 The proposed catalytic cycle starts with oxidative addition of H2 to a Co(0) species generating a Co(II)–dihydride, followed by substrate insertion to give an alkyl intermediate. The inter-mediate may undergo direct reductive elimination to yield the product alkane and regenerate Co(0) (Fig. 2, path (a)) or may proceed via β-hydrogen elimination to form an alkene regioisomer, followed by substrate reinsertion and reductive elimination (Fig. 2, path (b)). Regardless if alkene isomerization occurs, the same product is formed and the overall pathway involves a cycling between Co(0) and Co(II) oxidation states. The role of the hydroxyl group was not considered in the previous analysis.14
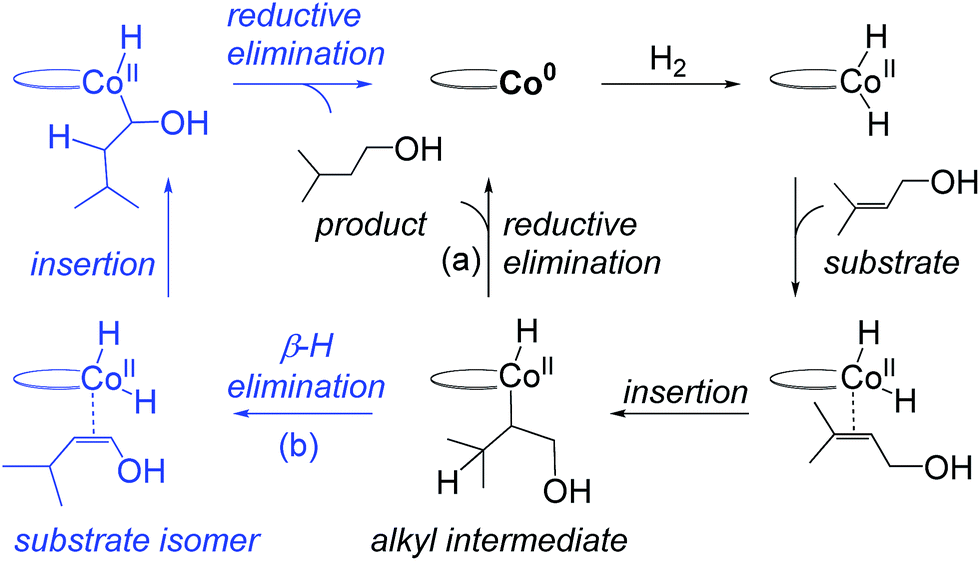 | ||
| Fig. 2 Previously proposed redox pathway for C1 (based on results in ref. 14). A Co(0) species oxidatively adds H2 to form a Co(II)–dihydride, followed by alkene insertion to form an alkyl intermediate. The alkyl undergoes reductive elimination (a) or β-H elimination (b) to yield the product or alkene isomers. | ||
Here we present computational and experimental results, which provide novel insights into the mechanism of C1-catalysed directed hydrogenation. On basis of our results, we propose that hydrogenation of hydroxylated alkenes occurs through a non-redox reaction pathway proceeding through a metallacycle, which is formed through activation of the hydroxyl group. The metallacycle mechanism correctly predicts the catalyst's preference for hydroxylated alkenes and the high diastereoselectivity observed in hydrogenation of terpinen-4-ol.
