DOI:
10.1039/C8SC01358F
(Edge Article)
Chem. Sci., 2018,
9, 6134-6142
High performance layer-by-layer Pt3Ni(Pt-skin)-modified Pd/C for the oxygen reduction reaction†
Received
23rd March 2018
, Accepted 26th June 2018
First published on 26th June 2018
Abstract
Bimetallic Pt–Ni with Pt on the outermost layer and an innermost layer enriched in Ni, referred to as Pt3Ni(Pt-skin), is a promising configuration of an electrocatalyst for the oxygen reduction reaction (ORR) in fuel cells. We prepare a core (Pd)/shell (Pt3Ni(Pt-skin)) catalyst (Pt3Ni(Pt-skin)/Pd/C) from Zn underpotential deposition (UPD) on a Ni UPD modified Pd/C catalyst, facilitating Pt atomic layer-by-layer growth on the Ni surface through the galvanic replacement process. Pt3Ni(Pt-skin)/Pd/C shows the best ORR performance, with a Pt specific activity of 16.7 mA cm−2 and Pt mass activity of 14.2 A mgPt−1, which are 90- and 156- fold improvements over commercial Pt/C catalysts. The Pt3Ni(Pt-skin) structure effectively inhibits Ni leaching to improve the durability in two accelerated durability test modes mimicking the catalyst lifetime and start-up/shut-down cycles.
Introduction
Cathodic oxygen reduction reaction ORR electrocatalysts play a crucial role in fuel cell performance.1–3 The Pt catalyzed ORR has sluggish kinetics and requires a high overpotential, causing lower Pt specific activity (jkPt, the catalytic activity normalized by the Pt electrochemically active surface area (ECSA)) and Pt mass activity (imPt, the catalytic activity per Pt mass). The high cost and the low durability of Pt ORR electrocatalysts remain a challenge for the widespread commercialization of fuel cells.4–18 Alloying Pt with another transition metal (Co, Ni, Fe, etc.) has attracted much attention in the design of advanced electrocatalysts, as this approach not only decreases the Pt content but also enhances the catalytic activity and durability.8,19–23 These designs include core–shell structured nanoparticles (NPs),24,25 as well as de-alloyed26–28 and porous structured NPs.6,26 The Pt alloys may tailor the electronic (affecting the Pt–OH bond energetics) and geometric structures (affecting the Pt–Pt bond distance and coordination number) to enhance the catalytic activity.8 The highest recorded jkPt values were achieved on single crystal surfaces or well-defined NPs with a specifically engineered facet structure and alloy composition. For example, Stamenkovic et al. found that single-crystal Pt3Ni (111) with Pt-skin had a jkPt value 10 times higher than the corresponding Pt (111) surface and 90 times higher than the commercial Pt/C catalysts used for the ORR.8 Pt3Ni octahedral NPs were shown to exhibit favorable microstructures for greatly enhanced activity in the ORR,28–30 but were still limited by their insufficient stability due to Ni leaching from the alloys and decreased ECSA from the agglomeration of the NPs during electrochemical cycling.31 Core–shell NPs represent a multi-metallic structure with tunable properties to enhance ORR catalytic activity.32–38 A promising structure to optimize imPt and Pt utilization is a thin shell or skin layers of Pt or Pt alloys over a non-Pt NP core. As reported by Adzic et al., Cu underpotential deposition (UPD) is used as a sacrificial coating on the core, followed by galvanic replacement (Gal) with noble metal ions for the final shell metal.7,39–41 UPD-Gal is one of the most successful methods to specifically coat Pt monolayers on different metals, but the commonly used Cu UPD (Cuu) limits the options of Pt coating substrates due to its high work function, ΦCu (∼4.94 eV).42 It is known that when the Φ of an electrodeposited metal, ΦM, is lower than that of the substrate metal, ΦS, UPD may occur at a potential more positive than the equilibrium potential. The Kolb–Gerischer equation, ΔE = 0.5ΔΦ (ΔE is the underpotential shift in V and ΔΦ is ΦM − ΦS of the electron in eV), has been used to evaluate the level of underpotential shift.43 Despite numerous attempts to synthesize Pt alloying nanocatalysts with Pt-skin surfaces on transition metals,28,44–50 it still remains a challenge to demonstrate their existence at the nanoscale. To resolve this issue, we attempted to improve the elegant UPD-Gal approach, also referred to as electrochemical atomic layer deposition (E-ALD) or electrochemical atomic layer epitaxy EC-ALE.51,52 In UPD-Gal, the UPD adlayer enables a type of surface limited reaction (SLR). SLRs occur only at the substrate or deposit surface and specifically form an atomic layer or a monolayer coverage. The “atomic layer” refers to a coverage less than a monolayer, a monolayer being a unit of deposit coverage. Zn UPD (Znu) was used to replace the Cuu in the UPD-Gal due to its lower value of ΦZn (∼3.95 eV) compared to ΦCu,42 and the more negative standard reduction potential, E0 (Zn2+/Zn) = −0.76 V.53 The lower ΦZn makes Znu occur on a greater variety of substrates, particularly Ni. Znu can therefore replace many metals that are nobler than Zn in the Gal process thanks to its more negative E0.
We report Znu assisted UPD-Gal (ZnUPD-Gal) in a Ni UPD (Niu) process of constructing a layer-by-layer Pt3Ni(Pt-skin) thin layer on a carbon-supported Pd electrocatalyst (Pd20/C) (20 wt% Pd on XC-72 Valcan carbon, E-TEK) (Pt3Ni(Pt-skin)/Pd20/C). The Pt3Ni(Pt-skin) structure retains the advantages of the ultra-thin layer structure and the synergetic effects of the Ni sublayer. Pt3Ni(Pt-skin)/Pd20/C possesses an ultra-high jkPt = 16.7 mA cm−2 and imPt = 14.2 A mgPt−1 (Pt loading = 2.97 μgPt cm−2), at 0.9 V vs. RHE, which are 90-fold and 156-fold improvements, respectively, over commercial Pt/C catalysts (0.185 mA cm−2 and 0.091 A mgPt−1, Pt loading = 24 μgPt cm−2). We also show that the perfect Pt3Ni(Pt-skin) structure effectively inhibits Ni leaching, significantly improving the durability of catalysts.
