
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic investigations into the cyclization and crystallization of benzobisoxazole-linked two-dimensional covalent organic frameworks†
David A.
Pyles
,
William H.
Coldren
,
Grace M.
Eder
,
Christopher M.
Hadad
 and
Psaras L.
McGrier
*
and
Psaras L.
McGrier
*
Department of Chemistry & Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA
First published on 25th June 2018
Abstract
Although many diverse covalent organic frameworks (COFs) have been synthesised over the past decade, our fundamental understanding of their nucleation and growth during the crystallization process has progressed slowly for many systems. In this work, we report the first in-depth mechanistic investigation detailing the role of nucleophilic catalysts during the formation of two distinct benzobisoxazole (BBO)-linked COFs. The BBO-COFs were constructed by reacting 1,3,5-tris(4-formylphenyl)benzene (TFPB) and 1,3,5-tris(4-formylphenyl)triazine (TFPT) C3-symmetric monomers with a C2-symmetric o-aminophenol substituted precursor using different nucleophiles (e.g. NaCN, NaN3, and NaSCH3). Our experimental and computational results demonstrate that the nucleophiles help initiate an oxidative dehydrogenation pathway by producing radical intermediates that are stabilized by a captodative effect. We also demonstrate that the electron deficient TFPT monomer not only aids in enhancing the crystallinity of the BBO-COFs but also participates in the delocalization of the radicals generated to help stabilize the intermediates.
Introduction
Covalent organic frameworks (COFs)1–4 are an advanced class of crystalline porous materials that have emerged as appealing candidates for applications related to gas storage,5,6 catalysis,7–9 optoelectronics,10–14 proton conduction,15,16 and energy storage.17–19 COFs offer the unique advantage of utilizing the geometry of various molecular building blocks to construct atomically precise two-dimensional (2D) and three-dimensional (3D) polymeric networks with high surface areas and tunable pore sizes. While the field has enjoyed a rapid increase of exceptional COF architectures with tailored functionalities over the past decade, there has been an increased effort of late to probe the underlying mechanisms that govern the nucleation and crystallization of COFs containing the most commonly used boronate ester20–23 and imine24–26 covalent linkages. Such investigations are believed to be vital for not only bypassing the tedious task of screening numerous reactions conditions (e.g., solvent concentrations, reaction times, temperature, etc.) to produce a desired framework, but also establishing a rational experimental protocol for generating high quality COF structures with improved efficiency and controlled morphologies.Recently, we reported a cyanide-catalyzed protocol to synthesize chemically stable benzobisoxazole-linked (BBO) COFs utilizing C3-symmetric formyl- and C2-symmetric o-aminophenol-substituted organic linkers.26 Upon doing so, we discovered that the addition of cyanide enables the construction of ordered polymeric materials with high surface areas. We hypothesized that the addition of cyanide induces ring closure and the formation of a benzoxazoline intermediate that then undergoes subsequent aerobic oxidation in the presence of air to form the BBO-COF. This result is consistent with what has been reported for the cyanide-catalyzed 5-exo-tet cyclization of benzo-fused azoles,28,29 which is based on Baldwin's rules30 for ring closure. We concluded that the non-covalent interactions between the adjacent π-systems during this cyclization process were significant enough to stabilize the stacking layers upon formation of the irreversible BBO-linkage giving rise to the ordered 2D BBO-COF structures. In contrast, there are only a few synthetic routes that have been reported to construct BBO-based porous materials: (1) the annealing of silyl protected prepolymers at high temperatures,31 and (2) the direct formation of aniline Schiff base precursors followed by subsequent dehydrogenation and cyclization.32 While both methods are effective, they exclusively lead to the formation of amorphous polymer networks. Since the second synthetic route is similar to our method with the exception of adding a nucleophile to catalyze the reaction, we began to question the active role of cyanide during the cyclization process. Since BBO-COFs could be utilized for developing 2D materials for organic electronics33 and sensory applications,34 a thorough investigation into the optimal reaction conditions needed to produce ordered BBO-based polymeric systems are warranted (Scheme 1).
