
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium catalyzed cascade cyclization featuring B–H and C–H activation: one-step construction of carborane-fused N-polyheterocycles†
Hairong
Lyu
,
Yangjian
Quan
 * and
Zuowei
Xie
*
* and
Zuowei
Xie
*
Department of Chemistry and State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong, China. E-mail: yjquan@cuhk.edu.hk; zxie@cuhk.edu.hk
First published on 29th June 2018
Abstract
A one-pot strategy for efficient and facile synthesis of C,B-substituted carborane-fused N-polyheterocycles is reported. A rhodium catalyzed cascade cyclization of carboranyl N-arylimines with vinyl ketones enables the effective construction of three new B–C and C–C bonds in one reaction. Both carboranyl B–H and aryl C–H bonds are sequentially activated, leading to a series of previously unavailable C,B-substituted carborane-fused cyclopenta[b]quinoline derivatives, for potential applications in pharmaceuticals and materials, in a step-economical manner. The successful isolation and structural identification of a key intermediate provide solid evidence for the reaction mechanism, involving a tandem sequence of regioselective B–H activation, alkene insertion, nucleophilic cyclization, C–H activation, nucleophilic cyclization, dehydration and oxidative aromatization.
Introduction
Modern chemistry strives for the generation of complex target molecules starting from readily available feedstocks. With the economical consideration related to resources, labour and time, synthetic protocols integrating multi-step procedures into a simple one-pot process have shown outstanding superiority. One important example is known as a cascade reaction, transforming simple starting materials into highly functionalized products without any isolation of the intermediates or alteration of the reaction conditions during the process, whose significance has been manifested by wide applications in the synthesis of bioactive pharmacophores and functional materials.1,2 Among these processes, transition metal catalyzed cascade cyclization has attracted growing research interest, as the development of metal catalyzed synthetic methodologies, including C–H activation has brought more and more diversities and possibilities in synthetic chemistry nowadays.3,4Icosahedral carboranes, a class of polyhedral boron–carbon molecular clusters, are often viewed as three-dimensional analogues to 2D-benzene.5 Their unique properties such as high boron content, variable electronic nature, and special σ-conjugation make them multifaceted building blocks in boron neutron capture therapy agents,6,7 pharmacophores,8,9 supramolecular design,10–13 nanomaterials,14–16 optoelectronics17–19 and organometallic/coordination chemistry.20–22 Recent research on incorporating a carborane moiety into π-conjugated molecules or replacing the phenyl/heterocyclic ring in known drugs by a carborane unit has provided a series of new optoelectronic materials17–19,23,24 and potent drug molecules.8,9,23,24 On the other hand, cyclopenta[b]quinoline scaffolds exist widely in natural products and pharmaceutical molecules, exhibiting valuable biological properties of antimalarial, anticancer and Alzheimer's disease inhibition.25–27 No hitherto reported methods are available to prepare carborane-fused cyclopenta[b]quinoline derivatives that may possess potential applications.
Recently, our group has reported transition-metal-catalyzed carboxylic-group-guided regioselective cage B–H alkenylation, arylation, alkynylation, amination, hydroxylation and halogenation of o-carborane.31–35,37,44 In view of recent advances in C–H activation4,28–30 and catalytic selective B–H functionalization of carboranes,31–46 we have combined both C–H/B–H activation in one reaction and report herein the first example of transition metal catalyzed cascade cyclization for one-pot synthesis of C,B-substituted carborane-fused cyclopenta[b]quinolines (Scheme 1).