Results and discussion
Computational results
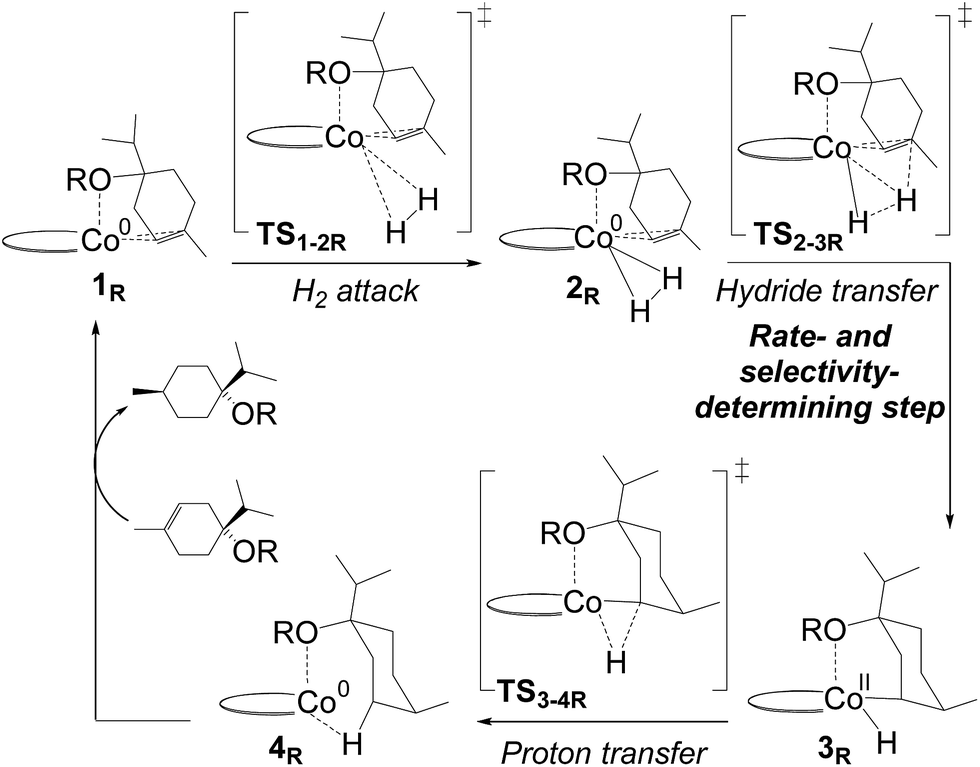 | ||
| Fig. 3 Co(0)–Co(II) redox mechanism computed for C1-catalysed hydrogenation of terpinen-4-ol (R = H) and its methoxy-derivative (R = CH3). Computed energies are shown in Fig. 4. Terpinen-4-ol prefers an alternative mechanism, Fig. 5. | ||
In order to evaluate the validity of the redox mechanism, we considered the substrate preference with this pathway. As previously determined in experiments, terpinen-4-ol (Fig. 1, entry 2) is the preferred substrate compared to its methoxy derivative (Fig. 1, entry 3). With the redox mechanism, the overall barriers are calculated to be +23.6 kcal mol−1 for terpinen-4-ol and +23.3 kcal mol−1 for the methoxy-derivative, indicating a slight preference for the methoxy-substrate (Fig. 4). This is in disagreement with the experimentally observed strong preference for the