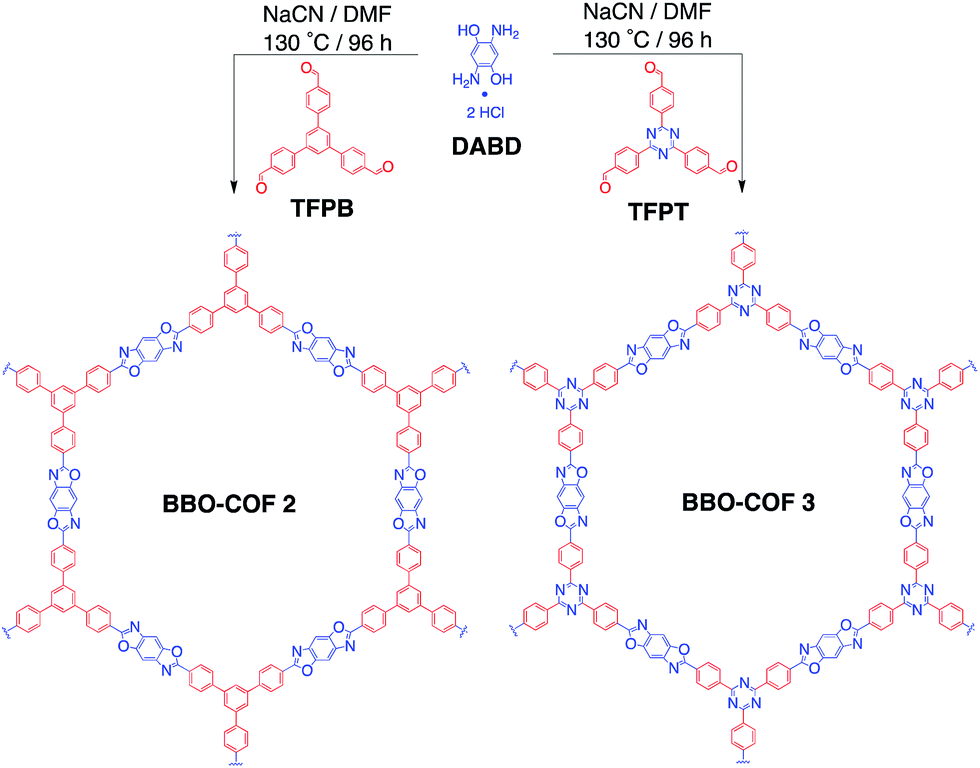 | ||
| Scheme 1 Cyanide-catalyzed synthesis of BBO-COF 2 and 3. | ||
Recently, Wang and Yu reported a density functional theory (DFT)35 study suggesting that cyanide does not actually promote the direct cyclization of benzoxazoles but, instead, assists with the oxidative dehydrogenation process by generating a radical intermediate that is stabilized through a captodative effect.36 The captodative effect is a process by which adjacent electron donating and electron withdrawing substituents are utilized to stabilize a radical center through resonance stabilization. This concept of push–pull resonance stabilization was embraced in the field of polymer science as a method to help improve the lack of control and reactivity of 1,1-disubstituted monomers used in radical polymerizations.37 It has also emerged as an alternative method for enhancing the conductivity of organic electronic devices.38 However, mechanistic studies detailing how the captodative effect and careful selection of organic linkers can support the nucleation and growth of ordered 2D polymeric systems has yet to be reported.
In an effort to validate the likelihood of a mechanistic pathway involving the captodative effect experimentally, we were curious to determine if other nucleophiles (e.g. NaN3, NaSCH3) could be used to catalyze the formation of the BBO-COFs. We were also interested in examining what impact an electron deficient organic linker like 1,3,5-tris(4-formylphenyl)triazine (TFPT) would have on the formation and crystallization of the BBO-linked COFs. Since triazine-based monomers are known for exhibiting high electron mobilities (thanks to their electron withdrawing nature),39 we hypothesized that this feature could assist with stabilizing the radical species generated during the oxidative dehydrogenation process.
