
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDewetting transitions coupled to K-channel activation in cytochrome c oxidase†
Shreyas
Supekar
and
Ville R. I.
Kaila
 *
*
Department Chemie, Technische Universität München, Lichtenbergstraße 4, D-85748 Garching, Germany. E-mail: ville.kaila@ch.tum.de
First published on 9th July 2018
Abstract
Cytochrome c oxidase (CcO) drives aerobic respiratory chains in all organisms by transducing the free energy from oxygen reduction into an electrochemical proton gradient across a biological membrane. CcO employs the so-called D- and K-channels for proton uptake, but the molecular mechanism for activation of the K-channel has remained elusive for decades. We show here by combining large-scale atomistic molecular simulations with graph-theoretical water network analysis, and hybrid quantum/classical (QM/MM) free energy calculations, that the K-channel is activated by formation of a reactive oxidized intermediate in the binuclear heme a3/CuB active site. This state induces electrostatic, hydration, and conformational changes that lower the barrier for proton transfer along the K-channel by dewetting pathways that connect the D-channel with the active site. Our combined results reconcile previous experimental findings and indicate that water dynamics plays a decisive role in the proton pumping machinery in CcO.
Introduction
Cytochrome c oxidase (CcO) functions as the terminal electron acceptor in respiratory chains of mitochondria and aerobic bacteria.1,2 CcO catalyses the reduction of oxygen to water and it employs the released free energy to pump four protons across the mitochondrial inner membrane or the bacterial cytoplasmic membrane.1,2 This establishes a proton motive force that is utilized to thermodynamically drive ATP synthesis and active transport.3CcO receives electrons from the soluble cytochrome c, and transfers them to oxygen, which is reduced to water in the binuclear heme a3/CuB active site (binuclear centre, BNC; Fig. 1A). Reduction of the BNC is coupled with uptake of protons via the D- and K-channels, named after the conserved Asp-91 and Lys-319 residues (bovine numbering is used if not otherwise stated, Fig. 1A). All four pumped protons and two chemical protons used for the oxygen reduction chemistry in the oxidative half of the catalytic cycle (Fig. 1, PM → OH) originate from the D-channel. In contrast, the K-channel is activated in the reductive half of the catalytic cycle (OH → R), and it is employed for the uptake of two chemical protons (Fig. 1). Despite extensive experimental4–8 and computational studies,9–14 the molecular mechanism for this channel-switching remains unclear.2,15,16 Interestingly, certain bacterial oxidases entirely lack the D-channel and utilize only their K-channel analogue to transfer both chemical and pumped protons.17–19
 | ||
| Fig. 1 Structure and function of cytochrome c oxidase (CcO). O2 reduction to H2O drives the electron transfers (blue arrow) to the binuclear centre (BNC: heme a3/CuB) and proton transfers (red arrow) to the BNC for oxygen chemistry and for pumping of protons to the positively charged (P) side across the membrane. Protons are taken up from the negatively charged (N) side of the membrane through conserved D- and K-channels, terminating at Glu-242 and Tyr-244, respectively. Water molecules occupying the channels are shown as red spheres. Hemes are shown in purple and the copper centres are depicted in orange. Inset: close-up of the structure and proton transfer (pT)/electron transfer (eT) pathways between Glu-242, Lys-319, the proton loading site (PLS),20–27 heme a, and heme a3/CuB. (B) Catalytic cycle of CcO showing the BNC and the catalytic intermediates. Protons (H+) marked in blue and red refer to chemical protons taken up from the D- and K-channels, respectively. Protons pumped across the membrane are indicated in black. | ||
The catalytic cycle of CcO is initiated from the reduced R state (aII3/CuI/Tyr-OH) that binds and splits dioxygen to yield the PM state (aIV3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2−/CuII–OH−/Tyr-O˙) (Fig. 1B). Subsequent reduction of the BNC results in the PR state (aIV3O2−/CuII–OH−/Tyr-O−),2 which is coupled with transfer of a (pumped) proton from the conserved Glu-242 to a transient proton-loading site (PLS), located in the vicinity of heme a3.20–28 Protonation of the PLS leads to transfer of a second (chemical) proton from Glu-242 to the BNC, which ejects the PLS proton to the positively charged side (P-side) of the membrane and yields the F state (aIV3O2−/CuII–H2O/Tyr-O−).2 Further reduction along with internal proton reorganization within the BNC yields the FR state (aIII3–OH−/CuII–OH−/Tyr-O−),29 which upon protonation decays to the oxidized OH state, following a similar sequence as in the PM → F transition.
O2−/CuII–OH−/Tyr-O˙) (Fig. 1B). Subsequent reduction of the BNC results in the PR state (aIV3O2−/CuII–OH−/Tyr-O−),2 which is coupled with transfer of a (pumped) proton from the conserved Glu-242 to a transient proton-loading site (PLS), located in the vicinity of heme a3.20–28 Protonation of the PLS leads to transfer of a second (chemical) proton from Glu-242 to the BNC, which ejects the PLS proton to the positively charged side (P-side) of the membrane and yields the F state (aIV3O2−/CuII–H2O/Tyr-O−).2 Further reduction along with internal proton reorganization within the BNC yields the FR state (aIII3–OH−/CuII–OH−/Tyr-O−),29 which upon protonation decays to the oxidized OH state, following a similar sequence as in the PM → F transition.
