DOI:
10.1039/C8SC01434E
(Edge Article)
Chem. Sci., 2018,
9, 5015-5020
A supramolecular radical cation: folding-enhanced electrostatic effect for promoting radical-mediated oxidation†
Received
28th March 2018
, Accepted 7th May 2018
First published on 16th May 2018
Abstract
We report a supramolecular strategy to promote radical-mediated Fenton oxidation by the rational design of a folded host–guest complex based on cucurbit[8]uril (CB[8]). In the supramolecular complex between CB[8] and a derivative of 1,4-diketopyrrolo[3,4-c]pyrrole (DPP), the carbonyl groups of CB[8] and the DPP moiety are brought together through the formation of a folded conformation. In this way, the electrostatic effect of the carbonyl groups of CB[8] is fully applied to highly improve the reactivity of the DPP radical cation, which is the key intermediate of Fenton oxidation. As a result, the Fenton oxidation is extraordinarily accelerated by over 100 times. It is anticipated that this strategy could be applied to other radical reactions and enrich the field of supramolecular radical chemistry in radical polymerization, photocatalysis, and organic radical battery and holds potential in supramolecular catalysis and biocatalysis.
Introduction
Supramolecular free radicals refers to radicals stabilized or activated by non-covalent interactions,1–4 which hold potential in the fields of spin-based materials5–12 and radical-mediated reactions.13–19 Various non-covalent interactions can be employed to modulate the reactivity of radicals, including hydrogen bonding,20,21 metal–ligand interactions,22,23 charge-transfer interactions,24,25 and host–guest interactions.26–29 Benefiting from the dynamic and reversible nature of non-covalent interactions, these supramolecular methods have distinctive advantages in many aspects, such as avoidance of tedious covalent synthesis, switchable properties and potential in constructing adaptive radical systems.30–32 However, the fabrication of an ultra-stable or highly activated supramolecular radical still remains difficult. Therefore, it is highly desirable to amplify the effect of non-covalent interactions and thus induce a drastic modulation on the activity of radicals by supramolecular strategies.
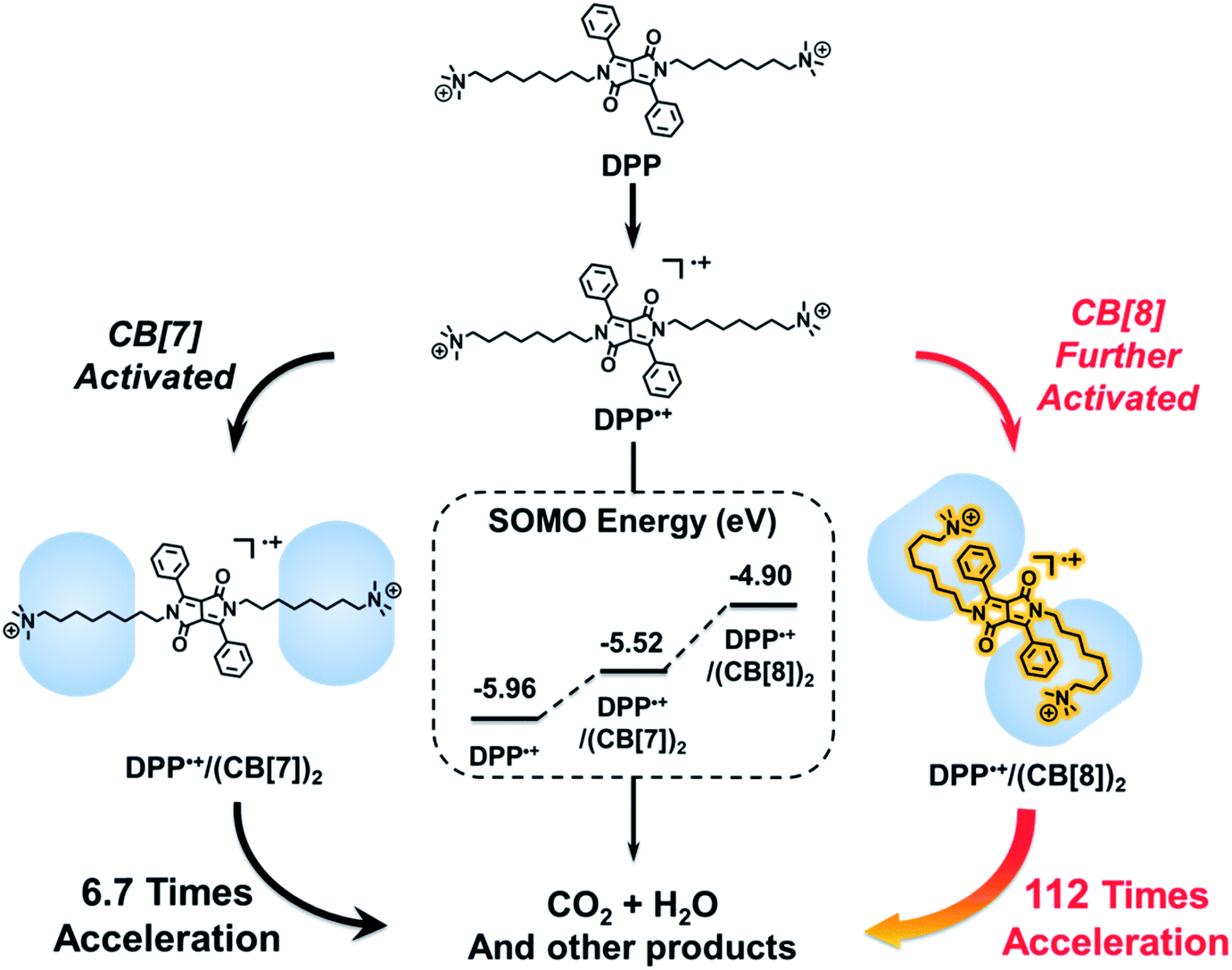
Recently, we demonstrated that the electrostatic effect of cucurbit[7]uril (CB[7]) can be used for activating radical cations.2 One might wonder if a highly activated supramolecular radical cation could be constructed by amplifying the electrostatic effect of cucurbituril. However, in the case of CB[7], this electrostatic effect is hard to further improve due to the binding mode between CB[7] and guests. CB[7] with a small cavity can only encapsulate one guest molecule, while cucurbit[8]uril (CB[8]) with a larger cavity can encapsulate