In this study, we first established the optimal cyanide-catalyzed reaction conditions for synthesizing BBO-COF 3 using TFPT and 2,5-diamino-1,4-benzenediol dihydrochloride (DABD) organic linkers.40 Afterwards, we investigated the ability of other good nucleophiles like NaN3 and NaSCH3 to initiate the formation of BBO-COF 3 and the previously synthesized BBO-COF 2. These studies were performed in conjunction with one another to determine if the electron withdrawing TFPT linker of BBO-COF 3 would exhibit any enhanced effect on the formation and crystallization of the COFs in comparison to the isostructural 1,3,5-tris(4-formylphenyl)benzene (TFPB) linker that was used to synthesize BBO-COF 2. We demonstrate that NaN3 and NaSCH3 are effective at catalyzing the formation of BBO-COFs, but NaCN is the only catalyst that promotes the stabilization of radical intermediates through the captodative effect. Interestingly, we also show that the electron withdrawing TFPT monomer not only enhances the crystallinity of BBO-COF 3, but also plays a significant role in stabilizing the radical intermediates generated during oxidative dehydrogenation. These results were validated using DFT calculations, including population and spin density analyses, along with powder X-ray diffraction (PXRD). We expect that this work will provide a rational protocol for constructing highly ordered BBO-based polymeric systems for practical applications.
Results and discussion
Synthesis and characterization of BBO-COF 3
BBO-COF 3 was synthesized by reacting TFPT with DABD in DMF at −15 °C for ∼3 h. Afterwards, the reaction mixture was slowly warmed to room temperature overnight before adding 1 equiv. of NaCN dissolved in 0.2 mL of methanol. The mixture was then stirred at 130 °C for four days in the presence of air. The product was obtained by filtration and washed with acetone to afford a light brown crystalline solid. BBO-COF 3 was purified by immersing the solids in methanol and acetone for 24 h to remove any unreacted monomers and dried under vacuum. Thermogravimetric analysis (TGA) revealed that BBO-COF 3 maintained more than 98% of its weight up to 470 °C (Fig. S32†).BBO-COF 3 was characterized using Fourier transform infrared (FT-IR) and 13C cross-polarization magic angle spinning (CP-MAS) spectroscopic analyses. The FT-IR spectrum revealed stretching modes at 1662 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 1120 cm−1 (C–O) confirming the formation of the benzoxazole ring (Fig. S11†). The TFPT linker exhibited two intense stretches at 1515 and 1360 cm−1, which correspond to the presence of the benzene and triazine moieties (Fig. S10†). The triazine ring and BBO-linkage were also confirmed by solid-state 13C CP-MAS NMR, displaying distinct resonances at 167.7, 160.5, and 146.4 ppm (Fig. S31†).
N) and 1120 cm−1 (C–O) confirming the formation of the benzoxazole ring (Fig. S11†). The TFPT linker exhibited two intense stretches at 1515 and 1360 cm−1, which correspond to the presence of the benzene and triazine moieties (Fig. S10†). The triazine ring and BBO-linkage were also confirmed by solid-state 13C CP-MAS NMR, displaying distinct resonances at 167.7, 160.5, and 146.4 ppm (Fig. S31†).
The porosity of BBO-COF 3 was evaluated using nitrogen gas adsorption measurements at 77 K (Fig. 1).