The OH → EH transition is also believed to follow a similar sequence as the PM → F and F → OH transitions, but instead, the K-channel is employed for uptake of a chemical proton in the one-electron reduced OH state (here the OH,R, aIII3–OH−/CuI–H2O Tyr-O−).15,20,30 This yields the EH state,8,31 which by reduction and further protonation from the K-channel yields the R state, completing the catalytic cycle. The overall pumping cycle thus results in consumption of four electrons from the P-side, four protons from the N-side, and one oxygen molecule, producing two water molecules and pumping of four protons across the membrane from the N-side to the P-side.2
In addition to the pumping OH state, CcO can also relax into a resting oxidized O state, but in contrast to the former, the reduction of the O state does not couple with proton pumping across the membrane.32 The exact molecular structure of this low-potential O form is unclear and it is spectroscopically indistinguishable from the activated OH state.33 Based on computational studies,9,11,34 it has been suggested that the high and low potential forms of the O state could be connected to the electronic equilibrium between CuB (CuII/I) and Tyr-244 (Tyr-O˙/Tyr-O−) that is modulated by the ligand state of CuB (H2O/OH−). We show here that this unique CuII/Tyr-O− state is important for regulating the hydration state of the channels.
The K-channel starts at residue Glu-62 (subunit II) at the N-side of the membrane and leads via Lys-319 and Thr-316 to the cross-linked Tyr-244 of CuB, providing a proton conduit from the negatively charged side (N-side) to the BNC (Fig. 1A).15,35 Mutation of Lys-319 drastically slows down the reduction of the fully oxidized state, whereas oxidation of the fully reduced state remains unaffected. This suggests that the K-channel provides chemical protons in the reductive half of the catalytic cycle.4,6 Interestingly, electrometric studies of that K319M mutant suggest that reduction of the BNC is coupled with an electrogenic charge movement within the K-channel31 that could arise from flipping Lys-319 towards the BNC, with the positive charge density facilitating the reduction of the BNC.
Molecular dynamics (MD) simulations of the K-channel have highlighted the role of hydrogen-bonded water molecules that are important for proton transfer reactions.10,14 Recent computational voltage studies36 have reinforced the Lys-319 “up”-flip assignment from electrometric studies31 that probe voltage changes associated with charge movement along the membrane axis. Moreover, continuum electrostatics calculations based on the X-ray structures of the oxidized and reduced forms of CcO suggested that Lys-319 is in its protonated form, whereas pKa calculations based on MD structures suggested that Lys-319 is deprotonated throughout the catalytic cycle, except in the one-electron reduced states of the BNC (e.g. PR), where Lys-319 was predicted to be protonated.13,14 Quantum chemical calculations also suggest that Lys-319 might modulate the pKa of Tyr-244.9,11,12,34 However, despite these important insights, the molecular mechanism behind the K-channel activation process still remains elusive.
In order to probe the K-channel activation process, we study here the P → F and OH → EH transitions (Fig. 1B) using multi-scale quantum and classical molecular simulations. To this end, we performed classical MD simulations of CcO embedded in a biological membrane environment on the microsecond timescales of each catalytic state, with the enzyme redox-states modelled based on quantum chemical calculations. Based on the MD simulations, we further characterized water networks connecting the D- and K-channels with the BNC using a graph-theoretical approach, and computed free energy profiles for the proton transfer process using DFT-based quantum mechanics/classical mechanics (QM/MM) free energy simulations.
Computational models
Classical MD simulations