two guests.33–36 Therefore, we attempted to use CB[8] for the formation of a ternary host–guest complex, which may break through the limitation of the host–guest chemistry of CB[7] and thus amplify the electrostatic effect. As a proof of concept, we chose the Fenton oxidation of a derivative of 1,4-diketopyrrolo[3,4-c]pyrrole (DPP) as a model reaction, in which the DPP radical cation (DPP˙+) is the key intermediate.37–39 After the introduction of CB[8], we expected that a folded host–guest complex based on DPP and CB[8] could be formed.40,41 As shown in Scheme 1, both the alkyl chain and the phenyl group of DPP could be encapsulated into the cavity of CB[8]. In this way, the spatial distance between the spin center of DPP˙+ and the carbonyl groups of cucurbituril could be shortened, and so the electrostatic effect of CB[8] could be strengthened. As a result, DPP˙+ might be activated by CB[8] and the Fenton oxidation could be accelerated through the formation of this highly activated intermediate.
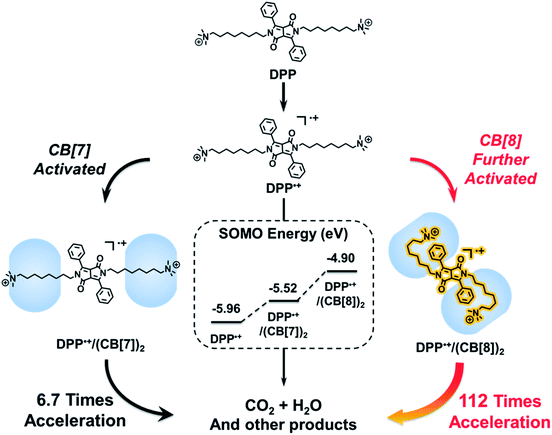 |
| | Scheme 1 Proposed mechanism of the significantly accelerated Fenton oxidation based on the highly activated DPP/(CB[8])2 radical cation with high singly occupied molecular orbital (SOMO) energy (vide infra). | |
Results and discussion
To test the above idea, we carried out experiments of the Fenton oxidation of DPP with and without the addition of CB[7] and CB[8], and studied their kinetics of oxidation by UV-Vis spectroscopy. First, we conducted the oxidation of DPP and the DPP/(CB[7])2 supramolecular complex at 0.1 mM. The concentration of Fe3+ and H2O2, the Fenton reagent, was fixed to 1.25 mM and 100 mM, respectively. As shown in Fig. 1, it took about 2 h and 10 min for the complete oxidation of DPP and DPP/(CB[7])2, respectively, while the oxidation of DPP/(CB[8])2 was completed in less than 2 min, which was too fast to be accurately measured by UV-Vis spectroscopy. Therefore, the concentrations of Fe3+ and H2O2 were decreased to 0.1 mM and 10 mM for further study. Along with the process of Fenton oxidation, the characteristic peak of the DPP chromophore at 457 nm would continually decrease to the baseline. As shown in Fig. 2a, it took about 32 h for DPP to achieve complete oxidation at 25 °C without the addition of cucurbituril. In the case of DPP/(CB[7])2, the reaction time was about 5 h (Fig. 2b) under similar conditions. For DPP/(CB[8])2, the time was about 15 min (Fig. 2c). A comparison of the time–conversion relationships is shown in Fig. 2d. For DPP/(CB[7])2, the oxidation rate was 6.7 times faster than that for DPP alone, and for DPP/(CB[8])2, the oxidation rate was 112 times faster. These data indicate that the introduction of CB[8] leads to a much greater acceleration than CB[7] in the Fenton oxidation of DPP.