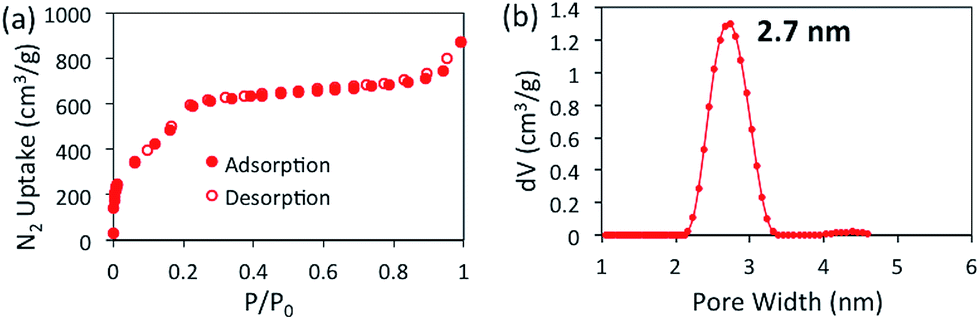 | ||
| Fig. 1 Nitrogen isotherm at 77 K (a) and NLDFT pore size distribution (b) for BBO-COF 3. | ||
BBO-COF 3 displays a type IV isotherm exhibiting a steep uptake at low pressure (P/P0 < 0.06) followed by a noticeable step between P/P0 = 0.06 and 0.22 which validates the mesoporosity of the material. The Brunauer–Emmett–Teller (BET) method was applied over the low-pressure region (0.01 < P/P0 < 0.22) of the isotherm to afford a surface area of 2039 m2 g−1. It is worth noting that this is the highest surface area reported to date for a benzoxazole-linked porous material.27,31,32 The total pore volume of BBO-COF 3 calculated at P/P0 = 0.989 provided a value of 1.22 cm3 g−1. The pore size distribution was estimated using nonlocal density functional theory (NLDFT) to provide an average pore size of 2.7 nm, which is close to the predicted value of 3.3 nm. In comparison, the isostructural BBO-COF 2 exhibited an average pore size of 1.8 nm. The enhanced pore size of BBO-COF 3 could be attributed to the planarity of the triazine TFPT, which allows for coplanarity between the vertex and linker units (Fig. S46 and S47†). This is in contrast to the out-of-plane twisting of the TFPB moieties, which can hinder the formation of vertically stacked eclipsed layers.
In contrast to the PXRD pattern of BBO-COF 2, BBO-COF 3 displays an enhancement in crystallinity (see Fig. S30†). We believe this enhancement is attributed to (1) the planarity of the TFPT units allowing for more efficient van der Waals interactions between the layers, and (2) the ability of the electron deficient triazine units to form donor–acceptor (DA) stacks with the adjacent TFPT aromatic rings along the c direction. Lotsch and coworkers have also observed this phenomenon for imine-linked COFs containing triazine moieties.41 Since DA interactions are often more favourable than non-covalent interactions between π-electron rich aromatic rings,42 this could also further explain why the offsets for BBO-COF 2 (15 Å) are more significant compared to BBO-COF 3 (6 Å).
BBO-COF 3 time-dependent growth studies
Before evaluating other nucleophiles as potential catalysts, we first wanted to ensure that the optimal cyanide-catalyzed reaction conditions for constructing BBO-COF 3 were established. Initially, we found that allowing the monomers to react at −15 °C under nitrogen for several hours before adding 1 eq. of cyanide and heating the reaction to 130 °C under air facilitated the precipitation of amorphous imine-linked porous polymers after reacting for one day. However, it was unclear if the reaction really required four days to produce the BBO-COFs.To address this issue, we monitored the growth of BBO-COF 3 over time. After reacting for one day, the isolated solid provided a low surface area of 704 m2 g−1 and a pore size of 1.7 nm indicating that the reaction requires a longer reaction time (Fig. 2). The IR spectrum displayed a broad stretch at 3343 cm−1 and a sharp stretch at 1693 cm−1, which are attributed to the OH stretch from the phenolic imine-linked intermediate and the aldehyde (CO) stretch from the TFPT linker, respectively (Fig. 3). Although the IR stretches for the intermediate and starting material begin to disappear after reacting for a few additional days, the surface area increases by two orders of magnitude to 1435 m2 g−1 after day two, but then surprisingly decreases to 1295 m2 g−1 on day three. This trend is echoed in the pore size data yielding a value of 2.7 nm on day two followed by the formation of multiple smaller pores at 2.6 and 2.1 nm on day three. Since the benzoxazole bond is irreversible, we believe that the dramatic changes in the surface area and pore size when transitioning from day-one to day-three could be attributed to the formation of phenolic imine-linked polymeric networks that are generated during the dynamic imine exchange process prior to the formation of the BBO linkage.25 PXRD patterns from the isolated solids also indicate that a four-day reaction period is required to attain the crystalline BBO-COF 3 structure (Fig. S20†). Allowing the reaction to proceed beyond the four-day reaction period did not lead to an additional increase in the surface area (Fig. S21†).