Results and discussion
Fig. 1 shows the CVs of Niu and Znu on Pd20/C@GC in an Ar-saturated 0.5 M Na2SO4 aqueous solution (Na2SO4aq). These voltammograms both show two pairs of redox waves, c1/a1 and c2/a2, which correspond to UPD/stripping and bulk deposition (OPD)/bulk stripping, respectively. These indicate that Niu or Znu can modify the Pd surface through controlled potential electrodeposition. Herein Znu was used to assist in the selective growth of Pt atomic layers on a given metallic surface, here Ni and Pt. Znu was further studied on Niu/Pd20/C@GC and Pt atomic layer covered Pd20/C@GC (Pt/Pd20/C@GC) (Fig. 1b). Niu/Pd20/C was from direct electrodeposition of Niu on Pd20/C in 0.5 M Na2SO4aq containing 20 mM NiSO4. Pt/Pd20/C was prepared by UPD-Gal to deposit a Pt atomic layer on Pd20/C. The Znu redox waves, c1/a1, on Pd20/C positively shifted from ∼0.18 V to ∼0.2 V and ∼0.3 V after the modification of the Niu and Pt layer, respectively. The changes in CVs are ascribed to Znu and are surface dependent. Although Znu on Pt has been reported in the literature,54 Znu on Ni has not been observed until now as the Niu was freshly produced without serious oxide or hydroxide surface inhibitors. The Znu on Pd, Ni, and Pt surfaces is related to the higher ΦS of these substrates, ΦPd (∼5.0 eV), ΦNi (∼4.91 eV) and ΦPt (∼5.4 eV), in comparison with the lower ΦZn (∼3.95 eV).42 Based on the Kolb–Gerischer equation, we could approximately calculate ΔE for the metal couples, Pd substrate/Zn, Ni substrate/Zn and Pt substrate/Zn, as ∼0.53 V, ∼0.48 V and ∼0.7 V, respectively. This provides grounds for realizing that Znu on Pd, Ni and Pt is possible. In comparison, Cuu cannot occur on Ni surfaces due to ΦCu (∼4.94 eV) being close to ΦNi. This implies that Znu is a more suitable candidate to promote the Pt atomic layer to specifically form on a Ni surface.
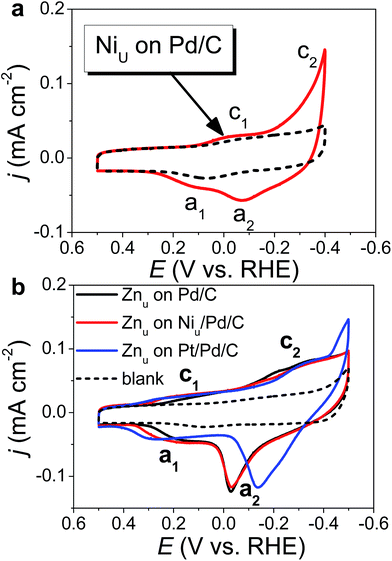 |
| | Fig. 1 CVs of (a) Niu on Pd20/C@GC and (b) Znu on Pd20/C@GC, Niu/Pd20/C@GC, and Niu/Pd20/C@GC recorded in Ar saturated 0.5 M Na2SO4aq (solid line) with and (dashed line) without (a) 20 mM NiSO4 and (b) 20 mM Zn(ClO4)2 at a sweep rate of 50 mV s−1. | |
Based on the findings of Znu on Ni, Pd and Pt surfaces and Niu on Pd and Pt surfaces, ZnUPD-Gal was used to prepare a Pt3Ni(Pt-skin)/Pd20/C electrocatalyst (Scheme 1). First, Niu was electrodeposited on the Pd surface of Pd20/C, followed by modification of Znu on Niu/Pd20/C. Subsequently, Znu was replaced by Pt in the Gal process, leading to Pt atomic layer covered Niu/Pd20/C (PtNi/Pd20/C). The repetitive ZnUPD-Gal continually introduced the second and the third Pt atomic layers onto PtNi/Pd20/C to obtain the desired electrocatalyst, Pt3Ni(Pt-skin)/Pd20/C. The micromorphology and elemental composition distribution of Pt3Ni(Pt-skin)/Pd20/C were examined by high-resolution transmission electron microscopy (HRTEM) combined with energy dispersive X-ray spectroscopy (EDS) (Fig. 2a and b and S1†). In Fig. S1a–c,† typical bright-field TEM images of pristine Pd20/C reveal a relatively uniform dispersion of Pd NP (Pdnano) with sizes of approximately 4.8–5.6 nm (diameter). After Pt3Ni(Pt-skin) modification, the growth of the particle size occurred as expected (diameter increased to ∼6.8 nm) (Fig. S1d–f†). The core of Pt3Ni(Pt-skin)/Pdnano shows clear fringe orientations of the Pd (111) single-crystal structure (Fig. 2a). The EDS line profile analysis shows the distribution of Pt, Ni, and Pd components in a single NP (Fig. 2b). The width of Pt3Ni(Pt-skin)/Pdnano examined was 4 nm, as designated by the red line in Fig. 2a. The line profile analysis validates the core–shell structure, which is a Pd core covered by an ultra-thin PtNi shell with a thickness of approximately 1.2 nm. The Pd composition is constant and high in the particle center, and decreases from the edge of the core to the particle surface. Most of the Ni component is located at the interface between the Pd core and PtNi shell. At the exterior of the PtNi shell, the Pt intensity is approximately 2.5 times that of Ni in the interior of the PtNi shell. This analysis demonstrates the formation of a core (Pd)/shell (Pt3Ni(Pt-skin)) structure. To extend the diversity of the ZnUPD-Gal with the Niu process, a three repeated PtNi layer covered Pd core ((PtNi)3/Pd20/C) was also prepared as a control example. The EDS line profile analysis of (PtNi)3/Pd20/C confirms that a Pd core/(PtNi)3 shell structure was successfully obtained using this process (Fig. S2 and S3†).