 | ||
| Scheme 1 Synthesis of carborane-fused cyclopenta[b]quinoline. | ||
Results and discussion
During the course of our study on imine-guided cage B–H activation, we accidently discovered a Rh-catalyzed cascade reaction of carboranyl N-4-chlorophenylimine (1a) with 2-butenone (2a) to generate the unexpected C,B-substituted o-carborane-fused tricyclic (3a), in which one cage B–H and one sp2 C–H bond were activated, accompanied by the construction of three new B–C and C–C bonds (Table 1). Under the optimal reaction conditions, 3a was obtained in 80% NMR yield. The choice of suitable additives was important for achieving a high reaction yield. The absence of Cu(OPiv)2 or replacement of it by Cu(OAc)2 led to a dramatic decrease of the yield (entries 1 and 2, Table 1) and the changes of the organic acid or silver salt proved to be less effective (entries 3–6, Table 1). Screening of reaction temperatures did not offer better results (entries 7 and 8, Table 1). Other solvents such as toluene and 1,2-dichloroethane were not compatible (entries 9 and 10, Table 1). The [Ru(p-cymene)Cl2]2 catalyst gave 3a in 36% yield, while only a trace amount of the target product was observed using [Cp*IrCl2]2 as the catalyst (entries 11 and 12, Table 1). Lowering the catalyst loading to 2.5 mol% resulted in a reduced yield of 70% (entry 13, Table 1). Reducing the amount of 2-butenone also decreased the yield of 3a (entries 14 and 15, Table 1).|
|
||
|---|---|---|
| Entry | Variations from the ‘standard’ conditions | Yield of 3a (%) |
| a Reaction conditions: 1a (0.05 mmol) and 2a (0.25 mmol) in 1.5 mL of solvent under argon in a closed flask; 1,4-DCB = 1,4-dichlorobutane; Cu(OPiv)2 = copper pivalate; AgSbF6 = silver hexafluoroantimonate(V); MesCOOH = 2,4,6-trimethylbenzoic acid; PivOH = pivalic acid; AgNTf2 = silver bis(trifluoromethanesulfonyl)imide; DCE = 1,2-dichloroethane; [Ir] = [Cp*IrCl2]2; [Ru] = [Ru(p-cymene)Cl2]2. Yield determined by 1H NMR spectroscopy using dibromomethane as an internal standard. | ||
| 1 | Without Cu(OPiv)2 | Trace |
| 2 | Cu(OAc)2 instead of Cu(OPiv)2 | 17 |
| 3 | Without MesCOOH | 68 |
| 4 | PivOH instead of MesCOOH | 74 |
| 5 | AgSbF6 (0.5 equiv.) | 53 |
| 6 | AgNTf2 instead of AgSbF6 | 9 |
| 7 | 80 °C instead of 90 °C | 34 |
| 8 | 100 °C instead of 90 °C | 74 |
| 9 | DCE instead of 1,4-DCB | Trace |
| 10 | Toluene instead of 1,4-DCB | — |
| 11 | [Ir] instead of [Rh] | Trace |
| 12 | [Ru] instead of [Rh] | 36 |
| 13 | [Rh] (2.5 mol%) | 70 |
| 14 | 2-Butenone (1.0 equiv.) | 33 |
| 15 | 2-Butenone (3.0 equiv.) | 62 |
| 16 | Cu(OPiv)2 (1.0 equiv.) | 49 |
| 17 | Cu(OPiv)2 (2.0 equiv.) | 71 |
| 18 | Under air | 39 |
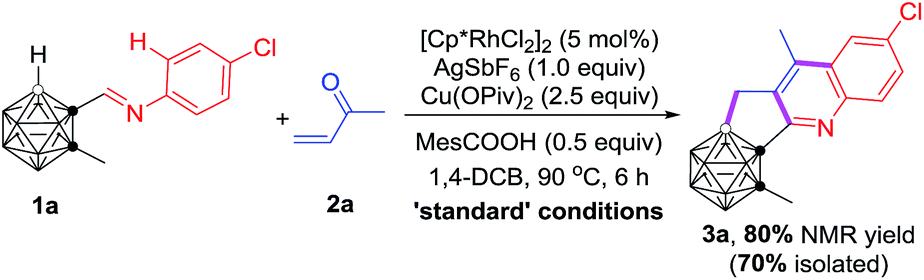
With the optimal reaction conditions in hand, the substrate scope of such cascade cyclization was subsequently examined (Table 2). A series of substituents at cage C(2) gave the corresponding products 3a–g in 64–74% isolated yields. The effects of the substituents of the phenyl ring on the reaction results were also evaluated. The chloro group at the meta-position of 1j afforded a 68% isolated yield of 3j, whereas the ortho-chlorinated substrate 1k afforded 3k in a much reduced yield of 39%, due probably to steric effects. Various functional groups at the para-position in 1 were compatible with this reaction, leading to 3l–r in moderate to good isolated yields. No obvious electronic effect was observed. Substrates with two functional groups on the phenyl ring gave very comparable yields (3s–v). It was noteworthy that such Rh-catalyzed cascade cyclization was tolerant of different halo groups, which could be readily used for further transformations. Naphthalene- and anthracene-containing substrates also worked, affording C,B-substituted carborane-fused polycyclic aromatics (3w–y), which may find valuable applications in materials science. Other vinyl ketones were also tested, and the corresponding products 3h, 3i and 3z were isolated in 63%, 35% and 38% yield, respectively, indicating that larger substituents reduced the yields of 3 probably because of steric reasons.