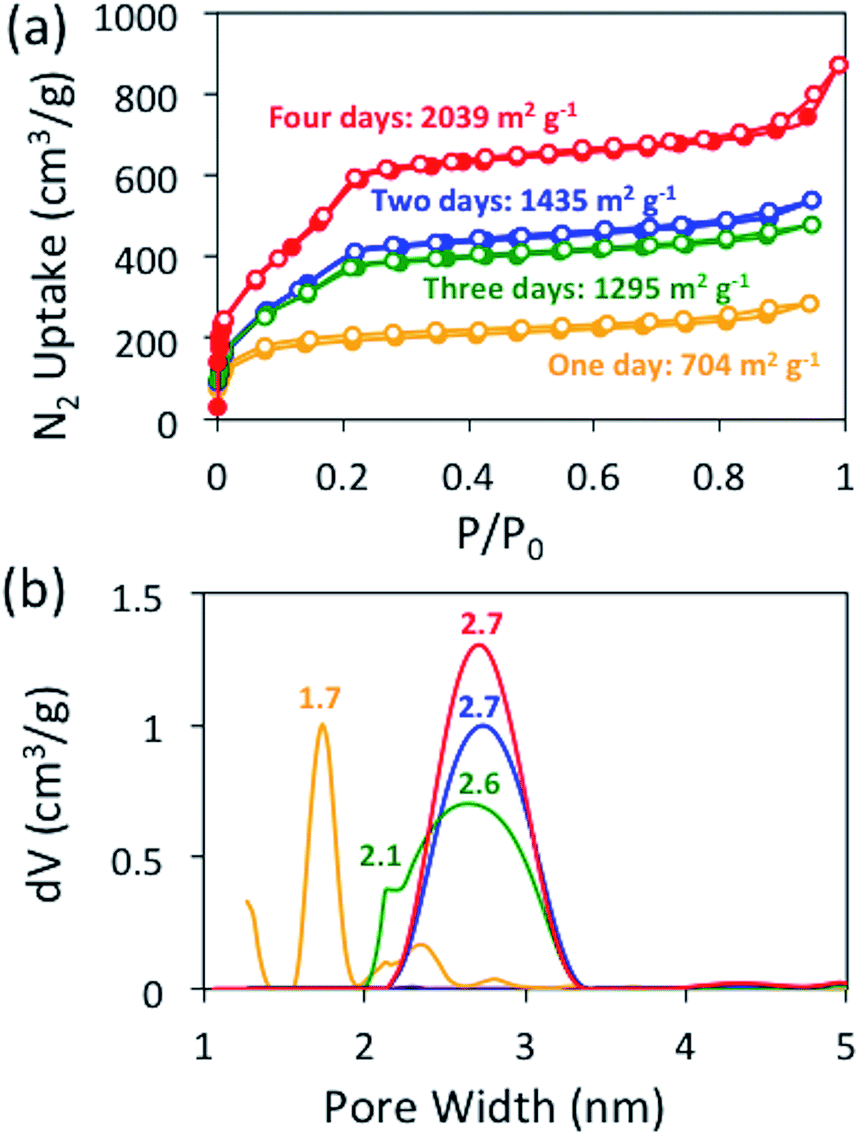 | ||
| Fig. 2 Nitrogen isotherms at 77 K (a) and NLDFT (b) pore size distributions for BBO-COF 3 over a four-day reaction period. | ||
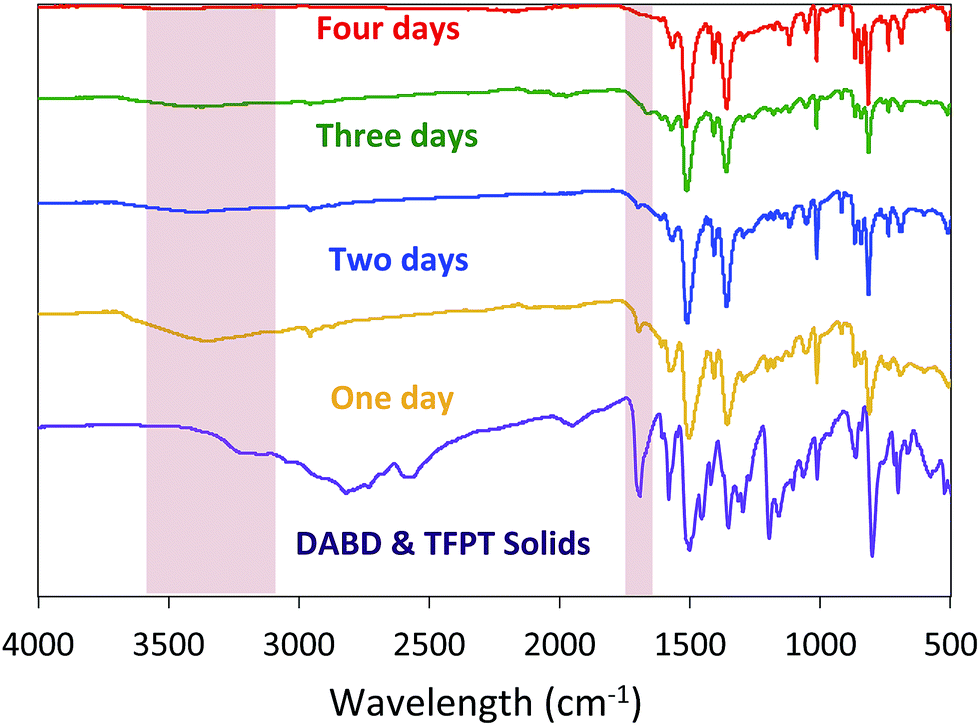 | ||
| Fig. 3 IR spectra of BBO-COF 3 highlighting the disappearance of the OH stretch from phenolic imine-linked intermediate and aldehyde stretch from the TFPT linker over a four-day reaction period. | ||
It is also worth noting that the BBO-linkage does not form without oxygen even under the optimal growth conditions (Fig. S6–S8 and S23–S25†).
BBO-COF formation with different nucleophiles
With the optimal reaction conditions established, we examined if other nucleophiles could be utilized to catalyze the formation of the BBO-COFs. Wang and Yu have shown that the activation energy for the direct 5-exo-tet cyclization of benzoxazole in the presence of NaCN has to overcome a high-energy barrier of 47.9 kcal mol−1,35 and suggested that the cyclization proceeds through a stepwise mechanism. DFT calculations revealed that after the cyanide attacks the imine carbon to convert the hybridization from sp2 to sp3 triplet- state oxygen then directly abstracts a hydrogen from the newly generated sp3 α-carbon to produce a radical that is stabilized through the captodative effect (Scheme 2). Surprisingly, the activation energy for this process was found to be ∼24.4 kcal mol−1 lower than the suggested direct cyanide-catalyzed cyclization pathway. Intrigued by these results, we were curious to examine if (1) BBO-COF 2 and 3 could be constructed using other good nucleophiles like NaN3 and NaSCH3, and (2) the electron deficient TFPT linker of BBO-COF 3 would have any effect on the cyclization and crystallization of the framework compared to BBO-COF 2, which contained the electron rich TFPB linker. | ||