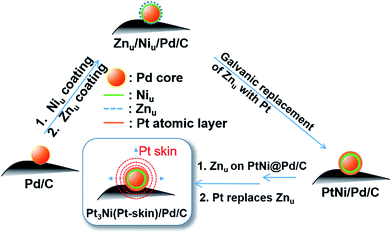 |
| | Scheme 1 The preparation process of Pt3Ni(Pt-skin)/Pd20/C electrocatalysts through ZnUPD-Gal on Niu. | |
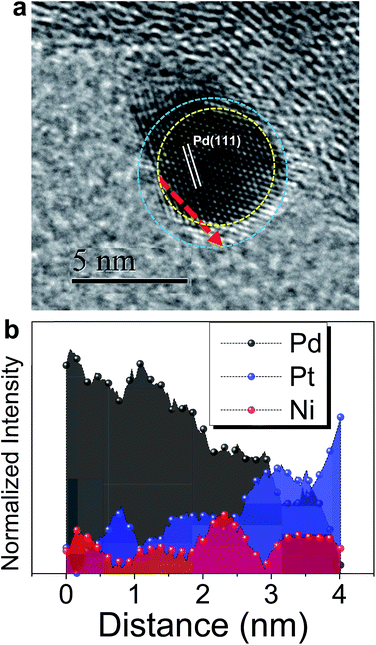 |
| | Fig. 2 (a) The representative HRTEM image of a core (Pd)/shell (Pt3Ni(Pt-skin)) nanoparticle; (b) the corresponding EDS line-scan profile along the red dashed line as shown in (a). | |
The CVs of H2SO4aq were used to track different modified layers grown on the Pd surface. The typical CV of Pd20/C shows redox peaks corresponding to the formation and removal of Pd hydroxide (Pd(OH)x) (0.6–1.0 V vs. RHE) and H absorption (Hab)/desorption (0.3–0.0 V vs. RHE) (Fig. 3a). In comparison with the CV of pristine Pd20/C, the first modified layer, Niu, on Pd20/C inhibits the formation of Pd(OH)x and Hab, causing the reduction of related redox charges on Niu/Pd20/C. The following Pt atomic layers were grown in turn on the Niu surface by ZnUPD-Gal. With the increase of the Pt layer, characteristic Pt redox waves, including the formation of Pt hydroxide (Pt(OH)x) occurred at a more negative potential (∼0.75 V vs. RHE) and hydrogen adsorption (Had)/desorption waves gradually grew. For the purpose of comparison, three Pt layer-covered Pd20/C@GC (Pt3/Pd20/C@GC) without Niu was also prepared using UPD-Gal. This indicated that the surface of Pt3/Pd20/C was mainly composed of Pt, due to the similar CV features of Pt3/Pd20/C and Pt/C. In comparison to the CV features of Pt3/Pd20/C, those of Pt3Ni(Pt-skin)/Pd20/C based on the same Pt content showed that the onset potential of Had shifted towards a more negative potential and the formation of Pt(OH)x occurred at a more positive potential. This is a consequence of the electronically modified structure of Pt for “Pt skin” surfaces by the subsurface Ni atoms, which leads to weakened interactions between Pt and adsorbates such as Had and surface hydroxides (OHad). This is also typical for the Pt3Ni(Pt-skin) structure.8,55 The Had integrated charge is a conventional approach in the estimation of ECSA (ECSAH).56 However, the suppression of Had on the Pt3Ni(Pt-skin) structure can substantially affect the accurate estimation of the real ECSA. The electro-oxidation of adsorbed carbon monoxide (COad), known as CO stripping, has been suggested as a complementary ECSA evaluation method (ECSACO).55,57Fig. 3b shows the voltammetric curves of CO stripping obtained for pristine Pd20/C, PtNi/Pd20/C, and Pt3Ni(Pt-skin)/Pd20/C. The oxidation of the COad takes place in a single peak whose peak potential shifts towards more negative values as the Pt content increases. The broad CO stripping peak for Pd20/C becomes sharper with increasing Pt content in the PtNi shell.58 These results suggest that the PtNi shell significantly weakens the interaction of Pd surface atoms with COad. Interestingly, the onset of CO stripping on Pt3Ni(Pt-skin)/Pd20/C is more negatively shifted than on Pt3/Pd/C, and the shape of the stripping peak is broader due to the weaker interaction of the Pt surface atoms with CO from the Ni sublayer. However, the similar charge of CO oxidation points to an equal coverage of CO. The specific ECSA (sECSA = ECSA/metal loading, m2 g−1) for Pt3Ni/Pd/C and Pt3/Pd/C electrocatalysts was evaluated from ECSAH (sECSAH) and ECSACO (sECSACO), respectively (Fig. 3b). Although the suppression of sECSAH on Pt3Ni/Pd/C (95.1 m2 g−1) was observed in comparison with that of Pt3/Pd/C (102.3 m2 g−1), sECSACO shows a similar value of ∼106 m2 g−1 on both Pt3Ni(Pt-skin)/Pd/C and Pt3/Pd/C.
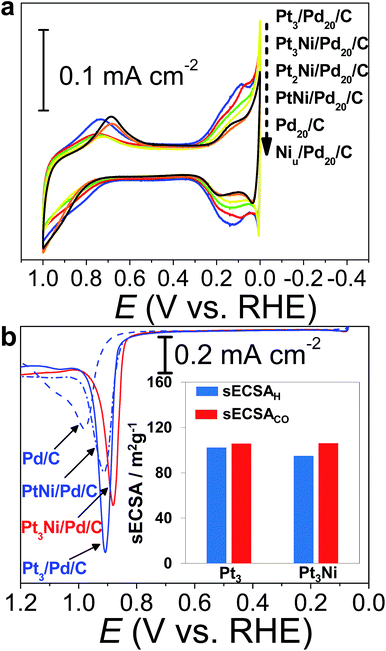 |
| | Fig. 3 Electrochemical surface characterization of electrocatalysts by using a GC electrode in 0.1 M H2SO4aq: (a) cyclic voltammograms of (blue) Pt3/Pd20/C, (red) Pt3Ni(Pt-skin)/Pd20/C, (green) Pt2Ni(Pt-skin)/Pd20/C, (yellow) PtNi(Pt-skin)/Pd20/C, (black) Pd20/C, and (orange) Niu/Pd20/C; (b) CO stripping curves. The inset shows (blue) sECSAH and (red) sECSACO for (Pt3) Pt3/Pd20/C and (Pt3Ni) Pt3Ni(Pt-skin)/Pd20/C. | |
The ORR polarization curves were obtained with Pd20/C, Ni/Pd20/C, Pt3/Pd20/C, PtNi/Pd20/C and Pt3Ni(Pt-skin)/Pd20/C electrocatalysts as thin films on the GC disc electrode of a RRDE in an O2-saturated 0.1 M HClO4 solution at 1600 rpm (Fig. 4a). The Pt ring electrode of the RRDE was potentiostated at 1.1 V to collect the ring current (ir) related to the H2O2 oxidation reaction. The polarization curves on the disc electrode displayed two distinguishable potential regions: well-defined diffusion limiting currents (iD) for the ORR below 0.7 V and a mixed kinetic-diffusion control region between 0.7 and 1.1 V. In both potential regions, ir was a rather small fraction of iD for all electrocatalysts, revealing that the ORR proceeds almost entirely through the 4e− reduction pathway. A quantitative presentation of the H2O2 production (current efficiency, χH2O2) was given using eqn (1):20
| | 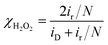 | (1) |
where
N is the collection efficiency of the RRDE. In the potential region of 0.05 <
E < 1 V, similarly small amounts of H
2O
2 were detected on the ring electrode from the Pd
20/C and Ni
u/Pd
20/C electrocatalysts, implying that Ni modification does not alter the reaction pathways. Pt-containing shells on the other three electrocatalysts (Pt
3/Pd
20/C, PtNi/Pd
20/C and Pt
3Ni(Pt-skin)/Pd
20/C) effectively inhibited the production of H
2O
2 during the ORR, since there was no detectable H
2O
2 on the ring electrode in the kinetically controlled potential region, implying that Pt atomic layers perfectly covered the outermost layer of Pt-containing shells. The half-wave potential of an ORR polarization curve,
E1/2, is often used to evaluate the electrocatalytic activity of a catalyst.