 |
| | Fig. 1 The comparison of Fenton oxidation time between DPP, DPP/(CB[7])2 and DPP/(CB[8])2 under the conditions of 0.1 mM DPP, 1.25 mM Fe3+ and 100 mM H2O2. | |
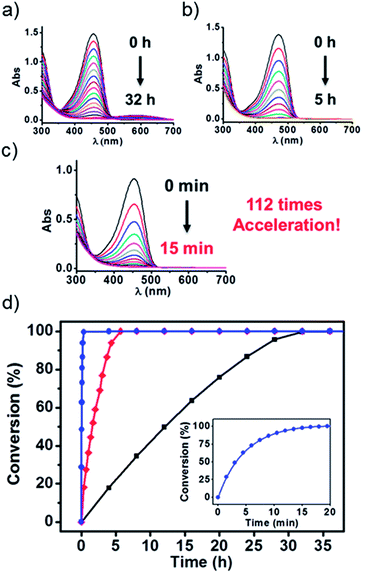 |
| | Fig. 2 UV-Vis spectroscopy was used to monitor the Fenton oxidation kinetics of (a) DPP, (b) DPP/(CB[7])2 and (c) DPP/(CB[8])2 at 25 °C (Fenton oxidation conditions: 0.1 mM DPP with 0.1 mM Fe3+ and 10 mM H2O2). (d) Time–conversion relationships of DPP ( ), DPP/(CB[7])2 ( ), DPP/(CB[7])2 ( ) and DPP/(CB[8])2 ( ) and DPP/(CB[8])2 ( ) at 25 °C. Inset: magnification of the time–conversion relationships of DPP/(CB[8])2. ) at 25 °C. Inset: magnification of the time–conversion relationships of DPP/(CB[8])2. | |
To understand the mechanism behind the acceleration, we studied the oxidation kinetics of DPP, DPP/(CB[7])2 and DPP/(CB[8])2 at the same concentration and different temperatures (Table S1†). According to the Arrhenius formula the apparent activation energies (Ea) were calculated with the linear fitting method using the values of half-life (t1/2) at different temperatures (Fig. 3). The t1/2 values were obtained directly from the time–conversion curves. As shown in Fig. 3, the apparent activation energy decreases by 35.2 kJ mol−1 after the introduction of CB[7], while a dramatic decrease of apparent activation energy by 47.6 kJ mol−1 is induced after the introduction of CB[8], which is responsible for the much more significant acceleration of Fenton oxidation than that achieved with CB[7].
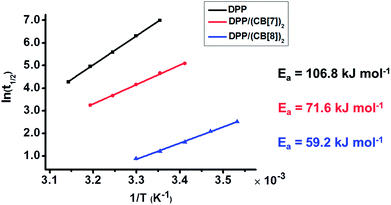 |
| | Fig. 3 Linear fitting of half-life (t1/2) to reaction temperature (T) gives the apparent activation energy (Ea) of the Fenton oxidation of DPP, DPP/(CB[7])2 and DPP/(CB[8])2. | |
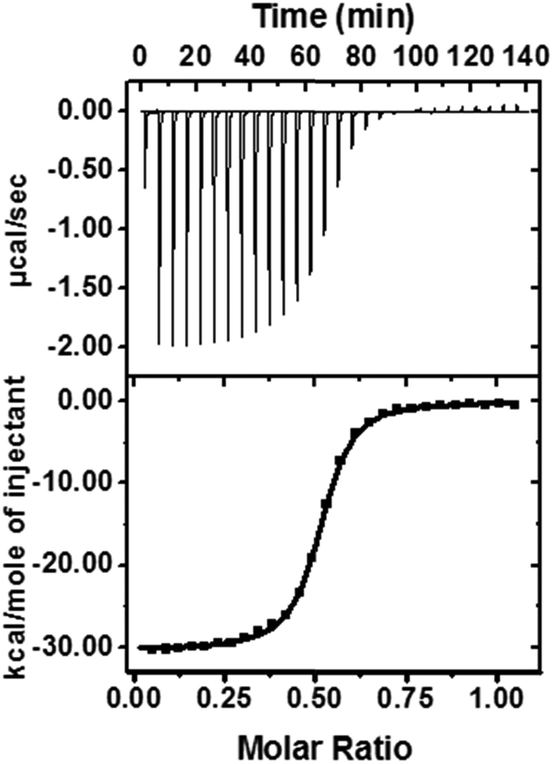
To reveal the nature of this high activation on the level of the supramolecular structure, we used isothermal titration calorimetry (ITC), 1H-NMR, and rotating frame Overhauser effect spectroscopy (ROESY) to identify the proposed structure of DPP/(CB[8])2 (Fig. 5a). ITC experiments were performed to obtain thermodynamic information on the host–guest complexation (Table 1 and Fig. 4). We confirmed that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 host–guest complex was formed after the introduction of CB[7] or CB[8]. As shown in Table 1, the larger binding constant of DPP/(CB[8])2 is mainly attributed to the larger binding enthalpy, suggesting that more high-energy water molecules are excluded from the cavity of CB[8] than CB[7].42 From this point of view, the binding mode of DPP/(CB[8])2 and DPP/(CB[7])2 might be different: not only the alkyl chain, but also another hydrophobic part (probably the phenyl group) could be encapsulated into the cavity of CB[8] to form a ternary host–guest complex. Meanwhile, the negative binding entropy of DPP/(CB[8])2 can be explained by the limited motion and rotation of the alkyl chain. Therefore, we can propose a folded host–guest complex of DPP/(CB[8])2, in which the alkyl chain and phenyl group are both encapsulated in the cavity of CB[8].
:2 host–guest complex was formed after the introduction of CB[7] or CB[8]. As shown in Table 1, the larger binding constant of DPP/(CB[8])2 is mainly attributed to the larger binding enthalpy, suggesting that more high-energy water molecules are excluded from the cavity of CB[8] than CB[7].42 From this point of view, the binding mode of DPP/(CB[8])2 and DPP/(CB[7])2 might be different: not only the alkyl chain, but also another hydrophobic part (probably the phenyl group) could be encapsulated into the cavity of CB[8] to form a ternary host–guest complex. Meanwhile, the negative binding entropy of DPP/(CB[8])2 can be explained by the limited motion and rotation of the alkyl chain. Therefore, we can propose a folded host–guest complex of DPP/(CB[8])2, in which the alkyl chain and phenyl group are both encapsulated in the cavity of CB[8].