alcohol substrate (Fig. 1). Analysis of the substrate selectivity indicates that C1-catalysed hydrogenation may not proceed through the previously proposed redox mechanism.
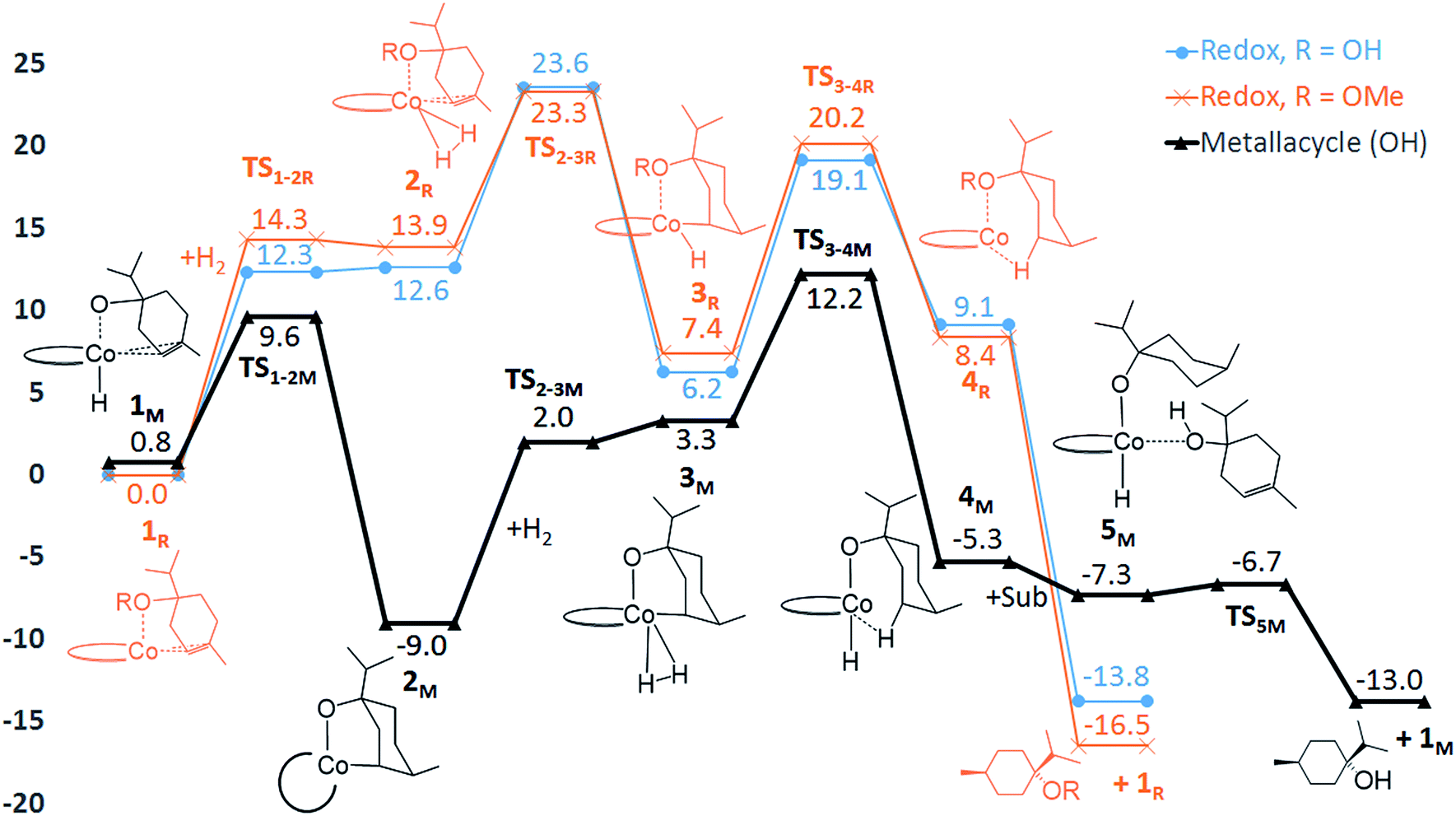 | ||
| Fig. 4 Computed free energies (kcal mol−1) for C1-catalysed hydrogenation. The redox mechanism is shown for substrates terpinen-4-ol (R = OH, blue line) and its methoxy derivative (R = OMe, orange line). The metallacycle mechanism is shown for terpinen-4-ol (black line). All energies are referenced to the 1R state of each substrate. | ||
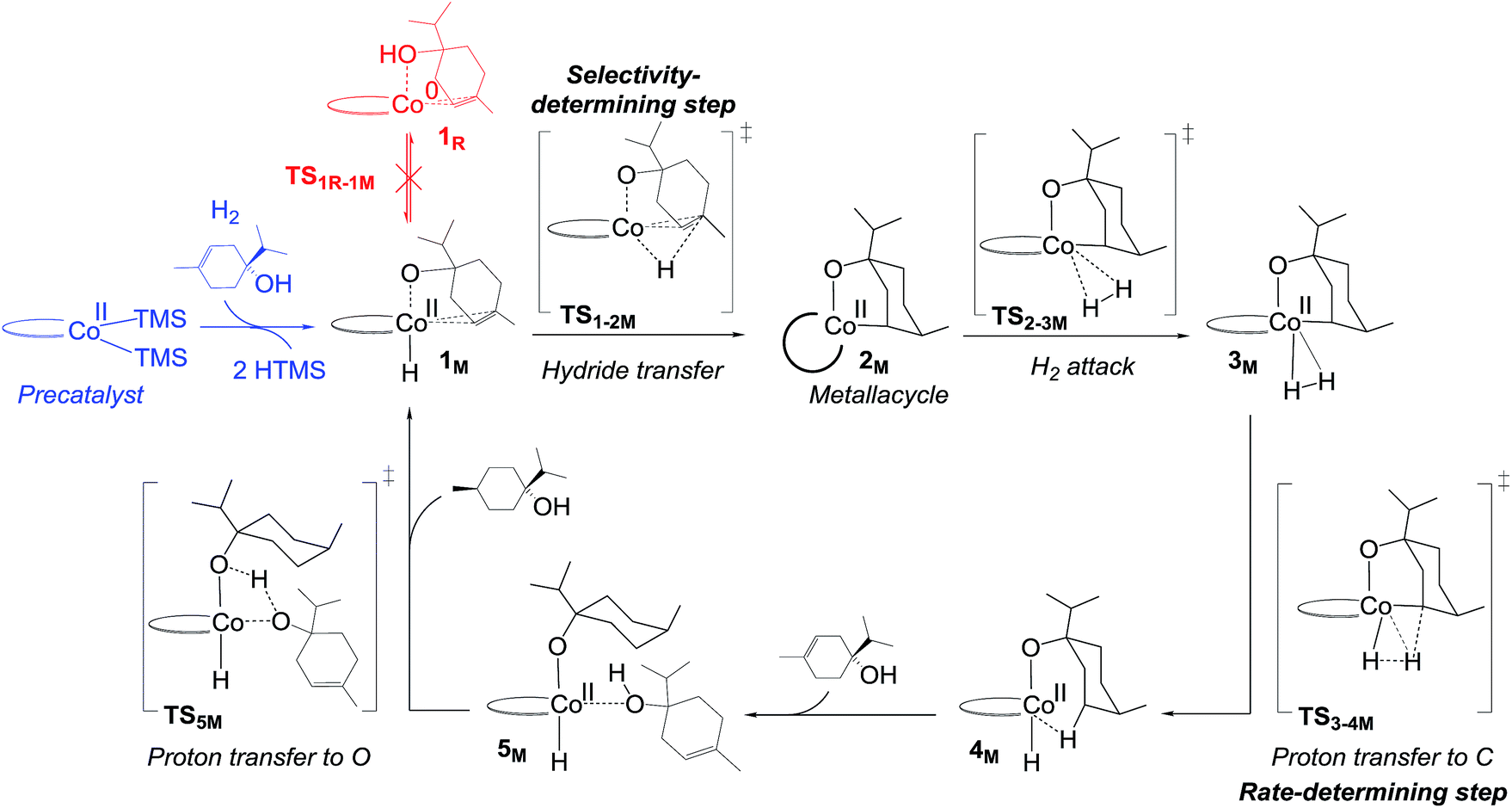 | ||
| Fig. 5 Non-redox metallacycle mechanism proposed for bis(phosphine)cobalt-catalysed hydrogenation of terpinen-4-ol. For details on the activation of the precatalyst, see Fig. S5, ESI.† | ||
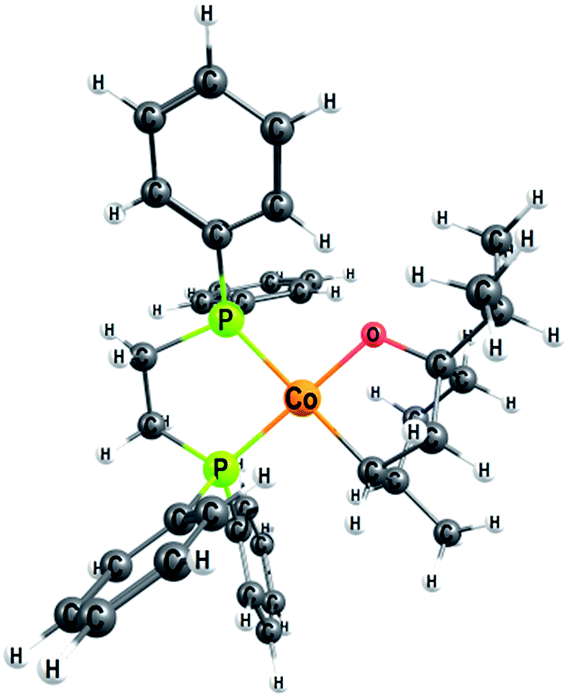 | ||
| Fig. 6 Optimised geometry of the metallacycle 2M, a Co(II) open shell species (S = 1/2) with a planar configuration around the cobalt atom. | ||
The calculated free energies show that the metallacycle 2M is facile to form and that it is the lowest-lying intermediate computed here (Fig. 4), implying that other reaction pathways must be referenced to 2M, even if the metallacycle constitutes an off-cycle species to these. The barrier for hydrogenation of terpinen-4-ol via the redox mechanism thus raises from 23.6 kcal mol−1 to 32.6 kcal mol−1 (TS2-3R relative to 2M, Fig. 4). The barrier for the metallacycle mechanism is instead 21.3 kcal mol−1 (TS3-4M relative to 2M, Fig. 4). Although DFT protocols may exhibit an error of some kcal mol−1,15 we consider a difference of 11.3 kcal mol−1 between the redox and the metallacycle mechanism to be more than significant to conclude that the metallacycle pathway is preferred for hydrogenation of terpinen-4-ol.