E1/2 increased in the following sequence: Pd
20/C ∼ Ni
u/Pd
20/C < Pt
3/Pd
20/C < PtNi/Pd
20/C ≪ Pt
3Ni(Pt-skin)/Pd
20/C. Pt
3Ni(Pt-skin)/Pd
20/C showed a marked positive shift in
E1/2 of 200 mV and 100 mV relative to Pd
20/C and Pt
3/Pd
20/C, respectively. These data show that the Pt
3Ni(Pt-skin) structure exhibits marked activity improvements over Pd
20/C and Pt
3/Pd
20/C catalysts.
Fig. 4b compares the Tafel plots for the specific activity (
jk) towards the ORR obtained by normalizing the kinetic current (
ik) to the ECSA
CO for Pt
3/Pd
20/C, PtNi/Pd
20/C and Pt
3Ni(Pt-skin)/Pd
20/C, at 1600 rpm in the cathodic sweep direction.
ik was obtained from the measured currents, corrected for mass transport according to
eqn (2).
59–61| | 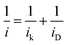 | (2) |
 |
| | Fig. 4 (a) ORR polarization curves of Pt3Ni(Pt-skin)/Pd20/C, PtNi/Pd20/C, Pt3/Pd20/C, Niu/Pd20/C, and commercial Pd20/C catalysts recorded at room temperature in an O2-saturated 0.1 M HClO4 aqueous solution with a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm. Inset: the corresponding H2O2 production current efficiency, χH2O2, from ring current (ir) during the ORR, ring potential = 1.1 V, collection efficiency: N = 0.2. (b) The corresponding Tafel plots. Inset: (ORR@0.85 V and ORR@0.9 V) mass activities and (ORRPt@0.85 V and ORRPt@0.9 V) Pt mass activities for (Pt3Ni) Pt3Ni(Pt-skin)/Pd20/C, (PtNi) PtNi/Pd20/C, and (Pt3) Pt3/Pd20/C measured at 0.85 V and 0.9 V. | |
Two Tafel slopes of Pt3/Pd20/C, −60/−119 mV dec−1, were similar to those observed on conventional Pt/C. The values of the Tafel slopes of Pt3Ni(Pt-skin)/Pd20/C, −70/−122 mV dec−1, showed a slight difference from those of Pt3/Pd20/C, probably owing to a different state and coverage of Pt(OH)x after the insertion of the Ni sublayer. When comparing jk for different electrocatalysts at 0.9 V, the specific activity of Pt3Ni(Pt-skin)/Pd20/C was 2.34 mA cm−2, which is much higher than those of Pt3/Pd20/C (∼0.06 mA cm−2) and PtNi/Pd20/C (∼0.16 mA cm−2). Considering the ECSACO contribution from Pt (ECSACO(Pt) = sECSACO × the mass of Pt (∼2.97 μgPt cm−2)), Pt3Ni(Pt-skin)/Pd20/C shows a very high specific activity jk of Pt (jkPt) 16.7 mA cm−2 at 0.9 V, which is 90-fold enhancement over commercial Pt/C catalysts (0.185 mA cm−2, Pt loading = 24 μg cm−2) and even higher than the possibly highest record, 10.3 mA cm−2, for octahedral Mo–Pt3Ni/C (the particle shape is close to spherical)62 and 11.5 mA cm−2, for jagged Pt nanowires from de-alloying PtNi nanowires with a totally different microstructure.31 The Pt content was obtained by measuring the charge associated with Znu (after correcting for the double layer charging) on Pd20/C and assuming that there was a one-to-one ratio between the UPD adlayer and the Pt atoms. The total amount of Pt on Pt3/Pd20/C and Pt3Ni(Pt-skin)/Pd20/C (∼2.97 μgPt cm−2) was much less than that on the commercial Pt/C (∼24 μgPt cm−2). This finding highlights the advantages of ultra-thin layer electrocatalysts, which contribute to not only the reduction of Pt content but also the enhancement of catalytic activity thanks to their high Pt utilization.39,40 The Pt mass activities (imPt) at 0.9 and 0.85 V were calculated based on the ORR polarization curves and the amount of Pt on the electrodes. The Pt3Ni(Pt-skin) structure retained the advantages of the ultra-thin layer structure and the synergetic effect of the Ni sublayer presented an ultra-high imPt of 14.2 A mgPt−1 based on Pt loading (2.97 μgPt cm−2) at 0.9 V. The imPt of the Pt3Ni(Pt-skin)/Pd20/C catalyst achieved a 37- and 156-fold improvement over Pt3/Pd20/C (0.381 A mgPt−1, ∼2.97 μgPt cm−2) and commercial Pt/C catalysts (0.091 A mgPt−1, ∼24 μgPt cm−2), respectively. The imPt is more than an order of magnitude greater than the U.S. Department of Energy's 2017 goal (0.44 A mgPt−1). The imPt of Pt3Ni(Pt-skin)/Pd20/C is even higher than those of the state-of-the-art Pt–Ni catalysts, including the recently reported PtNi nanoframe catalysts (5.7 A mgPt−1),28 Mo–Pt3Ni/C (6.98 A mgPt−1, Pt loading = 4.08 μgPt cm−2)62 and jagged Pt nanowires (13.6 A mgPt−1, Pt loading = 2.2 μgPt cm−2) (Table 1).31 Two accelerated durability test (ADT) modes further examined the Pt3Ni(Pt-skin)/Pd20/C lifetime (Fig. 5).