Table 1 The thermodynamic information on DPP/(CB[7])2 and DPP/(CB[8])2 obtained by ITC
|
|
Molar ratio (DPP:CB[n]) |
Binding constant (M−1) |
Binding enthalpy (kJ mol−1) |
Binding entropy (J mol−1 K−1) |
| DPP/(CB[7])2 |
1:2 |
6.4 × 105 |
−19.3 |
46.5 |
| DPP/(CB[8])2 |
1:2 |
3.4 × 106 |
−63.8 |
−87.9 |
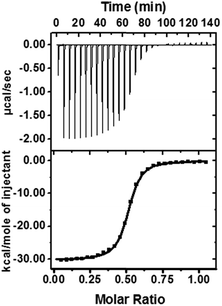 |
| | Fig. 4 ITC data and fitting curve for the titration of DPP (0.20 mM) into CB[8] (0.040 mM). | |
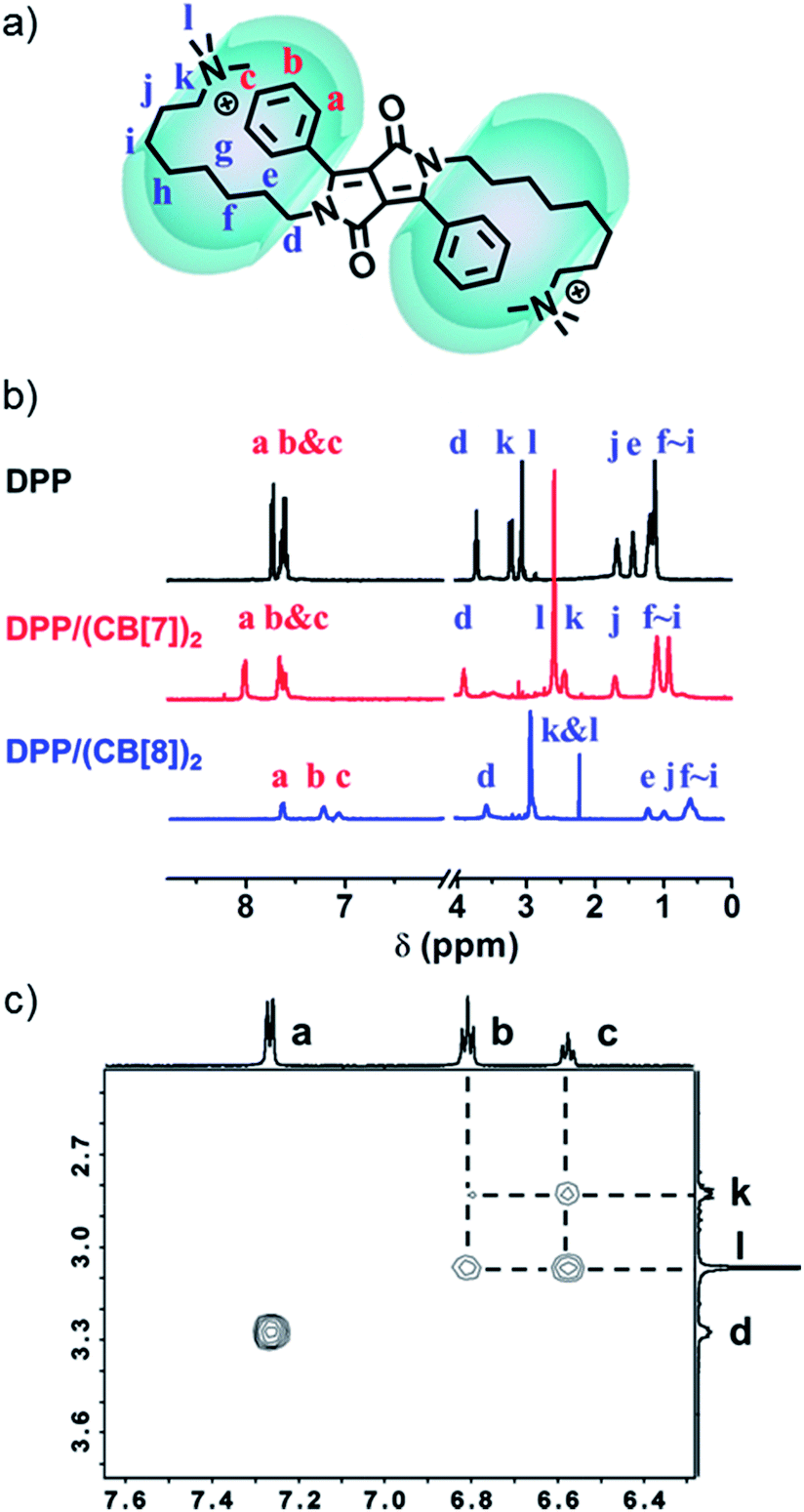
NMR spectroscopy was used to confirm the proposed supramolecular structure of DPP/(CB[8])2. As shown in the 1H-NMR spectra (Fig. 5b), after the addition of CB[8], the signals of protons a–c, which belong to the protons on phenyl groups, shifted upfield. Moreover, the signals of protons d–l, ascribed to the protons on alkyl chains, also shifted upfield. These changes of chemical shifts before and after the addition of CB[8] suggest that both of the phenyl groups and alkyl chains are encapsulated in the cavity of the CB[8] host. As a comparison, the signals of protons a–c shifted downfield after the introduction of CB[7]. The supramolecular structure was further investigated by ROESY. Four cross-peaks between protons b and c and protons k and l were observed, as shown in Fig. 5c. This indicates that the phenyl groups and the quaternary ammonium groups are spatially close to each other. As a comparison, in the ROESY spectrum of DPP without CB[8], the four cross-peaks were not observed (Fig. S12†). Therefore, the folded host–guest complex of DPP/(CB[8])2 is supported by 1H-NMR and ROESY.