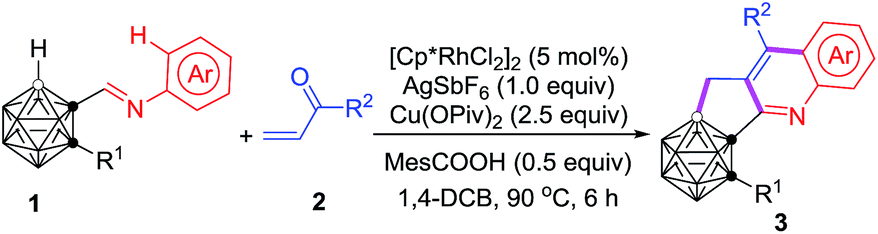
Compound 3m can be further functionalized through its C–Br bond (Scheme 2). Buchwald–Hartwig cross-coupling of 3m with carbazole in the presence of 5 mol% Pd2(dba)3 (dba = dibenzylideneacetone), 8 mol% PtBu3 and 4.5 equiv. of LiOtBu gave the corresponding product 4m in 95% isolated yield. With a catalytic system of 5 mol% PdCl2(PPh3)2 and 10 mol% CuI, Sonogashira coupling of 3m with phenylacetylene afforded the alkynylated product 5m in a yield of 96%. A Pd(dppf)Cl2 (dppf = 1,1′-bis(diphenylphosphino)ferrocene) catalyzed borylation of 3m with B2pin2 (pin = pinacolato) generated 6m in 89% isolated yield. In the presence of 10 mol% PdCl2(PPh3)2 and 4.0 equiv. of K2CO3, the Heck reaction of 3m with styrene offered 7m in 70% isolated yield. In addition, the homo-coupling product 8m was readily prepared in 83% isolated yield by treatment of 3m with 1.0 equiv. of the Ni(0) complex in situ generated from NiCl2 and Zn.
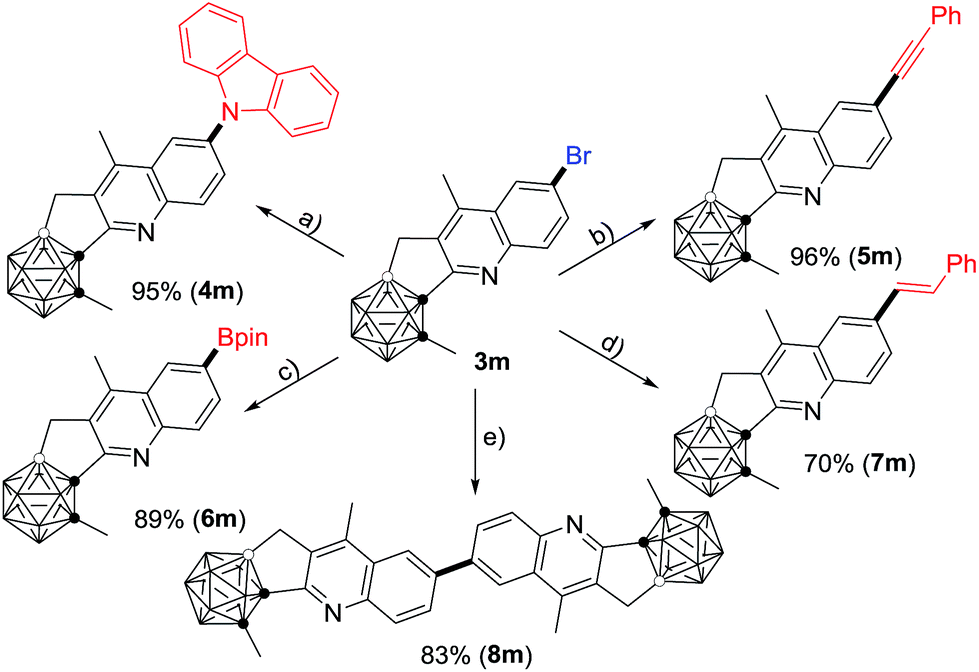 | ||
| Scheme 2 Transformations of 3m. (a) Carbazole (1.2 equiv.), Pd2(dba)3 (5 mol%), P(tBu)3 (8 mol%), LiOtBu (4.5 equiv.), o-xylene, 140 °C, 24 h; (b) phenylacetylene (1.2 equiv.), PdCl2(PPh3)2 (5 mol%), CuI (10 mol%), Et3N (5.0 equiv.), DMF, 60 °C, 16 h; (c) B2pin2 (1.1 equiv.), Pd(dppf)Cl2 (10 mol%), KOAc (3.0 equiv.), toluene, 90 °C, 18 h; (d) styrene (1.2 equiv.), PdCl2(PPh3)2 (10 mol%), K2CO3 (4.0 equiv.), toluene, 130 °C, 12 h; (e) NiCl2 (1.0 equiv.), PPh3 (4.0 equiv.), Zn (1.0 equiv.), DMF, 50 °C, 12 h. | ||
Compounds 1, 3 and 4–8m were fully characterized by 1H, 13C, and 11B NMR spectroscopy as well as high-resolution mass spectrometry (see the ESI for detail†). The molecular structures of 3l and 3w were further confirmed by single-crystal X-ray analyses.