Table 1 Performance of the Pt3Ni(Pt-skin)/Pd20/C catalyst and some representative results with high performance from recent published studiesa
| Catalysts |
Pt loading (μg cm−2) |
sECSACO (m2 g−1) |
j
kPt (mA cm−2) |
i
mPt (A mgPt−1) |
|
|
N/A: not available.
|
| Pt3Ni(Pt-skin)/Pd20/C |
2.97 |
106 |
16.7 |
14.2 |
This work |
| Mo–Pt3Ni/C |
4.08 |
83.9 |
8.2 |
6.98 |
Ref. 62
|
| Pt3Ni/C nanoframes |
N/A |
N/A |
1.48 |
5.7 |
Ref. 28
|
| Jagged Pt nanowires |
2.2 |
118 |
11.5 |
13.6 |
Ref. 31
|
| Commercial Pt/C |
24 |
91 |
0.185 |
0.091 |
This work |
 |
| | Fig. 5 ORR polarization curves of the Pt3Ni(Pt-skin)/Pd20/C catalyst on a GC RDE (0.196 cm2, ∼2.97 μg cm−2 Pt loading) (dashed line) before and (solid line) after accelerated durability tests include (a) ADTs: a linear potential polarization from 0.6 to 1.0 V vs. RHE at a scan rate of 50 mV s−1 for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and (b) ADTon–off: a linear potential polarization from 1.0 to 1.5 V vs. RHE for 20000 cycles at a scan rate of 500 mV s−1 in O2-saturated 0.1 M HClO4. Insets: the corresponding Tafel plots. (c) and (d) (solid pattern) sECSACO, and (slash pattern) Pt mass activities for (blue) before and (red) after accelerated durability tests. 000 cycles and (b) ADTon–off: a linear potential polarization from 1.0 to 1.5 V vs. RHE for 20000 cycles at a scan rate of 500 mV s−1 in O2-saturated 0.1 M HClO4. Insets: the corresponding Tafel plots. (c) and (d) (solid pattern) sECSACO, and (slash pattern) Pt mass activities for (blue) before and (red) after accelerated durability tests. | |
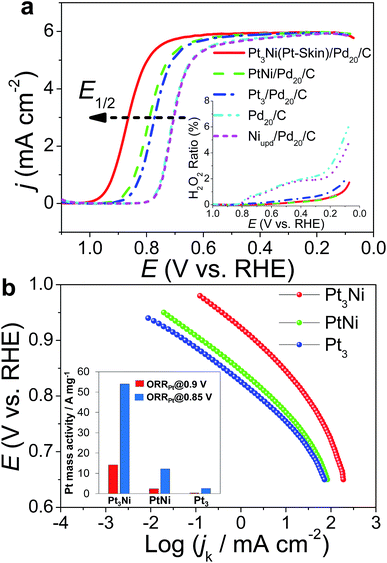
One was a commonly used test mode (ADTs) which applied a linear potential sweep from 0.6 to 1.0 V vs. RHE at a scan rate of 50 mV s−1 for 20000 cycles in O2-saturated 0.1 M HClO4. Recently, another ADT mode (ADTon–off), start-up/shut-down cycles, has gradually received attention.63,64 The ADTon–off performed a linear potential sweep from 1.0 to 1.5 V vs. RHE for 20000 cycles at a scan rate of 500 mV s−1 in an O2-saturated 0.1 M HClO4. After ADTs, Pt3Ni(Pt-skin)/Pd20/C retained its ECSA and high activity (Fig. 5a and c), exhibiting only an ∼2 mV shift for its E1/2. This result is also consistent with the pioneering work by Adzic's group in which the modification of Ptnano surfaces with Au clusters through UPD-Gal improved the stability of catalysts.7 Our previous studies also demonstrated that nonspecific noble metal cluster (Pt, Pd or Au) modification through UPD-Gal enhanced the interaction between Ptnano and the carbon support to improve catalyst durability during ADTs.11 Interestingly, Pt3Ni(Pt-skin)/Pd20/C shows the highest durability during ADTon–off (Fig. 5b and d). In the ADTon–off, Pt dissolution and Ni leaching were serious issues for typical Pt/C or PtNi catalysts owing to their operation at a higher anodic potential. The superior stability of Pt3Ni(Pt-skin)/Pd20/C during the ADTon–off may be a result of the perfect Pt3Ni(Pt-skin) structure. The Pt-skin located on the exterior of the PtNi shell effectively inhibits the dissolution of the Pd core and interior Ni sublayer leaching. The issue of Pt dissolution may have been improved due to the enlargement of the particle sizes of Pt3Ni(Pt-skin)/Pdnano in comparison with pristine Pdnano (from ∼5 nm to ∼6.8 nm). It has previously been demonstrated that the Pt dissolution is significantly dependent on particle size and environmental acidity.65 A larger particle size (>5 nm) could both reduce the trend of Pt dissolution and its activity. However, the ultra-thin layer Pt3Ni(Pt-skin) modification not only stabilizes Pt from particle size enlargement but also significantly enhances catalytic activity owing to the full exposure of Pt active sites on the surface of Pd/C.
Conclusions
In conclusion, considering that the Pt3Ni(Pt-skin) structure exhibited the highest catalytic activity ever recorded, in this work, the challenge of preparing a nanocatalyst mimicking the Pt3Ni(Pt-skin) structure was mitigated by growing a Niu covered Pdnano core using the ZnUPD-Gal procedure. ZnUPD-Gal is an improvement to common CuUPD-Gal that offers more options of coating surfaces and facilitates more metal growth in the Gal process owing to the intrinsic lower ΦZn and more negative E0 (Zn2+/Zn). The Pt3Ni(Pt-skin) thin layer with ultra-low Pt loading presents not only a breakthrough in Pt specific activity and Pt mass activity, both of which were superior to those of state-of-the-art Pt–Ni catalysts, but also in high durability owing to the perfect Pt3Ni(Pt-skin) construction.