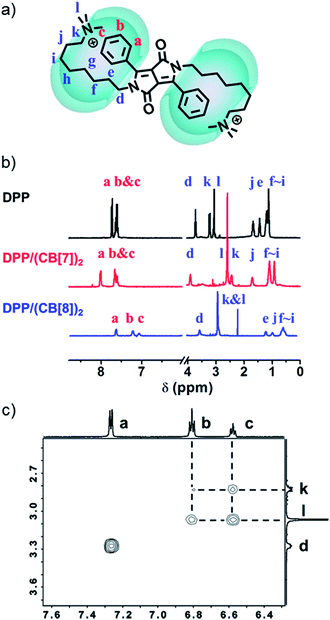 |
| | Fig. 5 (a) Supramolecular structure of DPP/(CB[8])2. (b) Partial 1H-NMR spectra (D2O) of DPP (1.0 mM), DPP/(CB[7])2 (1.0 mM) and DPP/(CB[8])2 (1.0 mM). (c) Partial ROESY 2D-NMR spectra of DPP/(CB[8])2 (1.0 mM in phosphate buffer, pH = 7.0). | |
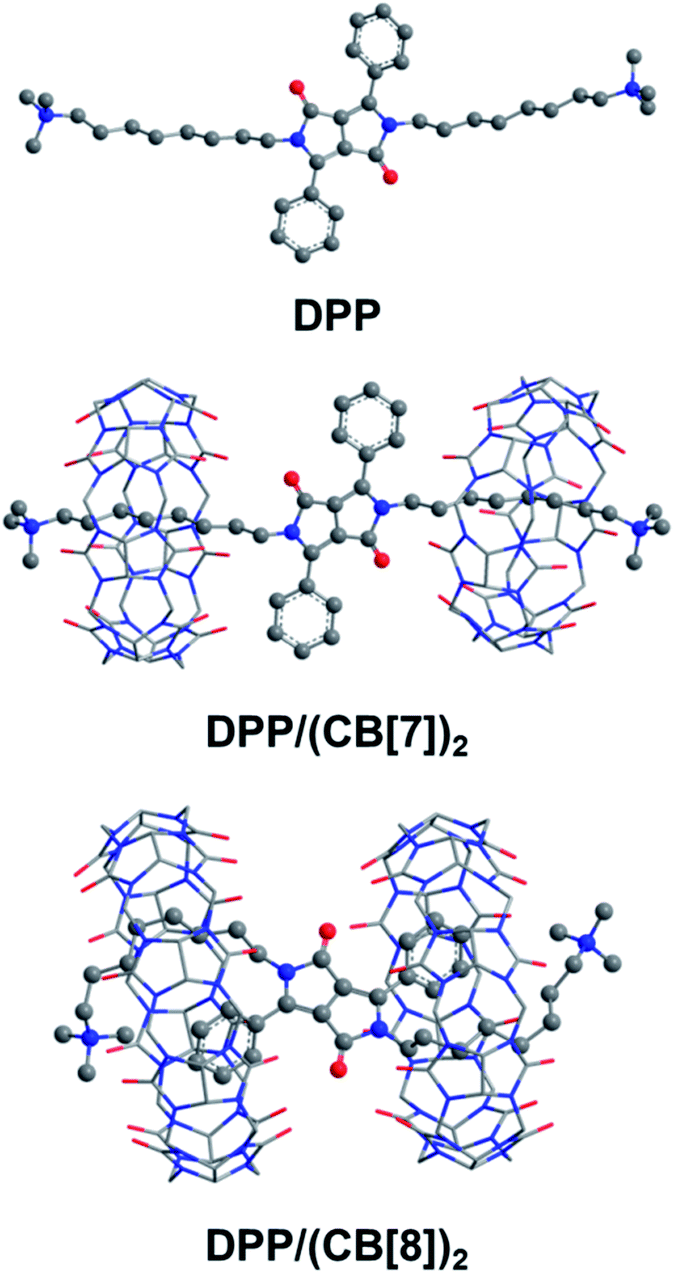
The folded host–guest complex of DPP/(CB[8])2 was also studied by theoretical chemistry modeling (see the ESI† for computational details). The structures of DPP, DPP/(CB[7])2 and DPP/(CB[8])2 from QM/MM calculations are shown in Fig. 6. The theoretical calculations predicted an extended dumbbell structure in the case of CB[7]. In contrast, the phenyl moieties and the alkyl moieties were encapsulated in the cavity of CB[8], with the alkyl chain bending from the sixth carbon atom at the tail end. Combining the theoretical and experimental evidence, the supramolecular structure of DPP/(CB[8])2 was confirmed. As indicated by the supramolecular structures, the spatial distance between the spin center of DPP˙+ and the carbonyl groups of cucurbituril is shorter in DPP/(CB[8])2 than in DPP/(CB[7])2. With the decreased spatial distance, the electrostatic effects of cucurbituril are enhanced, thus inducing further activation of DPP˙+ and a more significant acceleration of Fenton oxidation.