To gain some insight into the reaction mechanism, several control experiments were carried out (Scheme 3). Quenching the reaction under conditions shown in Scheme 3a after 30 min led to the isolation of an intermediate D in 28% yield, and 3a in 15% yield. The molecular structure of D was identified by single-crystal X-ray analysis and various spectroscopic data. The isolation of D clearly indicated that the carboranyl B–H bond was preferentially activated over the aryl C–H bond. Under the optimal reaction conditions, compound D was converted to product 3a in 95% NMR yield (Scheme 3b). In contrast, a trace amount of 3a was observed in the absence of the Rh-catalyst (Scheme 3c), which suggested that the C–H activation is most likely initiated by the reactive Rh(III) center, although the direct Friedel–Crafts cyclization pathway cannot be absolutely ruled out. The absence of silver salt resulted in a trace amount of 3a (Scheme 3d), whereas a yield of 43% for 3a was obtained without the addition of the copper salt (Scheme 3e). Furthermore, 2 equiv. of AgSbF6 led to 3a in 84% yield in the absence of copper salt (Scheme 3f). These results showed that the silver salt may serve as the active oxidant for oxidative aromatization to afford the final product, meanwhile the copper salt is considered as the auxiliary oxidant. On the other hand, the measured KIE (KIE = kinetic isotope effect) value kH/kD = 1.7 indicated that the C–H activation may not be involved in the