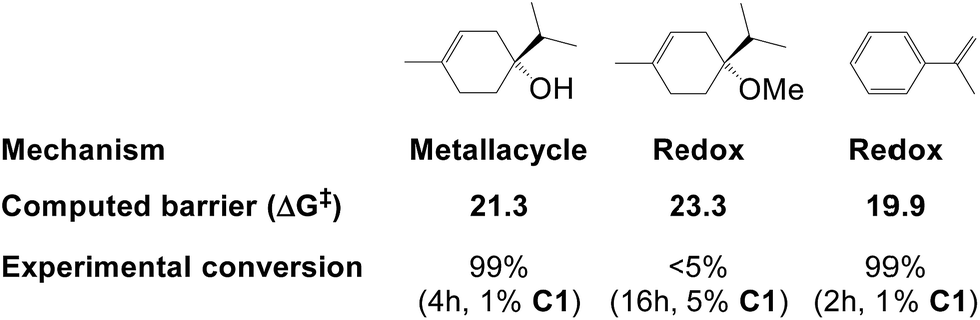 | ||
| Fig. 7 Hydrogenation barriers (kcal mol−1) computed here for three substrates and their experimental conversion (from ref. 2f). | ||
Hydrogenation of terpinen-4-ol preferably occurs via the metallacycle pathway, with a computed barrier of 21.3 kcal mol−1. Redox hydrogenation of the methoxy-derivative of terpinen-4-ol has a computed barrier of 23.3 kcal mol−1, whereas the lowest computed barrier for redox hydrogenation of α-methylstyrene is 19.9 kcal mol−1 (four possible pathways were modelled, see ESI, Fig. S7†). These results are in excellent agreement with experiment (Fig. 1), which showed 99% conversion of terpinen-4-ol and α-methylstyrene (after 4 and 2 hours, respectively), whereas the methoxy substrate gave <5% conversion after 16 hours. The barrier difference of 2 kcal mol−1 between terpinen-4-ol (barrier 21.3 kcal mol−1) and its methoxy derivative (barrier 23.3 kcal mol−1) translates roughly to a ratio of 97 to 3 (Table S1, ESI†), in good agreement with the experimental result of 99% to <5% conversion.
:0.2 (Fig. 1). This indicates that the hydroxyl group may have a directing effect that favours formation of the cis-isomer. We have here evaluated if the two discussed mechanisms, the metallacycle mechanism and the alternative redox pathway, are able to reproduce the experimentally observed diastereoselectivity.
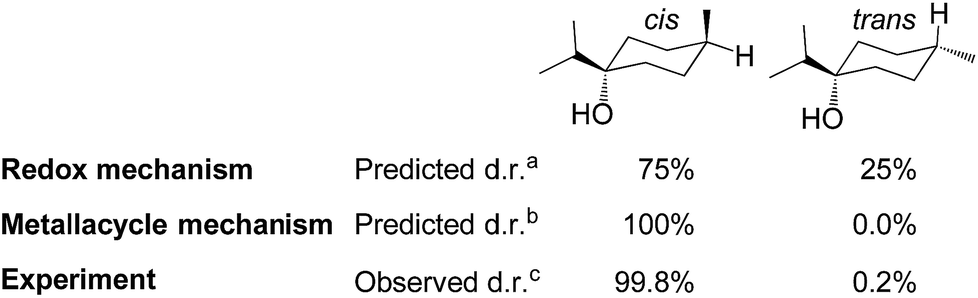 | ||
| Fig. 8 Computed diastereoselectivity of C1-catalysed hydrogenation of terpinen-4-ol, assuming a redox or metallacycle mechanism, and comparison to experiment (aTable S2, ESI, bTable S3, ESI,†cdata from ref. 2f). | ||
In the redox mechanism as depicted in Fig. 3, the diastereoselectivity is determined in the insertion step (TS2-3R), where the OH substituent is oriented towards cobalt, leading to the cis-diastereomer. For formation of the trans-diastereomer, we have analysed four pathways (see ESI, Fig. S8–S11†), and find that the lowest pathway proceeds via β-hydrogen elimination (Fig. S10, ESI†). The overall barriers for formation of the cis and trans-diastereomers via a redox pathway differ by 0.6 kcal mol−1, which corresponds to a predicted d.r. of 75(cis):25(trans) (Fig. 8, Table S2 ESI†).