| Scheme 2 Proposed mechanism for the stepwise oxidative dehydrogenation pathway to form the BBO-linkage using NaCN as a catalyst. | ||
The formation of BBO-COF 2 and 3 using NaN3 and NaSCH3 as catalysts was evaluated using nitrogen gas adsorption isotherms, IR spectroscopy, and PXRD analysis (Fig. 4). Since the direct cyclization of benzoxazoles only requires ∼8.4 kcal mol−1,35 we also monitored the formation of the BBO-COFs without the addition of a nucleophile. The activated solids for BBO-COF 2 provided surface area values of 1033 and 236 m2 g−1 for NaN3 and NaSCH3, respectively, whereas the reaction without catalyst yielded a surface area of 169 m2 g−1. Interestingly, NaSCH3 and NaN3 yielded pore sizes that were closer to the predicted value than NaCN (Fig. S5†). The IR spectra for BBO-COF 2 revealed that the reaction with NaSCH3 exhibited broad OH stretches at ∼3300 cm−1 indicating that the isolated solids still contain some of the unreacted phenolic imine-linked intermediate (Fig. 4b). The PXRD data revealed that NaN3 and NaCN were the only catalysts that produced crystalline samples of BBO-COF 2 while NaSCH3 afforded an amorphous porous polymer (Fig. 4c). In contrast to BBO-COF 2, the activated solids for BBO-COF 3 yielded surface areas of 1697, 1439, and 386 m2 g−1 for NaSCH3, NaN3, and the reaction with no catalyst, respectively (Fig. 4d). The BBO-COF 3 synthesized without a catalyst contained two pores at 1.7 and 2.6 nm, while the others displayed one distinct