Experimental
Chemicals
Carbon-supported palladium (Pd20/C) (20 wt% Pd on XC-72 Valcan carbon (E-TEK)), Pt20/C (20 wt% Pt on XC-72 Valcan carbon (E-TEK)), 70% HClO4 (JT-Baker), 95–98% H2SO4 (Aldrich), 98% NiSO4·6H2O (Aldrich), 98% Zn(ClO4)2·6H2O (Aldrich), 99.7% Cu(NO3)2 (JT-Baker), 99.9% K2PtCl4 (Alfa), and 5% Nafion (NF) perfluorinated resin in a mixture of lower aliphatic alcohols and water (Aldrich) were used as received.
Electrochemical experiments
The electrochemical experiments were conducted using a CHI 760C potentiostat/galvanostat and a three-electrode electrochemical cell. To prevent Cl− interference from the typical Ag/AgCl reference electrode, Hg/HgSO4 (0.5 M H2SO4) was used as the reference electrode and Pt wire was used as the counter electrode. The potentials were measured against the reference electrode and converted to the reversible hydrogen electrode (RHE) reference scale by ERHE = EHg/HgSO4 + 0.68 + 0.059pHelectrolyte. All potentials in this paper were stated with reference to the RHE. A glassy carbon (GC) electrode (Pine, 5.0 mm diameter, 0.196 cm2 for the fabrication of electrocatalysts or the examination of electrocatalyst performance for the ORR) served as the substrate electrode for the Pd20/C electrocatalyst suspension. The Pd20/C catalyst suspension was prepared by mixing 10 mg of commercial Pd20/C electrocatalyst powder in 5 mL of deionized water (with specific resistivity = 18.2 MΩ cm), followed by the gradual addition of 1 ml of isopropyl alcohol and 20 min of ultrasonication to obtain a Pd20/C suspension. The Pd20/C@GC electrode was fabricated using a drop coating procedure. Briefly, a GC electrode was polished successively with 1.0, 0.3, and 0.05 μm alumina powder cloth (Buchler) followed by sonication in deionized water and drying prior to use. 2–7 μl of the Pd20/C suspension was pipetted onto the surface of the GC electrode as a circle with a geometric area of 0.01–0.05 cm2. Pd20/C@GC was obtained after drying under an Ar flow at room temperature (∼28 °C) for solvent evaporation.
Preparation of Niu/Pd20/C, PtNi/Pd20/C, Pt3Ni(Pt-skin)/Pd20/C, Pt3/Pd20/C, and (PtNi)3/Pd20/C electrocatalysts
Pd20/C@GCE was used as a working electrode to prepare relative catalysts. The microstructure of the post-modified Pd20/C electrocatalysts was examined by the electrochemical characterization and image analysis. High-resolution transmission electron microscopy (TEM) images (HRTEM) and elemental composition micro-distribution of post-modified Pd20/C electrocatalysts were obtained using a JEOL JEM-2100F field emission TEM (FE-TEM) equipped with an energy dispersive X-ray spectrometer (EDS).
Preparation of Niu/Pd20/C.
The Ni underpotential deposition (Niu) covered Pd20/C@GC (Niu/Pd20/C@GC) electrocatalyst was prepared by direct electrodeposition of Niu on the Pd surface of the Pd20/C@GC electrode from a 0.5 M Na2SO4 aqueous solution (Na2SO4aq) containing 20 mM NiSO4. The potential was stopped at −0.1 V vs. RHE for 20 min to allow the Niu to completely cover the entire Pd surface. All operations were carried out in a three-electrode electrochemical cell under an Ar atmosphere.
Preparation of PtNi/Pd20/C.
The Pt atomic layer was prepared by ZnUPD-Gal on the Ni surface of the as-prepared Niu/Pd20/C@GC from 0.5 M Na2SO4aq containing 20 mM Zn(ClO4)2. The potential was stopped at 0.05 V vs. RHE for 10 min to allow the Znu to completely cover the whole surface of the Ni surface. All of these operations were carried out in a three-electrode electrochemical cell under an Ar atmosphere. The as-prepared Znu modified Niu/Pd20/C@GC was rinsed with deionized water and immersed in a 0.2 M H2SO4 solution containing 1.0 mM K2PtCl4 for about 5 min to displace the Znu with Pt and to obtain Pt atomic layer modified Niu/Pd20/C@GC (PtNi/Pd20/C@GC).
Preparation of Pt3Ni(Pt-skin)/Pd20/C.
The repetitive ZnUPD-Gal process was continually used to introduce the second and the third Pt atomic layers onto the as-prepared PtNi/Pd20/C@GC to obtain Pt3Ni(Pt-skin)/Pd20/C@GC.
Preparation of Pt3/Pd20/C.
The repetitive ZnUPD-Gal process continually introduced three Pt atomic layers on Pd20/C@GC to obtain Pt3/Pd20/C@GC.
Preparation of (PtNi)3/Pd20/C.
To extend the diversity of the ZnUPD-Gal on Niu, a Pt atomic layer and Niu repetitively overlapped shell, which was three repeated PtNi layers on a Pd core ((PtNi)3/Pd20/C), was prepared as a control example. Niu was electrodeposited on the Pt surface of the as-prepared PtNi/Pd20/C@GC from 0.5 M Na2SO4aq containing 20 mM NiSO4 (Niu/PtNi/Pd20/C@GC) at a controlled potential of −0.05 V vs. RHE for 20 min to allow the Niu to completely cover the whole surface of the Pt surface. The Znu was electrodeposited on the as-prepared Niu/PtNi/Pd20/C@GC from 0.5 M Na2SO4aq containing 20 mM Zn(ClO4)2, while controlling the potential at 0.05 V vs. RHE for 10 min. Subsequently, the Znu was replaced by more noble Pt in the Gal process, leading to Pt atomic layer covered Niu/PtNi/Pd20/C@GC ((PtNi)2/Pd20/C@GC). The ZnUPD-Gal on Niu was used to introduce the third PtNi layer on (PtNi)2/Pd20/C@GC and obtain (PtNi)3/Pd20/C@GC.