In the metallacycle mechanism, the diastereoselectivity is determined in the hydride transfer step TS1-2M (Fig. 5). Due to the strong coordination of the alkoxide oxygen, hydride addition inevitably has to lead to formation of only the cis-product (Fig. 8), in excellent agreement with the experimentally observed high d.r. of 99.8(cis):0.2(trans). Formation of the trans-diastereomer would require cleavage of the cobalt–alkoxide bond, which energetically is very costly (Table S3 ESI†). We propose that in the experimental reaction, the cis-diastereomer is formed via the energetically preferred metallacycle mechanism, whereas the very small amount of observed trans-diastereomer (0.2%, Fig. 1) must be formed via other mechanisms. Our computational results provide a rationale for the high diastereoselectivity of the cobalt complex C1. Interestingly, the Crabtree iridium catalyst provides the same diastereoselectivity with terpinen-4-ol.16 However, the mechanistic details of said system are not known so far.
Experimental results
Hydrogenation of deuterium-labelled terpinen-4-ol (C10H8-OD) afforded 1,2-H2 alkane with no deuterium incorporation into the alkene double bond (Fig. S12, ESI†), consistent with the proposed metallacycle mechanism, where proton transfer from a second substrate to the cobalt alkoxide turns over the catalytic cycle (TS5M, Fig. 5).Our attempts to obtain the metallacycle intermediate 2M from different synthetic routes and to characterize it by X-ray crystallography were unsuccessful due to strong interference from the thermodynamically more accessible bis(ligand)cobalt species formed during isolation of cobalt complexes.17 However, indirect experimental evidence suggests that a reaction between the precatalyst dppeCo(CH2SiMe3)2 (C1) and terpinen-4-ol does occur (Fig. 9). Specifically, adding five equivalents of terpinen-4-ol to C1 in benzene-d6 at room temperature resulted in protonolysis of the cobalt alkyl. The volatile component of the reaction was distilled and analysed by 1H-NMR after 12 hours, and the protonolysis product SiMe4 was observed.18 Integration suggests approximately half of the alkyl groups in C1 reacted with terpinen-4-ol (Fig. S13, ESI†). This experimental observation supports the formation of a mono-alkoxide intermediate, in agreement with the mono-alkoxide species expected during the metallacycle mechanism (1M, Fig. 5). Further treatment of the non-volatile component (containing the assumed mono-alkoxy complex) with TMSI resulted in formation of TMS-terpinen-4-ol (Fig. 9), providing additional support for formation of an alkoxide species.
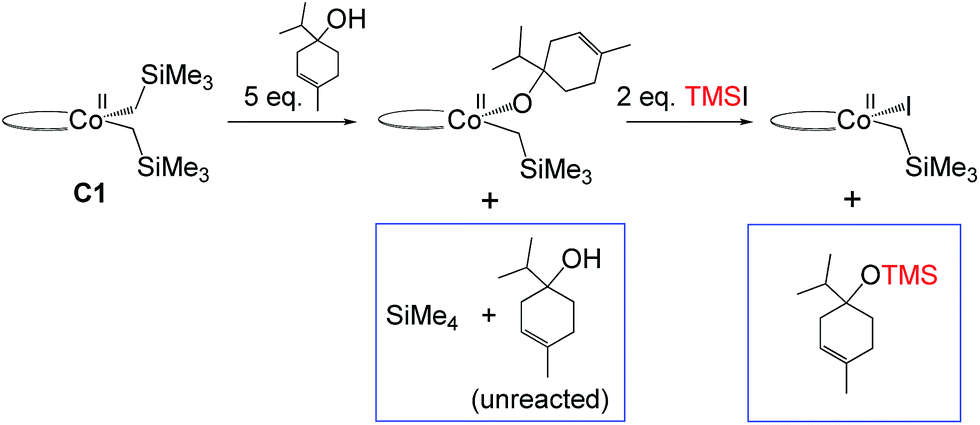 | ||
| Fig. 9 Indirect evidence for formation of an alkoxide intermediate. Components in boxes were identified via NMR (see ESI†). | ||
Treating precatalyst C1 with H2 in the absence of substrate unveiled a catalyst deactivation pathway, which involves formation of a catalytically inactive (dppe)2Co species (Fig. S14, ESI†). This observation suggests an essential role of the substrate in maintaining the catalyst in an active form.