The X-ray structure of subunits I–II bovine CcO [PDB ID: 1V54 (ref. 37)] was embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane and was solvated with TIP3P water to yield a lipid-membrane-water-ion environment. The BNC was modelled in the PM (aIV3O2−/CuII–OH−/Tyr-O˙), PR (aIV3O2−/CuII–OH−/Tyr-O−), OH (aIII3–OH−/CuII–OH2/Tyr-O−) and OH,R (aIII3–OH−/CuI–OH2/Tyr-O−) states using the CHARMM36 force field52a and in-house DFT based parameters of the co-factors.52b 500 ns MD simulations of each state were performed with NAMD 2.9–2.12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 at T = 310 K using a 2 fs integration timestep, and by treating long-range electrostatics using the particle mesh Ewald approach. To probe the protonic connectivities from Glu-242 and Lys-319 to the BNC, we calculated the shortest water-mediated pathway using Dijkstra's algorithm,39 and evaluated the longest (“rate-limiting”) distance, ζ, along the shortest path (see the ESI and ESI-Fig. 13†). The employed protonation states are given in ESI-Table 2† and also described in the extended Methods section of the ESI.† Notably, Lys-319 was modelled in both protonated (Fig. 2) and deprotonated (neutral, ESI-Fig. 10†) states, and Glu-242 and Asp-364 in their protonated states (see the ESI†). Continuum electrostatic titration profiles were obtained by solving the linearized Poisson–Boltzmann equation with MEAD.40 To this end, the protein was described using partial atomic charges, embedded in an inhomogeneous dielectric continuum with dielectric constants of ε = 4 for the protein, a dielectric membrane slab with ε = 2, and ε = 80 for the water. The probe radius was 1.4 Å and the ionic strength was 100 mM potassium chloride. In the PB calculations, all internal protein-bound water molecules were replaced by a polarizable medium with ε = 80. Monte-Carlo pKa evaluations based on the MD trajectories were performed with Karlsberg.41 The solvent accessible volume in the K-channel was calculated using Hollow.42
38 at T = 310 K using a 2 fs integration timestep, and by treating long-range electrostatics using the particle mesh Ewald approach. To probe the protonic connectivities from Glu-242 and Lys-319 to the BNC, we calculated the shortest water-mediated pathway using Dijkstra's algorithm,39 and evaluated the longest (“rate-limiting”) distance, ζ, along the shortest path (see the ESI and ESI-Fig. 13†). The employed protonation states are given in ESI-Table 2† and also described in the extended Methods section of the ESI.† Notably, Lys-319 was modelled in both protonated (Fig. 2) and deprotonated (neutral, ESI-Fig. 10†) states, and Glu-242 and Asp-364 in their protonated states (see the ESI†). Continuum electrostatic titration profiles were obtained by solving the linearized Poisson–Boltzmann equation with MEAD.40 To this end, the protein was described using partial atomic charges, embedded in an inhomogeneous dielectric continuum with dielectric constants of ε = 4 for the protein, a dielectric membrane slab with ε = 2, and ε = 80 for the water. The probe radius was 1.4 Å and the ionic strength was 100 mM potassium chloride. In the PB calculations, all internal protein-bound water molecules were replaced by a polarizable medium with ε = 80. Monte-Carlo pKa evaluations based on the MD trajectories were performed with Karlsberg.41 The solvent accessible volume in the K-channel was calculated using Hollow.42
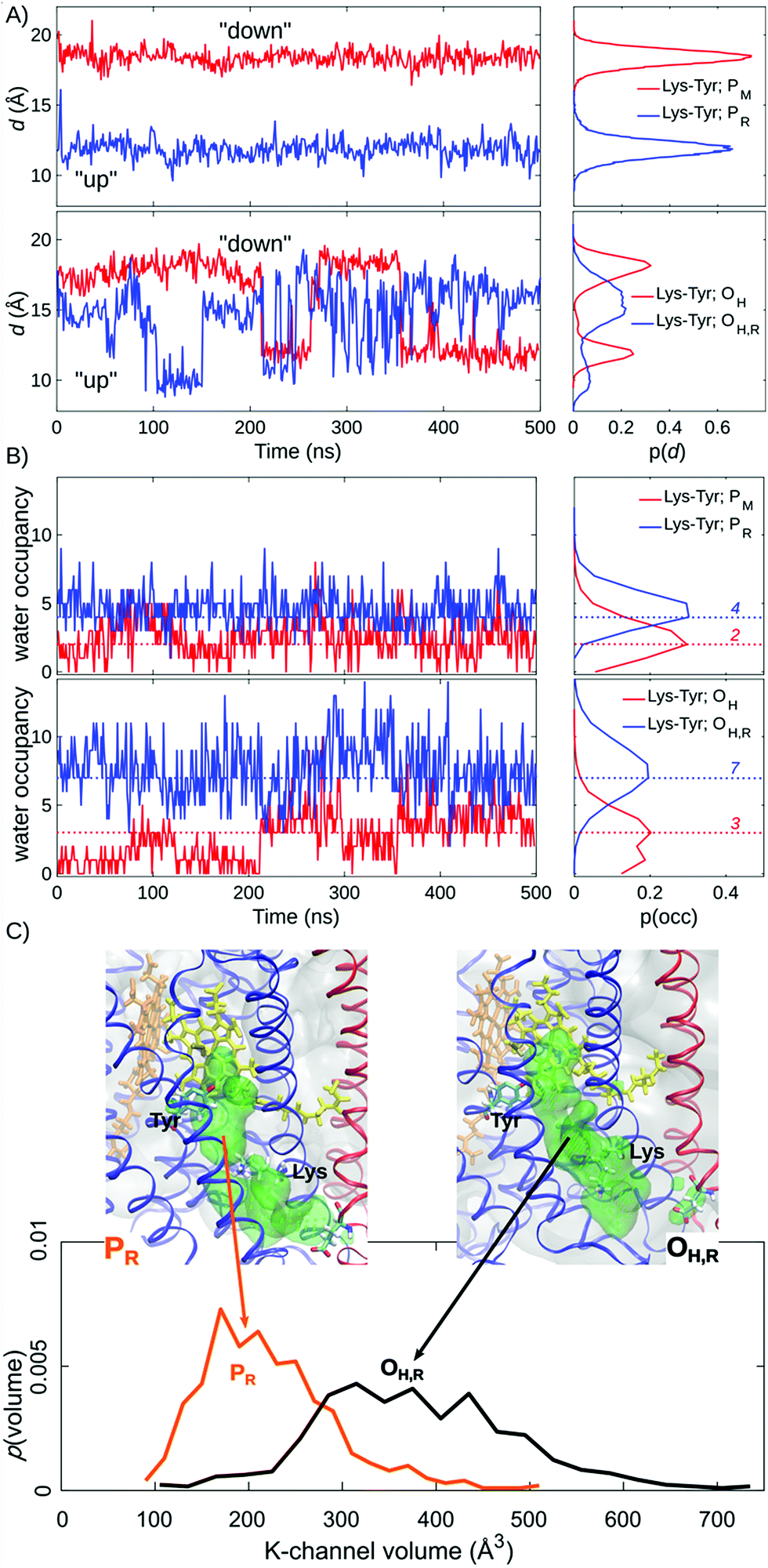 | ||
| Fig. 2 Lys-319 sidechain dynamics, water occupancy in the K-channel above Lys-319 and K-channel volume (in Å3) from 500 ns of MD simulations. (A) Distances between Lys-319 and Tyr-244 from simulations of PM, PR, OH, and OH,R states. (B) Water occupancies in the K-channel between Lys-319 and Tyr-244 from simulations of PM, PR, OH, and OH,R states. Modal water occupancies are indicated as dotted lines. (C) Water accessible volume in the K-channel for the PR and OH,R states. | ||
DFT calculations
Quantum chemical clusters were optimized at the D3-BP86 level43–45 using the multipole accelerated resolution of identity approximation (RI-MARIJ).46 The protein environment was modelled using a polarizable dielectric medium with ε = 4 using the conductor-like screening model (COSMO).47 The models included heme a3 (without the propionic groups), CuB, and Tyr-244, His-290, His-291, His-240, His-376, Thr-316 and Lys-319, as well as 10–15 water molecules obtained from the MD simulations (see below). The amino acids were cut at the Cβ-positions, which were fixed during the structure optimization to account for protein strain. Single point energy calculations of the optimized structures were performed using the D3-B3LYP functional.43,48,49 In all models, def2-TZVP (Fe, Cu) and def2-SVP basis sets (C, H, O, N) were employed for geometry optimizations, while def2-TZVP basis sets were employed for single point energy calculations.50 The DFT calculations were performed with TURBOMOLE v. 6.6–7.1.51QM/MM free energies