Electrochemical characterization of the ORR
A commercial (Pine Instruments) rotating ring-disk electrode (RRDE) was used with a Pt ring and an interchangeable disk. The disk electrode was a GC rod modified by a thin layer of as-prepared electrocatalyst covered by a Nafion polymer thin film used as the working electrode for ORR measurements. The as-prepared working electrode was electrochemically cleaned using a cycling potential between 0.0 and 1.0 V (vs. RHE) 10 times in Ar-purged 0.1 M H2SO4, except for Niu/Pd20/C owing to Niu leaching. The electrochemical surface areas (ECSA) of the electrocatalysts were determined by measuring the areas (charges) under the hydrogen adsorption peaks (Had) (ECSAH) and the electro-oxidation of adsorbed carbon monoxide (COad), otherwise known as CO stripping (ECSACO), respectively, in the cyclic voltammograms. A conversion factor of 0.21 mC cm−2 was used to determine ECSAH. For CO stripping experiments, CO was adsorbed onto the pre-cleaned electrode by holding the potential at 0.05 V for 10 min in CO saturated 0.1 M HClO4 solution. The CO stripping curve was taken after purging with Ar for 30 min. A conversion factor of 0.42 mC cm−2 was used to determine ECSACO. The specific ECSA values (sECSA = ECSA/metal loading, m2 g−1) of the electrocatalysts were evaluated from ECSAH (sECSAH) and ECSACO (sECSACO). When considering the ECSACO contribution from Pt (ECSACO(Pt) = sECSACO × the mass of Pt (∼2.97 μgPt cm−2 for Pt3Ni(Pt-skin)/Pd20/C and Pt3/Pd20/C)), the Pt content was obtained by measuring the charge associated with Znu (after correcting for the double layer charging) on Pd20/C and assuming that there was a one-to-one ratio between the UPD adlayer and Pt atoms. ORR experiments were performed in oxygen-saturated 0.1 M HClO4 aqueous solution. The solution was purged for at least 30 min to ensure oxygen saturation. The ORR electrochemical experiments were conducted in a three-electrode electrochemical cell. Hg/HgSO4 (0.5 M H2SO4) and a Pt wire were used as the reference and counter electrodes, respectively; however, all potentials are quoted with respect to a RHE. The scan rate was 0.01 V s−1. A Pine Model AFMSR electrode rotator controlled the electrode rotation for the ORR electrochemical experiments. During measurement of the polarization curves for the ORR on the disk electrode, the Pt ring electrode was potentiostated at 1.1 V vs. RHE, a potential where the peroxide oxidation reaction is under pure diffusion control; the collection efficiency, N, for the ring-disk assembly was ∼0.2.
Accelerated durability test (ADT, linear potential scanning)
The durability of the Pt3Ni(Pt-skin)/Pd20/C catalyst was investigated in two ADT modes. In the ambient temperature potential polarization, Pt3Ni(Pt-skin)/Pd20/C@GC was used as the working electrode.
Accelerated durability tests related to catalyst lifetime (ADTs).
The ADTs were conducted by linear potential sweeping from 0.6 to 1.0 V vs. RHE at a scan rate of 50 mV s−1 for 20000 cycles in an O2-saturated 0.1 M HClO4aq at room temperature.
Accelerated durability test related start-up/shut-down cycles (ADTon–off).
The ADTon–off was conducted by linear potential sweeping from 1.0 to 1.5 V vs. RHE for 20000 cycles at a scan rate of 500 mV s−1 in O2-saturated 0.1 M HClO4aq at room temperature.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Ministry of Science and Technology of the Republic of China, Taiwan.
Notes and references
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef PubMed.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. F. Chi, J. Y. Liu, S. I. Choi, J. Park, J. A. Herron, Z. X. Xie, M. Mavrikakis and Y. N. Xia, Science, 2015, 349, 412–416 CrossRef PubMed.
- Y. J. Wang, N. N. Zhao, B. Z. Fang, H. Li, X. T. T. Bi and H. J. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef PubMed.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302–1305 CrossRef PubMed.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef PubMed.
- D. J. Berger, Science, 1999, 286, 49 CrossRef.
- J. F. Huang and W. Y. Chen, Chem. Commun., 2015, 51, 12052–12055 RSC.
- J. F. Huang and H. Y. Hsiao, ACS Appl. Mater. Interfaces, 2016, 8, 33749–33754 CrossRef PubMed.
- J. F. Huang and H. W. Yang, Anal. Chem., 2016, 88, 6403–6409 CrossRef PubMed.
- J.-F. Huang and W.-R. Chang, J. Mater. Chem., 2012, 22, 17961–17966 RSC.
- F. Calle-Vallejo, M. D. Pohl, D. Reinisch, D. Loffreda, P. Sautet and A. S. Bandarenka, Chem. Sci., 2017, 8, 2283–2289 RSC.
- A. Fortunelli, W. A. Goddard, L. Sementa, G. Barcaro, F. R. Negreiros and A. Jaramillo-Botero, Chem. Sci., 2015, 6, 3915–3925 RSC.
- H. P. Liu, P. Zhong, K. Liu, L. Han, H. Q. Zheng, Y. D. Yin and C. B. Gao, Chem. Sci., 2018, 9, 398–404 RSC.
- D. F. Yancey, L. Zhang, R. M. Crooks and G. Henkelman, Chem. Sci., 2012, 3, 1033–1040 RSC.
- J. F. Huang and H. Y. Chen, Angew. Chem., Int. Ed., 2012, 51, 1684–1688 CrossRef PubMed.
- M. K. Carpenter, T. E. Moylan, R. S. Kukreja, M. H. Atwan and M. M. Tessema, J. Am. Chem. Soc., 2012, 134, 8535–8542 CrossRef PubMed.
- V. Stamenkovic, T. J. Schmidt, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2002, 106, 11970–11979 CrossRef.
- L. Z. Bu, Q. Shao, E. Bin, J. Guo, J. L. Yao and X. Q. Huang, J. Am. Chem. Soc., 2017, 139, 9576–9582 CrossRef PubMed.
- M. Zhou, H. L. Wang, M. Vara, Z. D. Hood, M. Luo, T. H. Yang, S. X. Bao, M. F. Chi, P. Xiao, Y. H. Zhang and Y. N. Xia, J. Am. Chem. Soc., 2016, 138, 12263–12270 CrossRef PubMed.
- N. Becknell, Y. J. Kang, C. Chen, J. Resasco, N. Kornienko, J. H. Guo, N. M. Markovic, G. A. Somorjai, V. R. Stamenkovic and P. D. Yang, J. Am. Chem. Soc., 2015, 137, 15817–15824 CrossRef PubMed.
- K. A. Kuttiyiel, Y. Choi, K. Sasaki, D. Su, S. M. Hwang, S. D. Yim, T. H. Yang, G. G. Park and R. R. Adzic, Nano Energy, 2016, 29, 261–267 CrossRef.
- H. Yang, Angew. Chem., Int. Ed., 2011, 50, 2674–2676 CrossRef PubMed.