pore size at ∼2.7 nm (Fig. S15†). NaSCH3 is the only nucleophile that exhibited an OH stretch at ∼3300 cm−1 signifying that a small portion of the polymer was not fully converted to the BBO-linkage (Fig. 4e). Surprisingly, all of the nucleophiles studied provided high quality samples of BBO-COF 3 with the lone exception being the reaction in which no catalyst was used (Fig. 4f). Although the complete formation of BBO-COF 2 and 3 seemed to vary upon the usage of NaSCH3 or NaN3, our studies suggest that NaCN is the most effective catalyst at producing crystalline BBO-COF materials. It should be noted that the reactions without catalyst generated amorphous porous polymers, and were unsuccessful at promoting the cyclization of the BBO-linkage even in the presence of oxygen. This also indicates that nucleophiles are critical for providing the sp3 hybridized α-carbon needed to initiate the stepwise oxidative dehydrogenation mechanistic pathway (Scheme 2).
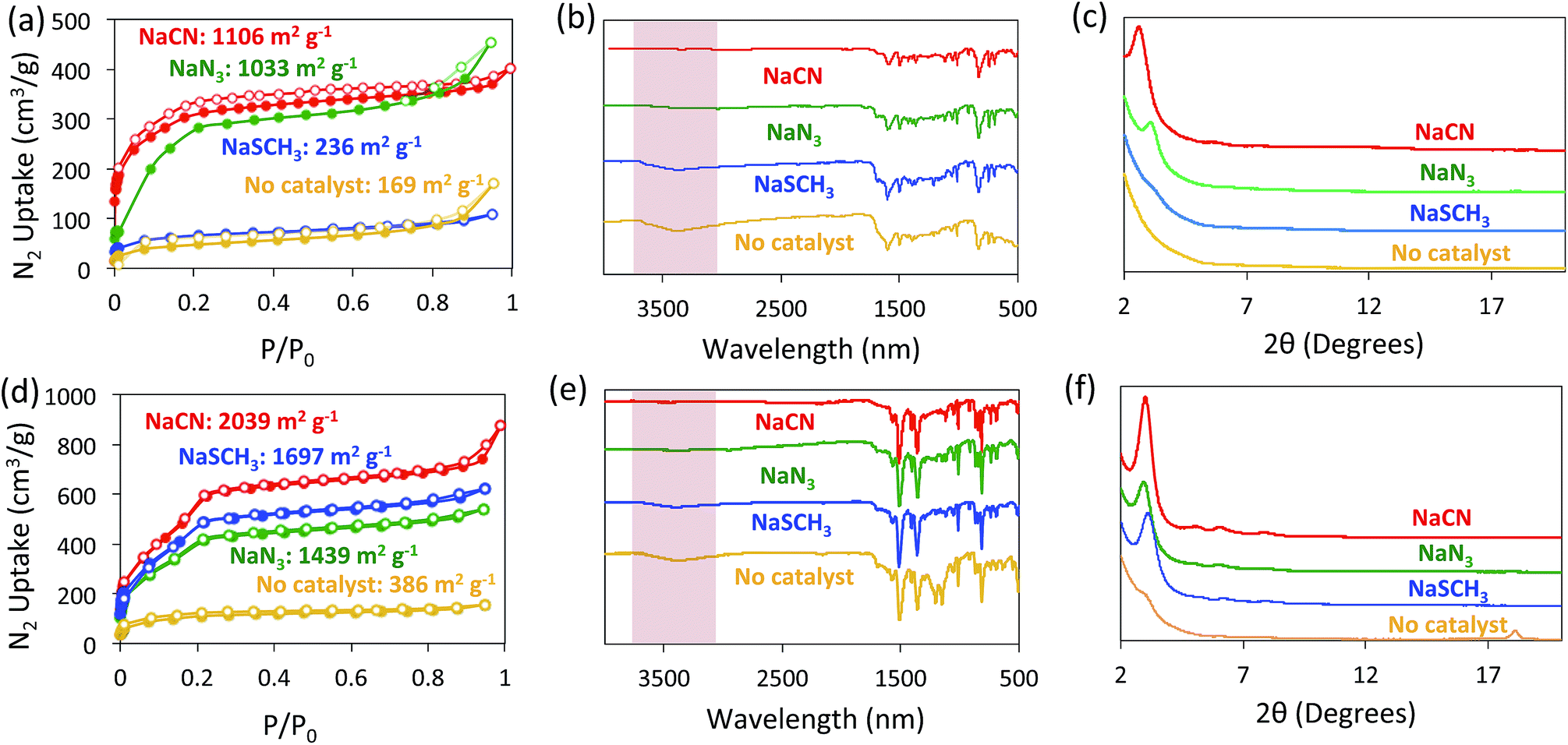 | ||
| Fig. 4 Nitrogen gas isotherms (a and d), IR spectra (b and e), and PXRD patterns (c and f) of BBO-COF 2 (top) and BBO-COF 3 (bottom). Each reaction was catalyzed by adding 1 eq. of NaCN (red), NaSCH3 (blue), and NaN3 (green). Reactions in which no catalyst was used are shown in orange. | ||
NPA population and spin density analysis – proposed mechanism
With this in mind, we wanted to validate that the radical generated at the α-carbon prior to the cyclization of the BBO-linkage is stabilized through the captodative effect. We were also curious to explore the role of the TFPB and TFPT linkers during the radical delocalization process. To