In QM/MM calculations, link atoms were introduced between the Cα and Cβ carbons of the amino acids in the QM region comprising ca. 120–130 atoms, while the remaining system was treated classically with the classical CHARMM36 force field.52a,b For the QM/MM reaction pathway optimization, the QM region was described with the D3-B3LYP functional43,48,49 and def2-TZVP (Fe, Cu)/def2-SVP(C, H, O, N) basis sets.50 QM/MM umbrella sampling (QM/MM US) simulations were performed with the D3-BP86 functional42–44 using the MARIJ approximation.46 For QM/MM US calculations, each restraint window along the reaction coordinate was simulated for 1 ps at T = 310 K. No cutoffs were employed in the electrostatic embedding scheme. Due to the high computational cost of DFT-based QM/MM free energy simulations, the dynamics sampling is shorter than in previous semi-empirical-QM/MM or reactive force field/MM calculations of CcO.22–25 Although the starting structures for the QM/MM simulations were obtained from the 500 ns MD simulations, we nevertheless expect large statistical uncertainties in the PMF due to the limited QM/MM-sampling. Our DFT-based QM/MM dynamics is not based on pre-parametrized potentials which is why we expect an energetically well-balanced description of chemical intermediates involved in the pT reactions to the BNC, which is challenging to accurately describe at the semi-empirical level. To sample the proton transfer path, the structures were subjected to harmonic restraints based on linear combination of all water O–H bond-distances between Glu-242 and CuB, and Lys-319 and Tyr-244 with a force constant of k = 500 kcal mol−1 Å−2 for QM/MM optimizations and k = 100 kcal mol−1 Å−2 for QM/MM MD US calculations. The CHARMM/TURBOMOLE53 interface was employed for the QM/MM calculations (see the ESI† for further simulation details).Results
Redox-state dependent lysine dynamics
Our MD simulations suggest that water molecules in the K-channel provide a hydrogen-bonded connectivity between Lys-319 and Tyr-244 via Thr-316, consistent with results from previous MD studies.10,12–14 In the PM state, the Lys-319 side-chain flips “down”, towards the N-side of the membrane, and remains in this conformation for the complete 500 ns simulation (Fig. 2A). In contrast, in the one-electron reduced PR state, Lys-319 flips “up” towards Tyr-244 (Fig. 2A). We observe that reduction of the BNC links to an average increase of two water molecules between Lys-319 and Tyr-244, suggesting that the charge state of the BNC might regulate the hydration of the proton channel (Fig. 2B). Similar electrostatic field effects have also been suggested to be involved in other redox-driven proton pumps, e.g., in respiratory complex I,54–56 and light-driven ion-pumps.57 The observed “up”-state of Lys-319 is also consistent with results from previous electrochemical experiments on the K319M mutant,31 suggesting that the “up” conformation can stabilize the reduced BNC, prior to its protonation, and also the oxidative phase where the K-channel is not employed for proton uptake.In the OH and OH,R intermediates, where the K-channel is employed for proton uptake,4,6,15,16,32 we find that the lysine side-chain flickers between the “up” and “down” conformations, suggesting that the flipping barrier is low (Fig. 2B, ESI-Fig. 1†). In these simulations, we also observe a significant increase in the water occupancy of the K-channel from four water molecules in the PR state to seven water molecules in the OH,R state (Fig. 2B, ESI-Fig. 2†), whereas the water occupancies in the PM and OH states resemble each other (Fig. 2). We also observe conformational changes in the tertiary structure of the surrounding helices VIII, IX, and X (subunit I), and helix II (subunit II), which make the K-channel wider to accommodate the increased hydration state (Fig. 2C, ESI-Fig. 3 and 4†) in the OH/H,R intermediates, but not in the PM/R intermediates. These findings are consistent with conformational changes reported in previous X-ray structures of CcO.58 Our pKa calculations at the Poisson–Boltzmann (PB) continuum electrostatics level with Monte Carlo (MC) sampling of possible protonation states suggest that Lys-319 remains predominantly protonated in the P and O states (ESI-Fig. 5,† but cf. also ref. 14). Moreover, based on experimental data from the OH → EH transition, it has been suggested that the Lys-319 “up” conformation might be a prerequisite for reduction of the BNC, which itself is coupled to PLS protonation.59 This further underpins the functional importance of Lys-319 in the proton pumping machinery of CcO.