rate-determining step (Scheme 3g). The addition of 1.0 equiv. of Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 as a radical scavenger did not affect the reaction result (Scheme 3h), suggesting that such cascade cyclization may not involve a radical process.
CH2 as a radical scavenger did not affect the reaction result (Scheme 3h), suggesting that such cascade cyclization may not involve a radical process.
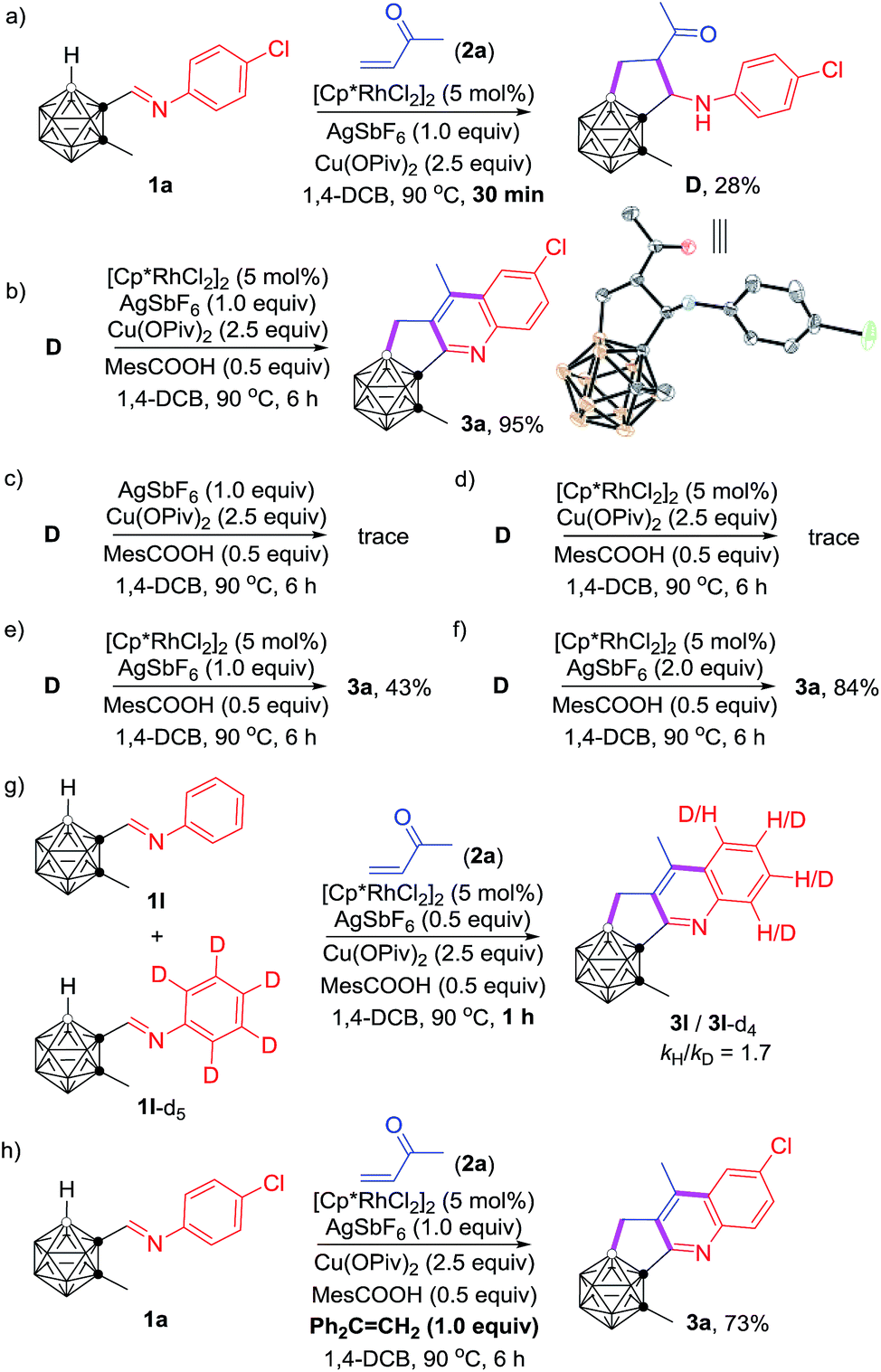 | ||
| Scheme 3 Mechanistic study. | ||
On the basis of the aforementioned control experiments, a plausible reaction mechanism is proposed in Scheme 4. Under the guidance of the imine directing group, electrophilic attack at the more electron-rich B(4/5)–H by the Rh(III) center generates a five-membered rhodacycle A.34 Alkene insertion into the cage B–Rh bond gives the intermediate B.37 Intramolecular nucleophilic cyclization47,48 of C–Rh with CN produces an intermediate C, which undergoes protonation to afford the intermediate D as well as regenerate the reactive Rh-catalyst. Subsequent Rh-mediated sp2 C–H activation29,30 occurs to form the eight-membered rhodacycle E. Intramolecular nucleophilic cyclization47,48 of C–Rh with CO affords the intermediate F. Protonation of F offers the intermediate G and the Rh-catalyst. Dehydration and oxidative aromatization of G give the final product 3. It was suggested that the formal oxidation state of Rh remained unchanged during the catalysis and the counterion may be PivO− or SbF6−.