- L. Gan, M. Heggen, R. O'Malley, B. Theobald and P. Strasser, Nano Lett., 2013, 13, 1131–1138 CrossRef PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef PubMed.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef PubMed.
- C. H. Cui, L. Gan, H. H. Li, S. H. Yu, M. Heggen and P. Strasser, Nano Lett., 2012, 12, 5885–5889 CrossRef PubMed.
- L. Cao and T. Mueller, J. Phys. Chem. C, 2015, 119, 17735–17747 CrossRef.
- M. F. Li, Z. P. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. H. Zhang, L. Gu, B. V. Merinov, Z. Y. Lin, E. B. Zhu, T. Yu, Q. Y. Jia, J. H. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. F. Duan, Science, 2016, 354, 1414–1419 CrossRef PubMed.
- S. H. Zhou, B. Varughese, B. Eichhorn, G. Jackson and K. McIlwrath, Angew. Chem., Int. Ed., 2005, 44, 4539–4543 CrossRef PubMed.
- D. Zhao and B. Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 4955–4959 CrossRef PubMed.
- K. Sasaki, J. X. Wang, H. Naohara, N. Marinkovic, K. More, H. Inada and R. R. Adzic, Electrochim. Acta, 2010, 55, 2645–2652 CrossRef.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- K. A. Kuttiyiel, K. Sasaki, Y. M. Choi, D. Su, P. Liu and R. R. Adzic, Nano Lett., 2012, 12, 6266–6271 CrossRef PubMed.
- M. B. Gawande, A. Goswami, T. Asefa, H. Z. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- S. I. Choi, M. H. Shao, N. Lu, A. Ruditskiy, H. C. Peng, J. Park, S. Guerrero, J. G. Wang, M. J. Kim and Y. N. Xia, ACS Nano, 2014, 8, 10363–10371 CrossRef PubMed.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef.
- M. B. Vukmirovic, J. Zhang, K. Sasaki, A. U. Nilekar, F. Uribe, M. Mavrikakis and R. R. Adzic, Electrochim. Acta, 2007, 52, 2257–2263 CrossRef.
- C. Koenigsmann, A. C. Santulli, K. P. Gong, M. B. Vukmirovic, W. P. Zhou, E. Sutter, S. S. Wong and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 9783–9795 CrossRef PubMed.
- S. Trasatti, J. Electroanal. Chem., 1971, 33, 351–377 CrossRef.
- D. M. Kolb, M. Przasnyski and H. Gerischer, J. Electroanal. Chem., 1974, 54, 25–38 CrossRef.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef PubMed.
- C. Wang, M. F. Chi, D. G. Li, D. Strmcnik, D. van der Vliett, G. F. Wang, V. Komanicky, K. C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef PubMed.
- C. H. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef PubMed.
- S. J. Guo, D. G. Li, H. Y. Zhu, S. Zhang, N. M. Markovic, V. R. Stamenkovic and S. H. Sun, Angew. Chem., Int. Ed., 2013, 52, 3465–3468 CrossRef PubMed.
- T. Fu, J. Fang, C. S. Wang and J. B. Zhao, J. Mater. Chem. A, 2016, 4, 8803–8811 RSC.
- S. Mezzavilla, C. Baldizzonev, A. C. Swertz, N. Hodnik, E. Pizzutilo, G. Polymeros, G. P. Keeley, J. Knossalla, M. Heggen, K. J. J. Mayrhofer and F. Schuth, ACS Catal., 2016, 6, 8058–8068 CrossRef.
- X. Zhao, S. Takao, K. Higashi, T. Kaneko, G. Samjeske, O. Sekizawa, T. Sakata, Y. Yoshida, T. Uruga and Y. Iwasawa, ACS Catal., 2017, 7, 4642–4654 CrossRef.
- B. W. Gregory and J. L. Stickney, J. Electroanal. Chem., 1991, 300, 543–561 CrossRef.
- Y. G. Kim, J. Y. Kim, D. Vairavapandian and J. L. Stickney, J. Phys. Chem. B, 2006, 110, 17998–18006 CrossRef PubMed.
-
J. R. Rumble, CRC Handbook of Chemistry and Physics, CRC Press, 98th edn, 2017 Search PubMed.
- M. D. Quaiyyum, A. Aramata, S. Moniwa, S. Taguchi and M. Enyo, J. Electroanal. Chem., 1994, 373, 61–66 CrossRef.
- D. F. van der Vliet, C. Wang, D. G. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2012, 51, 3139–3142 CrossRef PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Method: Fundamentals and Applications, John Wiley & Son, New York, 2nd edn, 2001 Search PubMed.
- M. H. Shao, J. H. Odell, S. I. Choi and Y. N. Xia, Electrochem. Commun., 2013, 31, 46–48 CrossRef.
- B. Alvarez, V. Climent, A. Rodes and J. M. Feliu, J. Electroanal. Chem., 2001, 497, 125–138 CrossRef.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 CrossRef PubMed.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, N. M. Markovic and P. N. Ross, Electrochim. Acta, 2002, 47, 3787–3798 CrossRef.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, V. Radmilovic, N. M. Markovic and P. N. Ross, J. Phys. Chem. B, 2002, 106, 4181–4191 CrossRef.
- X. Q. Huang, Z. P. Zhao, L. Cao, Y. Chen, E. B. Zhu, Z. Y. Lin, M. F. Li, A. M. Yan, A. Zettl, Y. M. Wang, X. F. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef PubMed.
- H. Schmies, A. Bergmann, J. Drnec, G. X. Wang, D. Teschner, S. Kuhl, D. J. S. Sandbeck, S. Cherevko, M. Gocyla, M. Shviro, M. Heggen, V. Ramani, R. E. Dunin-Borkowski, K. J. J. Mayrhofer and P. Strasser, Adv. Energy Mater., 2018, 8, 13 Search PubMed.
- K. Higashi, G. Samjeske, S. Takao, T. Kaneko, O. Sekizawa, T. Uruga and Y. Iwasawa, J. Phys. Chem. C, 2017, 121, 22164–22177 CrossRef.
- L. Tang, B. Han, K. Persson, C. Friesen, T. He, K. Sieradzki and G. Ceder, J. Am. Chem. Soc., 2010, 132, 596–600 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images and HRTEM-EDS mapping images. See DOI: 10.1039/c8sc01358f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Po-Kai
Tseng
* and
Po-Kai
Tseng