Proton-pathway connectivities regulate the channel activity
The increased K-channel hydration, subsequent conformational changes, and flipping of Lys-319 suggest that the BNC might become more accessible from the K-channel upon formation of the OH,R state. In order to probe the water-mediated protonic networks in the D- and K-channels, we employed a graph-theoretical approach for evaluating protonic connectives along the water-mediated pathways, which are important in catalysing Grotthuss-type pT reactions in proteins.60 To this end, the longest (“rate-limiting”) distance along the shortest hydrogen-bonded pathway, ζ, connecting the proton donor (Lys-319 or Glu-242) and proton acceptor (CuB, heme a3, and Tyr-244) was evaluated over the MD trajectories (see the ESI†).The ζ-reaction coordinate (see the ESI†) employed here should be considered as a qualitative measure of the hydration state and connectivity between the donor and acceptor paths. Previous studies suggest that proton pathways are not directly linked to the proton transfer barriers.22–25 However, if the proton donor and acceptor groups are distant as in CcO, and not connected by a water chain, as indicated by a high ζ value >4–5 Å, the proton transfer barriers are also significantly higher.2 These observations are also consistent with recent reactive force field calculations by Liang et al.25
We observe a persistent hydrogen-bonded connectivity along the K-channel between Lys-319 and Tyr-244 in the PR, OH, and OH,R states with a high population of configurations with ζ < 2.5 Å, but no connectivity between Lys-319 and Tyr-244 in the simulations of the PM state, possibly due to the “down” conformation of Lys-319 (Fig. 3, ESI-Fig. 6†). In stark contrast, the connectivity from the terminal D-channel residue, Glu-242 to the BNC is persistent in the P states, whereas no hydrogen-bonded connectivity is observed between these sites in the OH or OH,R states (Fig. 3, ESI-Fig. 7†). The electric field originating from the PR state strongly increases the hydrogen-bonded connectivity between Glu-242 and CuB, consistent with previous studies,2,61 but we also sample hydrogen-bonded connections between Glu-242 and CuB in the PM state, suggesting that the orientation of the water chain alone is unlikely to serve as a gate for premature proton transfer reactions to the BNC. We also observe that the water occupancy of the non-polar cavity connecting Glu-242 with the BNC sharply drops in the OH,R state, which could explain the lack of connectivity, whereas the water occupancy increases in the PR state, favouring the formation of water wires (Fig. 3C, ESI-Fig. 7 and 8†). The free energy for non-polar cavity hydration near Glu-242 was recently probed by Son et al.62 for the Rhodbacter sphaeroides (Rs) CcO. In the PR state, our simulations predict a somewhat higher cavity hydration in comparison to the Rs-CcO simulations that might arise from modelling CcO from a different organism. Moreover, here we have not considered the PLS protonation21–26 or Glu-242 deprotonation64–67 reactions in the classical simulations, which may also affect the cavity hydration state.67 Although our study focuses on the connections between the D-/K-channels and the BNC, we note that hydrogen-bonded pathways to the PLS also remains open when 3–4 water molecules are present in the non-polar cavity (cf.ref. 21). The MD timescales employed here are longer than in previous work, but our cavity hydration profiles are nevertheless similar to previous simulations of the Bos taurus enzyme.2,21,61
 | ||
| Fig. 3 Longest non-bonded connectivity along the shortest path connecting the proton donor and proton acceptor, ζ, calculated using Dijkstra's algorithm in graph theory. (A) Water-mediated connectivity from Glu-242 to CuB in PM, PR, OH, and OH,R states. Thresholds for hydrogen-bonding connectivity are shown in magenta (2.5 Å) and yellow (4.0 Å) dashed lines. (B) Water-mediated connectivity from Lys-319 to Tyr-244 in PM, PR, OH, and OH,R states. (C) Water occupancy in the non-polar cavity above Glu-242 in PM, PR, OH, and OH,R states. | ||
The dehydration of the non-polar cavity in the OH states might arise from the weak aquo-ligand of CuB that is unable to fully stabilize the water pathway from Glu-242, an effect which is expected to be even more pronounced upon dissociation of the aquo-ligand. As discussed above, it should be noted that the exact structure of the OH state is still unclear, but previous studies have suggested that heme a3 is coordinated by a hydroxo ligand, and CuB has a trigonal planar geometry, weakly interacting with a water molecule.11,34,63 Moreover, FTIR experiments suggest that Tyr-244 is deprotonated in the OH state.30 Our calculations also suggest that the dominant pathway forms between Glu-242 and CuB, but not between Glu-242 and heme a3 (ESI-Fig. 7†), and also upon formation of the FR state, whose ζ profiles resemble those computed for the PR state (ESI-Fig. 9†). These findings thus indicate that the dewetting of the non-polar cavity between the D-channel terminus (Glu-242) and the BNC in the reductive phase could trigger the activation of the K-channel.