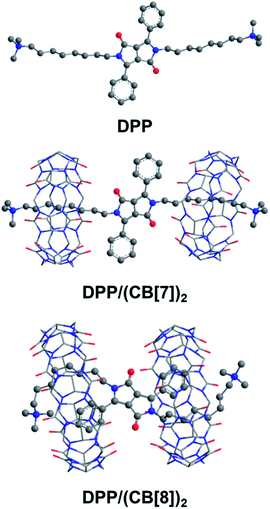 |
| | Fig. 6 The structures of DPP, DPP/(CB[7])2 and DPP/(CB[8])2 optimized by the QM/MM method. The water environment and counter-ions are omitted here for clarity. | |
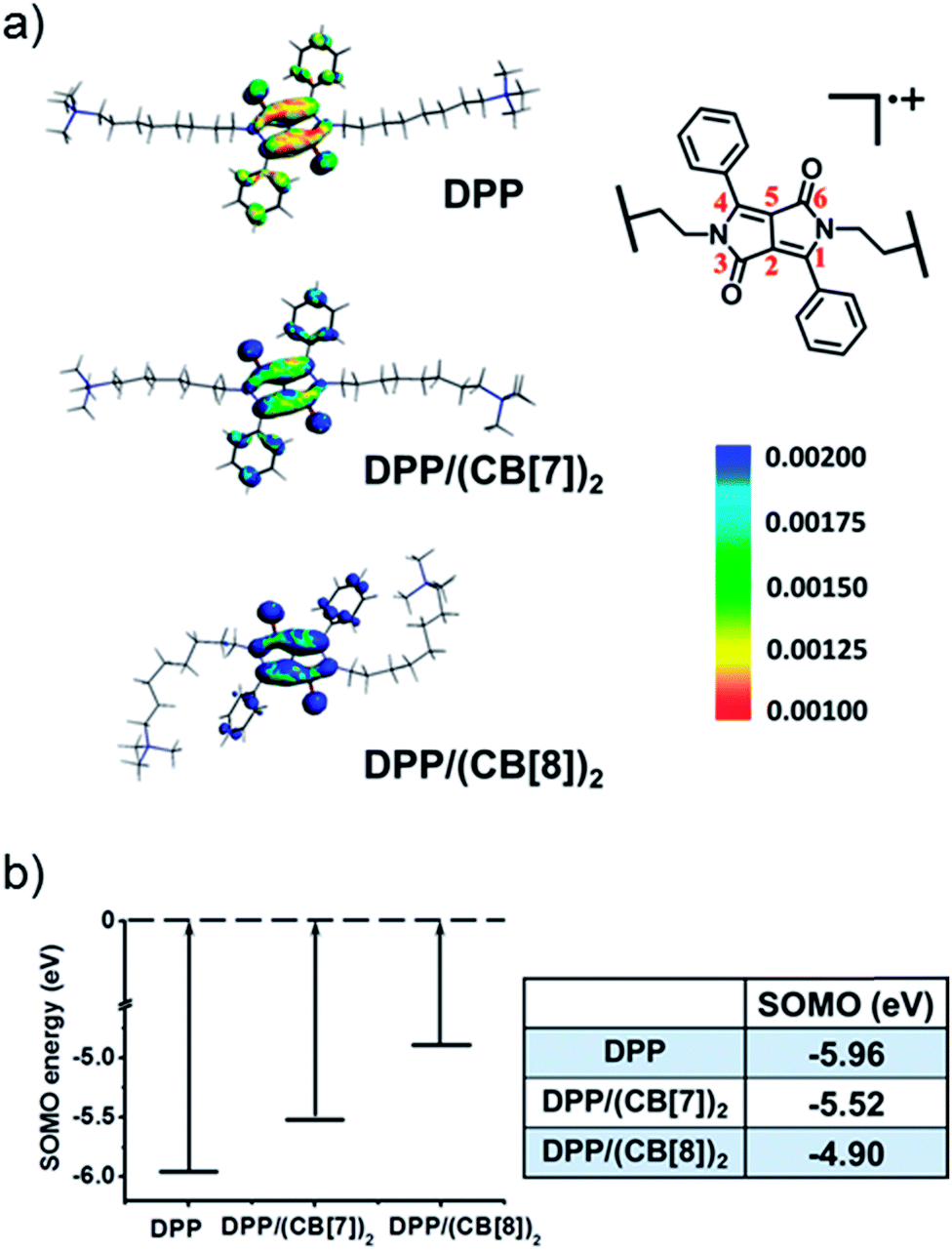
To understand the mechanism of the supramolecular activation and evaluate the electrostatic effect of cucurbituril on the DPP radical cation, we further carried out density functional theory (DFT) calculations on DPP, DPP/(CB[7])2 and DPP/(CB[8])2. Based on “free valence index” theory,43,44 we calculated Mulliken spin distributions of the aromatic carbons of the central conjugated part (C1–C6 as shown in Fig. 7a) in different cases to evaluate the reactivity of π radicals. According to this theory, the reactivity of DPP˙+ increases with the localization of spin density. From the calculation data, the degree of spin density localization increases in the following order: DPP < DPP/(CB[7])2 < DPP/(CB[8])2, which means that the single electron of DPP˙+ is the most reactive upon introduction of CB[8]. To evaluate the reactivity of DPP˙+ more quantitatively, we calculated the one-electron energy of the SOMO (singly occupied molecular orbital) of the DPP˙+ in different statuses (Fig. 7b). The SOMO energies of naked DPP˙+, DPP˙+/(CB[7])2, and DPP˙+/(CB[8])2 were calculated to be −5.96, −5.52, and −4.90 eV, respectively. This shows clearly that the electrostatic effect of the environment helps to dictate the SOMO energy and the single electron localization of the cationic radical, thus modulating the radical activity. As a result, DPP˙+/(CB[8])2 has the highest SOMO energy owing to the increased electrostatic potential, which is responsible for the highest chemical activity of the single electron and the capacity to be further oxidized. In other words, assisted by the strongest electrostatic effect, the single electron of DPP˙+/(CB[8])2 is mostly localized and has the highest SOMO energy, thus making it the most reactive radical cation.45,46
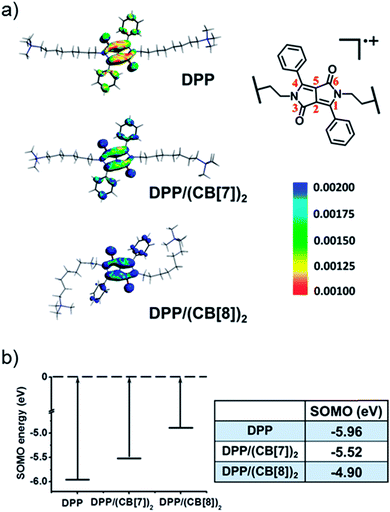 |
| | Fig. 7 (a) The spin densities and (b) calculated SOMO energies of the DPP radical cation in DPP, DPP/(CB[7])2 and DPP/(CB[8])2. | |
Conclusions
In conclusion, we provide a distinctive example of the rational design of a supramolecular structure for modulating the reactivity of organic radicals with high efficiency. Through the well-designed folded supramolecular structure based on CB[8], the electrostatic effect of the carbonyl groups of cucurbituril is greatly enhanced, which further activates the radical intermediate and induces an ultrafast Fenton oxidation reaction. This strategy could be extended to other organic radicals and applied to promoting other kinds of radical-mediated reactions. It is anticipated that this line of research could enrich the field of electrostatics-assisted supramolecular radical chemistry and provide a new perspective in supramolecular catalysis and biocatalysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported financially by the National Natural Science Foundation of China (91527000 and 21434004). We are grateful to Xixi Liang for her help with EPR measurements. We thank Zehuan Huang for helpful discussions.