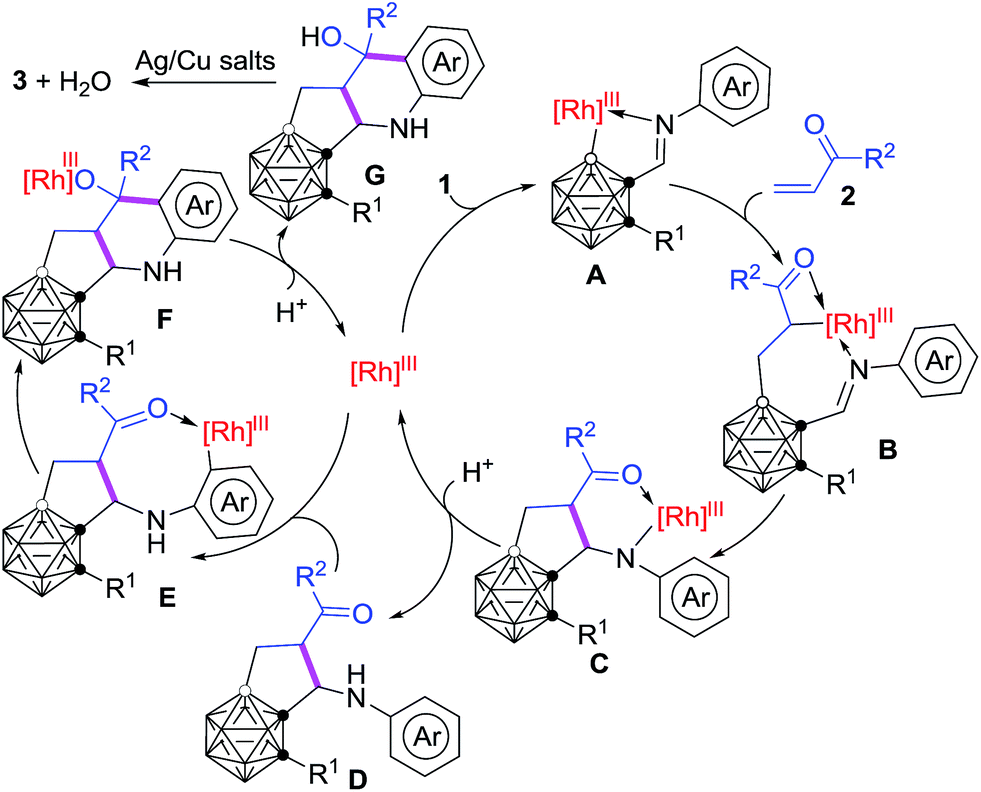 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, a regioselective and efficient Rh(III)-catalyzed cascade cyclization of carboranyl N-arylimines with vinyl ketones has been achieved, leading to the facile synthesis of a wide variety of C,B-substituted carborane-fused N-polyheterocycles, which cannot be prepared by any other methods. In a simple one-pot process, cage B–H and aryl C–H bonds are activated compatibly along with the formation of three new B–C and C–C bonds. This work represents the first example of transition metal catalyzed cascade one-pot construction of polycycles in carborane chemistry, which may also provide useful reference for crafting the quinoline framework in organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Research Grants Council of The Hong Kong Special Administration Region (Project No. 14304115 to Z. X. and 14305017 to Y. Q.) and NSFC/RGC Joint Research Scheme (N_CUHK442/14 to Z. X.).Notes and references
- Domino Reactions in Organic Synthesis, ed. L. F. Tietze, G. Brasche and K. M. Gericke, Wiley-VCH, Weinheim, 2006 Search PubMed.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef PubMed.
- Catalytic Cascade Reactions, ed. P.-F. Xu and W. Wang, Wiley-VCH, Weinheim, 2013 Search PubMed.
- Topics in Current Chemistry: C-H Activation, ed. J.-Q. Yu and Z.-J. Shi), Springer-Verlag, Berlin, 2010, vol. 292 Search PubMed.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191 CrossRef PubMed.
- M. F. Hawthorne, Angew. Chem., Int. Ed., 1993, 32, 950 CrossRef.
- M. F. Hawthorne and A. Maderna, Chem. Rev., 1999, 99, 3421 CrossRef PubMed.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701 CrossRef PubMed.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035 CrossRef PubMed.
- H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131 CrossRef PubMed.
- M. Koshino, T. Tanaka, N. Solin, K. Suenaga, H. Isobe and E. Nakamura, Science, 2007, 316, 853 CrossRef PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578 CrossRef PubMed.
- T. L. Chan and Z. Xie, Chem. Sci., 2018, 9, 2284 RSC.
- C. J. Villagómez, T. Sasaki, J. M. Tour and L. Grill, J. Am. Chem. Soc., 2010, 132, 16848 CrossRef PubMed.
- (a) E. Q. Qian, A. I. Wixtrom, J. C. Axtell, A. Saebi, P. Rehak, Y. Han, E. H. Moully, D. Mosallaei, S. Chow, M. Messina, J.-Y. Wang, A. T. Royappa, A. L. Rheingold, H. D. Maynard, P. Kral and A. M. Spokoyny, Nat. Chem., 2017, 9, 333 CrossRef PubMed; (b) A. C. Serino, M. E. Anderson, L. M. A. Saleh, R. M. Dziedzic, H. Mills, L. K. Heidenreich, A. M. Spokoyny and P. S. Weiss, ACS Appl. Mater. Interfaces, 2017, 9, 34592 CrossRef PubMed.