To address the dynamics of Lys-319 after pT to the BNC, we performed additional 500 ns MD simulations with Lys-319 in its neutral form, starting from the MD simulations of the PR state with Lys-319 flipped towards the BNC. We find that in the neutral state, Lys-319 flips “down” towards the N-side of the membrane in the one-electron reduced states, PR and OH,R, whereas there is an increased population in the “up”-flipped population in the PM and OH states (ESI-Fig. 10A†). Interestingly, we also observe a strong reduction of the K-channel hydration in all four states with the neutral lysine, suggesting that the charged state of Lys-319 might modulate the hydration state of the K-channel (ESI-Fig. 10B†), similar to that in complex I54–56 and bacterial ion-pumps.57 The sidechain dynamics of the neutral Lys-319 and the consequent loss of a hydrogen-bonded connectivity could help in preventing the chemical proton from leaking backwards towards the N-side. This could increase the overall efficiency of the pump, similar to our previously proposed glutamate-gate that might prevent leaks along the D-channel64 (cf. also ref. 65–67). The observed hydration/dehydration effects in the K-channel might be further linked to the two experimentally known forms of the oxidized state: the “resting”, non-pumping O state, and the physiologically relevant, pumping OH state. As discussed above, these states are spectroscopically indistinguishable,33 but it is known that the resting O state can be activated by reduction and re-oxidation to yield the activated OH state. This leads to synthesis of new water molecules in the BNC, which could in turn lead to hydration of the D- and K-channels, and the transient proton-loading site to sustain the proton pumping function.21 Moreover, wetting/dewetting transitions in the non-polar cavity above Glu-242 have been shown to play a key role in the proton pumping machinery of CcO in the P → F transition.62,67
Energetics of proton transfer from the K-channel
In order to probe the energetics for the pT process, we constructed quantum chemical models with ca. 160–200 atoms, comprising the BNC and its nearby surroundings based on the MD structures. The models were optimized at the density functional theory (DFT) level. The calculations suggest that transferring the proton from Lys-319 to Tyr-244 in the OH,R state is stabilized by ca. 3 kcal mol−1 as compared to the PR state (ESI-Fig. 11†), which might be linked to the unique electronic equilibrium between Cu[I]/Tyr-O˙ and Cu[II]/Tyr-O− in the OH → OH,R transition.11,29,34 The DFT calculations also suggest that the intrinsic proton affinity of the CuB hydroxo ligand in the PR state is up to ca. 6 pK-units higher in comparison to the proton affinities of Tyr-244 and ca. 14 pK-units higher compared to the oxo ligand of heme a3. In the OH,R state, Tyr-244 has a ca. 4 pK-units higher proton affinity than the oxygenous ligand of heme a3 (ESI-Table 1†), suggesting that Tyr-244 is the likely proton acceptor. Nonetheless, the absence of the protonic connectivity between both Glu-242 and Tyr-244 or heme a3 in the OH,R state further supports the view that the proton is taken up from the K-channel instead (Fig. 3, ESI-Fig. 7†). Previous DFT calculations29 suggest that the BNC could take up an extra proton in the absence of continuous electron flow. This significantly lowers the redox potential, which in turn does not support the proton pumping activity. Our models of the OH state are consistent with the ligand state of these calculations, and further suggest that the electric field arising from a ligand substitution could drastically affect the hydration dynamics and proton transfer energetics.Free energy profiles for proton transfer along the D- and K-channels
To probe free energies for the pT reaction from Lys-319 to Tyr-244 and Glu-242 to CuB in the presence of dynamic protein surroundings, we performed QM(DFT)/MM umbrella sampling (US) simulations. To this end, we computed the free energy surface for the pT process along water chains connecting the proton donor and acceptor that were formed during the classical MD simulations (ζ < 2.5 Å). We obtain free energy barriers of ca. 13 kcal mol−1 and 11 kcal mol−1 for the PR and OH,R states, respectively. The profiles indicate that the pT is unlikely from the K-channel in the PR state as the product state is not stabilized, whereas pT is feasible from the D-channel in the PR state, and from the K-channel in the OH,R state (Fig. 4). This suggests that the K-channel is both thermodynamically and kinetically preferred for pT in the OH,R state, while the D-channel is kinetically and thermodynamically likely to supply chemical protons in the PR state. The free energy barriers of 13 and 11 kcal mol−1 for pT in the PR and OH,R states are consistent with the pT rates inferred from experimental electrometric amplitudes with rate-constants of 0.8 ms (ca. 14 kcal mol−1),20 and by considering that reaction barriers are often somewhat underestimated at the DFT/GGA level. However, to further explain why the K-channel is not employed in the PR state, we also studied the energetics for the pT between Lys-319 and Tyr-244 and compared it to the pT profiles between Glu-242 and CuB based on MD structures with good hydrogen-bonded connectivities (ζ < 2.5 Å). A similar trend is also reflected in our QM/MM reaction pathway optimizations suggesting that the pT energy barrier along the K-channel (Lys-319 → Tyr-244) is ca. 5–8 kcal mol−1 higher than that for a pT from the D-channel (Glu-242 → CuB) in the PR state (ESI-Fig. 13†). Comparison of the free energy and energy profiles suggests that there could be an entropic contribution of up to 8 kcal mol−1 in the process. Although similar entropic estimates have also been found previously,68,69 it is nevertheless likely that these estimates arise, at least in part, from non-equilibrium protein relaxation effects, which are not accurately captured in picosecond timescale QM/MM calculations. Our combined QM/MM results thus suggest that the D-channel supplies the chemical protons in the P → F transition due to low kinetic barriers and stable product states, whereas lack of connectivity between the D-channel and the BNC in the O → E transition might trigger the activation of the K-channel for delivery of the chemical protons. | ||
| Fig. 4 QM/MM-MD free energy profiles for proton transfer from Glu-242 to CuB in the PR state and from Lys-319 to Tyr-244 in the PR and OH,R states. Lighter traces indicate the statistical error for the free energy profile. Snapshots of the reactants and products for the proton transfer profiles from Glu-242 to CuB in the PR state and from Lys-319 to Tyr-244 in the OH,R state shown in the bottom panel (see ESI-Fig. 12 for more snapshots†). | ||
Our work predicts that the wetting/dewetting transition is mediated by the redox and ligand states of the BNC, which in turn regulate the proton transfer kinetics along the D- and K-channels. Our putative model could be experimentally validated by kinetically accelerating proton uptake along the K-channel that is expected to rescue the catalytic activity of CcO with a blocked D-channel. This could be achieved by stabilizing an open conformation of the K-channel by removing bulky residues at the hydrophobic gate area close to Lys-319, and/or by levelling the pKa difference between the Lys-319 and Tyr-244, e.g., by replacing the former by a tyrosine residue. In addition to the crystallographic studies that support the subtle helix motions predicted to couple with the K-channel opening, this effect could also be probed, for example, in fluorescence resonance energy transfer (FRET)-experiments by labelling the helices surrounding the K-channel.