Notes and references
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342 RSC.
- Y. Jiao, W.-L. Li, J.-F. Xu, G. Wang, J. Li, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8933 CrossRef PubMed.
- Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975 RSC.
- Y. Yang, P. He, Y. Wang, H. Bai, S. Wang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16239 CrossRef PubMed.
- Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botrosbc and J. F. Stoddart, Chem. Sci., 2017, 8, 2562 RSC.
- H. M. Yamamoto, Y. Kosaka, R. Maeda, J. Yamaura, A. Nakao, T. Nakamura and R. Kato, ACS Nano, 2008, 2, 143 CrossRef PubMed.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485 CrossRef PubMed.
- P. Spenst, R. M. Young, M. R. Wasielewski and F. Würthner, Chem. Sci., 2016, 7, 5428 RSC.
- J. Wu, C. Tao, Y. Li, J. Li and J. Yu, Chem. Sci., 2015, 6, 2922 RSC.
- L. Zhang, T.-Y. Zhou, J. Tian, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 4715 RSC.
- Y. Liu, J. Shi, Y. Chen and C.-F. Ke, Angew. Chem., Int. Ed., 2008, 47, 7293 CrossRef PubMed.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814 CrossRef PubMed.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef PubMed.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef PubMed.
- N. J. Turro, Acc. Chem. Res., 2000, 33, 637 CrossRef PubMed.
- G. Feng, P. Cheng, W. Yan, M. Boronat, X. Li, J.-H. Su, J. Wang, Y. Li, A. Corma, R. Xu and J. Yu, Science, 2016, 351, 6278 CrossRef PubMed.
- J. M. Stauber, P. Muller, Y. Dai, G. Wu, D. G. Nocera and C. C. Cummins, Chem. Sci., 2016, 7, 6928 RSC.
- B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. De Cola, Angew. Chem., Int. Ed., 2010, 49, 6881 CrossRef PubMed.
- W. R. Leow, W. K. H. Ng, T. Peng, X. Liu, B. Li, W. Shi, Y. Lum, X. Wang, X. Lang, S. Li, N. Mathews, J. W. Ager, T.
C. Sum, H. Hirao and X. Chen, J. Am. Chem. Soc., 2017, 139, 269 CrossRef PubMed.
- E. Breinlinger, A. Niemz and V. M. Rotello, J. Am. Chem. Soc., 1995, 117, 5379 CrossRef.
- R. G. Hicks, M. T. Lemaire, L. Öhrström, J. F. Richardson, L. K. Thompson and Z. Xu, J. Am. Chem. Soc., 2001, 123, 7154 CrossRef PubMed.
- N. Doslik, T. Sixt and W. Kaim, Angew. Chem., Int. Ed., 1998, 37, 2403 CrossRef PubMed.
- M. S. Rodríguez-Morgade, T. Torres, C. Atienza-Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145 CrossRef PubMed.
- M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559 CrossRef PubMed.
- Y. Jiao, J.-F. Xu, Z. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22635 Search PubMed.
- W. Ong, M. Gómez-Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791 CrossRef PubMed.
- W. S. Jeon, H.-J. Kim, C. Leeb and K. Kim, Chem. Commun., 2002, 1828 RSC.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384 CrossRef PubMed.
- C. R. Benson, E. M. Fatila, S. Lee, M. G. Marzo, M. Pink, M. B. Mills, K. E. Preuss and A. H. Flood, J. Am. Chem. Soc., 2016, 138, 15057 CrossRef PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382 CrossRef PubMed.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. d. Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394 RSC.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052 CrossRef PubMed.