Conclusions
We have here reported a new mechanism for bis(phosphine)-cobalt-catalysed hydrogenation of hydroxylated alkenes, proceeding through an energetically low-lying metallacycle intermediate (2M, Fig. 5). In the computational analysis, the metallacycle mechanism correctly predicts the preference for hydroxylated alkenes (Fig. 7) and the high diastereomeric ratio observed in hydrogenation of terpinen-4-ol (Fig. 8). The mechanism is further supported by experimental investigations providing indirect evidence of a cobalt-terpinen-4-ol alkoxide intermediate (Co-OR, Fig. 9).Our computational analysis further shows that a previously proposed redox mechanism14 may be valid for non-hydroxylated substrates, but is unable to explain the activating effect and diastereoselectivity of hydroxylated alkenes. As also reported for iron-catalysed hydrogenation of carbonyl substrates,19 we have here shown that known substrate selectivities provide a straightforward tool for testing the validity of proposed mechanisms, and we suggest to always asses these important parameters in computational studies of reaction pathways.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
HZ and PJC acknowledge the U.S. National Science Foundation (NSF) Grant Opportunities for Academic Liaison with Industry (GOALI) grant (CHE-1265988). KHH acknowledges the Research Council of Norway for a FRINATEK grant (No. 231706) and a Centre of Excellence Grant (No. 262695), the Tromsø Research Foundation (No. TFS2016KHH) and Notur – The Norwegian Metacenter for Computational Science for grants of computer time (No. nn9330k and nn4654k).Notes and references
- Q. Knijnenburg, A. D. Horton, H. V. D. Heijden, T. M. Kooistra, D. G. H. Hetterscheid, J. M. M. Smits, B. D. Bruin, P. H. M. Budzelaar and A. W. Gal, J. Mol. Catal. A: Chem., 2005, 232, 151–159 CrossRef CAS.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed; (b) A. M. Archer, M. W. Bouwkamp, M. P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269–4278 CrossRef CAS; (c) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760–1764 CrossRef CAS PubMed; (d) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS PubMed; (e) R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, S. Debeer and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 13168–13184 CrossRef CAS PubMed; (f) M. R. Friedfeld, G. W. Margulieux, B. A. Schaefer and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 13178–13181 CrossRef CAS PubMed.
- (a) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed; (b) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS PubMed.
- (a) R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., 2011, 50, 10122–10126 CrossRef; (b) R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., 2011, 50, 2168–2172 CrossRef.
- (a) G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed; (b) G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC; (c) G. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668–8681 CrossRef CAS PubMed.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed.
- S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- (a) J. F. Sonnenberg, A. J. Lough and R. H. Morris, Organometallics, 2014, 33, 6452–6465 CrossRef CAS; (b) J. F. Sonnenberg, K. Y. Wan, P. E. Sues and R. H. Morris, ACS Catal., 2017, 7, 316–326 CrossRef CAS; (c) S. A. M. Smith, P. O. Lagaditis, A. Lüpke, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2017, 23, 7212–7216 CrossRef CAS PubMed.
- K. Tokmic, C. R. Markus, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 11907–11913 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- J. Chen, C. Chen, C. Ji and Z. Lu, Org. Lett., 2016, 18, 1594–1597 CrossRef CAS PubMed.
- J. H. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260–272 RSC.
- M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed.
- X. Ma and M. Lei, J. Org. Chem., 2017, 82, 2703–2712 CrossRef CAS PubMed.
- K. H. Hopmann, Organometallics, 2016, 35, 3795–3807 CrossRef CAS.
- R. H. Crabtree and M. W. Davis, J. Org. Chem., 1986, 51, 2655–2660 CrossRef CAS.
- R. Ciancanelli, B. C. Noll, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2002, 124, 2984–2992 CrossRef CAS PubMed.
- Although this reaction appears slow (12 h), it should be noted that this is in agreement with the computed barrier for this reaction in absence of H2, 32.3 kcal mol−1, which is 3 kcal mol−1 higher than activation of the catalyst in presence of H2 (29.2 kcal mol−1, ESI, Fig. S5†).
- G. Morello and K. H. Hopmann, ACS Catal., 2017, 7, 5847–5855 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational and experimental details, calculated reaction profiles, optimised coordinates, NMR and X-ray data. See DOI: 10.1039/c8sc01315b |
| This journal is © The Royal Society of Chemistry 2018 |