Conclusions
We have studied here the molecular mechanism for the K-channel activation in CcO by combined MD simulations, graph-theoretical analysis of hydrogen-bonded networks, and QM/MM free energy simulations. Our combined results suggest that formation of a reactive Cu[I]/Tyr-O− state upon reduction of the OH intermediate increases the proton affinity of Tyr-244, and leads to an increase in the hydration levels of the K-channel that provides an effective hydrogen-bonded connectivity to the BNC. These hydration changes are supported by tertiary structural changes around the K-channel. The increased proton affinities and the elevated hydration state subsequently lower the pT barriers along the K-channel. Importantly, our simulations also suggest that the D-channel cannot provide a proton to the BNC in the reductive phase due to a loss of water-mediated connectivity to the BNC from the D-channel. Consistent with electrometric studies that probe voltage changes associated with charge movement along the membrane axis,31 we also find that Lys-319 flips towards the BNC upon its reduction in the PM → PR transition that could stabilize electron transfer from heme a. The combined results thus suggest that Glu-242, the terminal residue of the D-channel is wired to the oxygenous ligand of CuB, whereas the Lys-319 of the K-channel is wired to Tyr-244 and further to heme a3. Upon formation of the aquo-ligand on CuB in the OH state, the connectivity from the D-channel weakens, and thus, the K-channel is kinetically selected for transfer of the protons to the BNC. In light of our present results, it is thus important to elucidate in future work how bacterial heme-copper oxidases employ their K-channel analogues for uptake of both chemical and pumped protons, and how their pumping stoichiometries compare to the corresponding canonical A-type oxidases.4,19Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Mårten Wikström for insightful discussions. This work was supported by the German Research Foundation (DFG). We acknowledge SuperMuc (grant: pr84gu) at the Leibniz Rechenzentrum for computing time.References
- M. K. F. Wikström, Nature, 1977, 266, 271–273 CrossRef.
- V. R. I. Kaila, M. I. Verkhovsky and M. Wikström, Chem. Rev., 2010, 110, 7062–7081 CrossRef PubMed.
- P. Mitchell, Nature, 1961, 191(4784), 144–148 CrossRef PubMed.
- P. Ädelroth, R. B. Gennis and P. Brzezinski, Biochemistry, 1998, 37, 2470–2476 CrossRef PubMed.
- C. Pecoraro, R. B. Gennis, T. V. Vygodina and A. A. Konstantinov, Biochemistry, 2001, 40, 9695–9708 CrossRef PubMed.
- D. Riegler, et al. , Biochim. Biophys. Acta, Bioenerg., 2005, 1706, 126–133 CrossRef PubMed.
- T. V. Vygodina, C. Pecoraro, D. Mitchell, R. B. Gennis and A. A. Konstantinov, Biochemistry, 1998, 37, 3053–3061 CrossRef PubMed.
- M. Ruitenberg, et al. , Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4632–4636 CrossRef PubMed.
- M. R. A. Blomberg, Biochemistry, 2016, 55, 489–500 CrossRef PubMed.
- R. I. Cukier, Biochim. Biophys. Acta, Bioenerg., 2005, 1706, 134–146 CrossRef PubMed.
- V. Sharma, K. D. Karlin and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16844–16849 CrossRef PubMed.
- V. Sharma and M. Wikström, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1111–1115 CrossRef PubMed.
- A. Tuukkanen, M. I. Verkhovsky, L. Laakkonen and M. Wikström, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1117–1121 CrossRef PubMed.
- A. L. Woelke, G. Galstyan and E.-W. Knapp, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1998–2003 CrossRef PubMed.
- A. A. Konstantinov, S. Siletsky, D. Mitchell, A. Kaulen and R. B. Gennis, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9085–9090 CrossRef.
- P. R. Rich and A. Marechal, J. R. Soc., Interface, 2013, 10, 20130183 CrossRef PubMed.
- H. Y. Chang, J. Hemp, Y. Chen, J. A. Fee and R. B. Gennis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16169–16173 CrossRef PubMed.
- H. Han, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14109–14114 CrossRef PubMed.
- V. Rauhamäki, M. Baumann, R. Soliymani, A. Puustinen and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16135–16140 CrossRef PubMed.
- I. Belevich, D. A. Bloch, N. Belevich, M. Wikström and M. I. Verkhovsky, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2685–2690 CrossRef PubMed.
- S. Supekar, A. P. Gamiz-Hernandez and V. R. I. Kaila, Angew. Chem., Int. Ed., 2016, 55, 11940–11944 CrossRef PubMed.
- P. Goyal, S. Yang and Q. Cui, Chem. Sci., 2015, 6, 826–841 RSC.