- C. Stoffelen and J. Huskens, Chem. Commun., 2013, 49, 6740 RSC.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899 RSC.
- J. Yao, Y. Cheng, M. Zhou, S. Zhao, S. Lin, X. Wang, J. Wu, S. Li and H. Wei, Chem. Sci., 2018, 9, 2927 RSC.
- S. Das, P. V. Kamat, S. Padmaja, V. Au and S. A. Madison, J. Chem. Soc., Perkin Trans. 2, 1999, 1219 RSC.
- Y. H. Ko, H. Kim, Y. Kim and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 4106 CrossRef PubMed.
- L. C. Smith, D. G. Leach, B. E. Blaylock, O. A. Ali and A. R. Urbach, J. Am. Chem. Soc., 2015, 137, 3663 CrossRef PubMed.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318 CrossRef PubMed.
- F. H. Burkitt, C. A. Coulson and H. C. Longuet-Higgins, Trans. Faraday Soc., 1951, 47, 553 RSC.
- C. A. Coulson, Discuss. Faraday Soc., 1947, 2, 9 RSC.
- K. Griesbaum, Angew. Chem., Int. Ed., 1970, 9, 273 CrossRef.
- J. M. Tedder, Angew. Chem., Int. Ed., 1982, 21, 401 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01434e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*

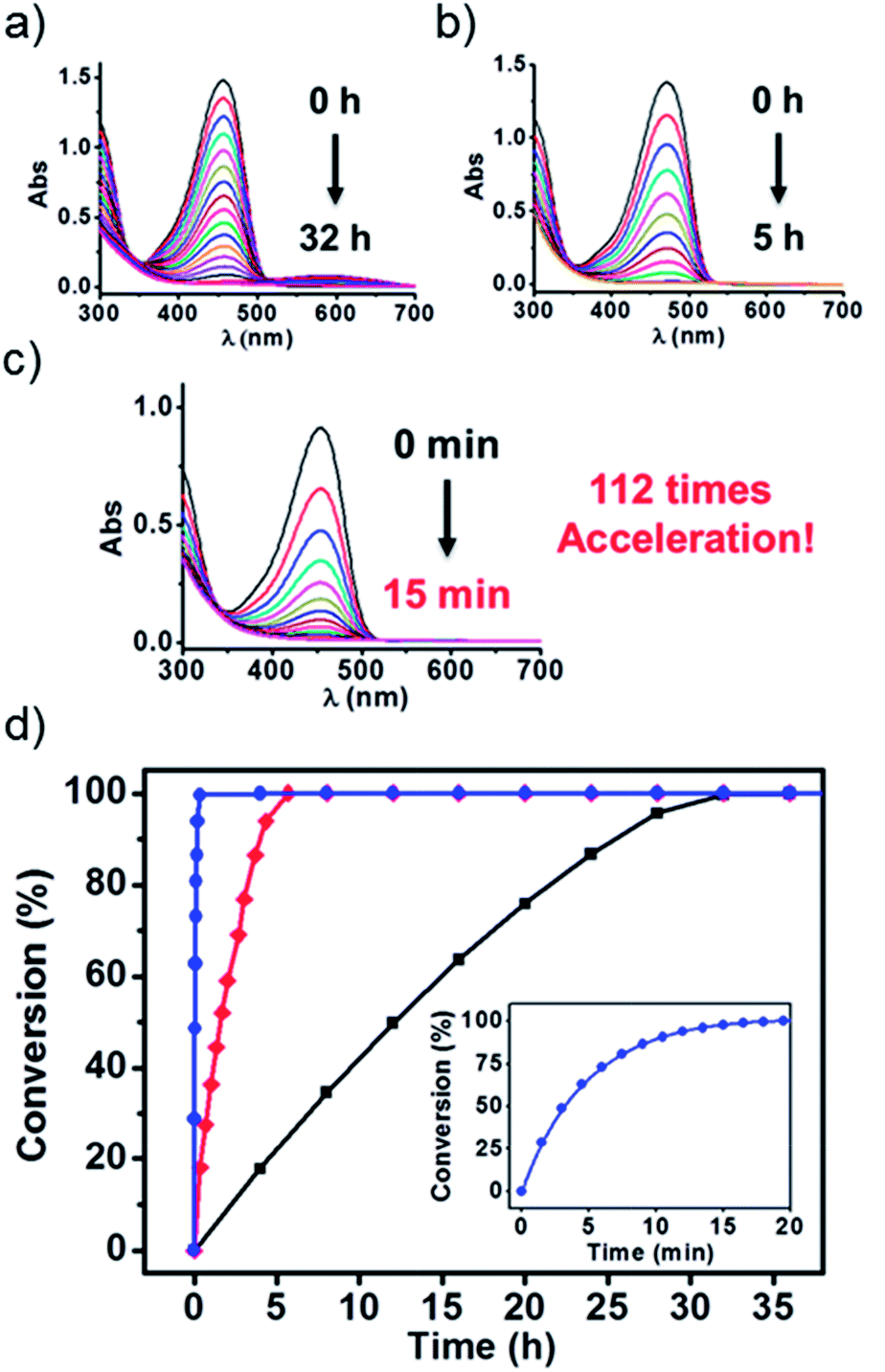
 ), DPP/(CB[7])2 (
), DPP/(CB[7])2 ( ) and DPP/(CB[8])2 (
) and DPP/(CB[8])2 ( ) at 25 °C. Inset: magnification of the time–conversion relationships of DPP/(CB[8])2.
) at 25 °C. Inset: magnification of the time–conversion relationships of DPP/(CB[8])2.