- A. Saha, E. Oleshkevich, C. Viñas and F. Teixidor, Adv. Mater., 2017, 29, 1704238 CrossRef PubMed.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070 RSC.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307 CrossRef PubMed.
- X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888 CrossRef PubMed.
- Z. Xie, Acc. Chem. Res., 2003, 36, 1 CrossRef PubMed.
- Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522 CrossRef.
- Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299 CrossRef PubMed.
- Carboranes, ed. R. N. Grimes, Elsevier, Oxford, UK, 3rd edn, 2016 Search PubMed.
- Boron Science: New Technologies and Applications, ed. N. S. Hosmane, Taylor & Francis Books/CRC, Boca Raton, FL, 2011 Search PubMed.
- J. M. Sanders, D. P. Clifford and R. E. Lutz, J. Med. Chem., 1971, 14, 1126 CrossRef PubMed.
- L. W. Deady, J. Desneves, A. J. Kaye, G. J. Finlay, B. C. Baguley and W. A. Denny, Bioorg. Med. Chem., 2000, 8, 977 CrossRef PubMed.
- M. Fernández, M. C. Carreiras, J. L. Marco and J. Caballero, J. Enzyme Inhib. Med. Chem., 2006, 21, 647 CrossRef PubMed.
- R. H. Crabtree and A. Lei, C-H Activation Special Issue, Chem. Rev., 2017, 117, 8481 CrossRef PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef PubMed.
- (a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef; (c) M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef PubMed.
- Y. Quan and Z. Xie, J. Am. Chem. Soc., 2014, 136, 15513 CrossRef PubMed.
- Y. Quan, C. Tang and Z. Xie, Chem. Sci., 2016, 7, 5838 RSC.
- Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 1295 CrossRef PubMed.
- H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 11840 CrossRef PubMed.
- H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2016, 138, 12727 CrossRef PubMed.
- R. Cheng, Z. Qiu and Z. Xie, Nat. Commun., 2017, 8, 14827 CrossRef PubMed.
- H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 10623 CrossRef PubMed.
- (a) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257 RSC; (b) K. Cao, T.-T. Xu, J. Wu, L. Jiang and J. Yang, Chem. Commun., 2016, 52, 11446 RSC.
- (a) Y. Zhang, Y. Sun, F. Lin, J. Liu and S. Duttwyler, Angew. Chem., Int. Ed., 2016, 55, 15609 CrossRef PubMed; (b) F. Lin, Y. Shen, Y. Zhang, Y. Sun, J. Liu and S. Duttwyler, Chem.–Eur. J., 2018, 24, 551 CrossRef PubMed.
- M. G. L. Mirabelli and L. G. Sneddon, J. Am. Chem. Soc., 1988, 110, 449 CrossRef.
- (a) X. Zhang, H. Zheng, J. Li, F. Xu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2017, 139, 14511 CrossRef PubMed; (b) X. Zhang and H. Yan, Chem. Sci., 2018, 9, 3964 RSC.
- R. M. Dziedzic, J. L. Martin, J. C. Axtell, L. M. A. Saleh, T.-C. Ong, Y.-F. Yang, M.-S. Messina, A. L. Rheingold, K. N Houk and A. M. Spokoyny, J. Am. Chem. Soc., 2017, 139, 7729 CrossRef PubMed.
- B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, J. Am. Chem. Soc., 2016, 138, 10531 CrossRef PubMed.
- Y. Quan, Z. Qiu and Z. Xie, Chem.–Eur. J., 2018, 24, 2795 CrossRef PubMed.
- (a) W.-B. Yu, P.-F. Cui, W.-X. Gao and G.-X. Jin, Coord. Chem. Rev., 2017, 350, 300 CrossRef; (b) B. J. Eleazer and D. V. Peryshkov, Comments Inorg. Chem., 2018 DOI:10.1080/02603594.2018.1465939.
- X. Zhang and H. Yan, Coord. Chem. Rev., 2017 DOI:10.1016/j.ccr.2017.11.006.
- F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef PubMed.
- Z. Shi, C. Grohmann and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5393 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, complete characterization data, NMR spectra, and CCDC numbers for 3l, 3w and D. CCDC 1823584–1823586. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01568f |
| This journal is © The Royal Society of Chemistry 2018 |