- A. V. Pisliakov, P. K. Sharma, Z. T. Chu, M. Haranczyk and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7726–7731 CrossRef PubMed.
- R. Liang, J. M. J. Swanson, M. Wikström and G. A. Voth, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5924–5929 CrossRef PubMed.
- R. Liang, J. M. J. Swanson, Y. Peng, M. Wikström and G. A. Voth, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7420–7425 CrossRef PubMed.
- J. Lu and M. R. Gunner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12414–12419 CrossRef PubMed.
- M. Svensson-Ek, J. Abramson, G. Larsson, S. Törnroth, P. Brzezinski and S. Iwata, J. Mol. Biol., 2002, 321, 329–339 CrossRef PubMed.
- M. Iwaki and P. R. Rich, J. Am. Chem. Soc., 2007, 129, 2923–2929 CrossRef PubMed.
- M. R. A. Blomberg and P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 364–376 CrossRef PubMed.
- E. A. Gorbikova, M. Wikström and M. I. Verkhovsky, J. Biol. Chem., 2008, 283, 34907–34912 CrossRef PubMed.
- H. Lepp, E. Svahn, K. Faxén and P. Brzezinski, Biochemistry, 2008, 47, 4929–4935 CrossRef PubMed.
- D. Bloch, I. Belevich, A. Jasaitis, C. Ribacka, A. Puustinen, M. I. Verkhovsky and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 529–533 CrossRef PubMed.
- D. Jancura, V. Berka, M. Antalik, J. Bagelova, R. B. Gennis, G. Palmer and M. Fabian, J. Biol. Chem., 2006, 281, 30319–30325 CrossRef PubMed.
- V. R. I. Kaila, M. P. Johansson, D. Sundholm, L. Laakkonen and M. Wikström, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 221–233 CrossRef.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660–669 CrossRef PubMed.
- I. Kim and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7810–7815 CrossRef PubMed.
- T. Tsukihara, et al. , Proc. Natl. Acad. Sci. U. S. A., 2003, 100(26), 15304–15309 CrossRef PubMed.
- J. C. Phillips, et al. , J. Comput. Chem., 2005, 26, 1781–1802 CrossRef PubMed.
- E. W. Dijkstra, Numer. Math., 1959, 1, 269–271 CrossRef.
- D. Bashford and K. Gerwert, J. Mol. Biol., 1992, 224, 473–486 CrossRef PubMed.
- (a) G. Kieseritzky and E. W. Knapp, Proteins, 2008, 71, 1335–1348 CrossRef PubMed; (b) B. Rabenstein and E. W. Knapp, Biophys. J., 2001, 80, 1141–1150 CrossRef PubMed.
- B. K. Ho and F. Gruswitz, BMC Struct. Biol., 2008, 8, 49 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 2, 799–805 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef.
- (a) A. D. MacKerell, et al. , J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed; (b) M. P. Johansson, V. R. I. Kaila and L. Laakkonen, J. Comput. Chem., 2008, 29, 753–767 CrossRef PubMed.
- S. Riahi and C. N. Rowley, J. Comput. Chem., 2014, 35, 2076–2086 CrossRef PubMed.
- A. Di Luca, A. P. Gamiz-Hernandez and V. R. I. Kaila, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6314–E6321 CrossRef PubMed.
- V. R. I. Kaila, M. Wikström and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6988–6993 CrossRef PubMed.
- V. R. I. Kaila, J. R. Soc., Interface, 2018, 15, 20170916 CrossRef PubMed.
- C. M. Suomivuori, A. P. Gamiz-Hernandez, D. Sundholm and V. R. I. Kaila, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7043–7048 CrossRef PubMed.
- L. Qin, et al. , Biochemistry, 2009, 48, 5121–5130 CrossRef PubMed.
- M. Wikström, V. Sharma, V. R. I. Kaila, J. P. Hosler and G. Hummer, Chem. Rev., 2015, 115, 2196–2221 CrossRef PubMed.
- F. Garczarek and K. Gerwert, Nature, 2005, 439, 109–112 CrossRef PubMed.
- V. Sharma, G. Enkavi, I. Vattulainen, T. Róg and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2040–2045 CrossRef PubMed.
- C. Y. Son, A. Yethiraj and Q. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8830–E8836 CrossRef PubMed.
- M. R. A. Blomberg and P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1173–1180 CrossRef PubMed.
- V. R. I. Kaila, M. I. Verkhovsky, G. Hummer and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6255–6259 CrossRef PubMed.
- A. L. Woelke, G. Galstyan, A. Galstyan, T. Meyer, J. Heberle and E. W. Knapp, J. Phys. Chem. B, 2013, 117, 12432–12441 CrossRef PubMed.
- B. M. Samudio, V. Couch and A. A. Stuchebrukhov, J. Phys. Chem. B, 2016, 120, 2095–2105 CrossRef PubMed.
- P. Goyal, J. Lu, S. Yang, M. R. Gunner and Q. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18886–18891 CrossRef PubMed.
- A.-L. Johansson, S. Chakrabarty, C. L. Berthold, M. Högbom, A. Warshel and P. Brzezinski, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 1083–1094 CrossRef PubMed.
- S. Narayan, D. L. Wyatt, D. S. Crumrine and S. Cukierman, Biophys. J., 2007, 93, 1571–1579 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation details, analysis of hydration dynamics, pKa calculations, DFT models, and QM/MM simulations. See DOI: 10.1039/c8sc01587b |
| This journal is © The Royal Society of Chemistry 2018 |