examine this, we performed DFT geometry optimizations (B3LYP/6-31+G*)43–46 along with natural population analysis (NPA, B3LYP/6-31+G*)47 charge density (population) and spin density calculations on the fragment cores of BBO-COF 2 and 3, as implemented in Gaussian 16.48 The results of these calculations are shown in Tables 1 and 2, respectively. Surprisingly, the calculations show that 100% of the single electron is delocalized entirely on the azide for both BBO-COF 2 and 3 indicating that these intermediates are not stabilized by the captodative effect. In the case of cyanide, the single electron for BBO-COF 2 is broadly delocalized on the nitrogen atom (25%), cyanide substituent (10%), and the TFPB linker (18%). In contrast, BBO-COF 3 displays similar percentages for the nitrogen atom and cyanide substituent, but exhibits a 7% increase in charge delocalization for the electron deficient TFPT linker. The calculations for cyanide confirm that (1) the radical intermediates generated are stabilized by the captodative effect for these particular systems, and (2) electron deficient substituents can also play a vital role in stabilizing radical intermediates that are generated during the formation of BBO-COFs. Lastly, only a small percentage of the single electron is delocalized on the SCH3 substituent for BBO-COF 2 and 3 possibly due to its electron donating nature, while 32% and 47% are delocalized on the TFPB and TFPT linkers, respectively. The higher charge delocalization for the latter could explain why the NaSCH3 catalyzed reaction for BBO-COF 3 produces a crystalline solid, while the reaction for BBO-COF 2 yields an amorphous powder.
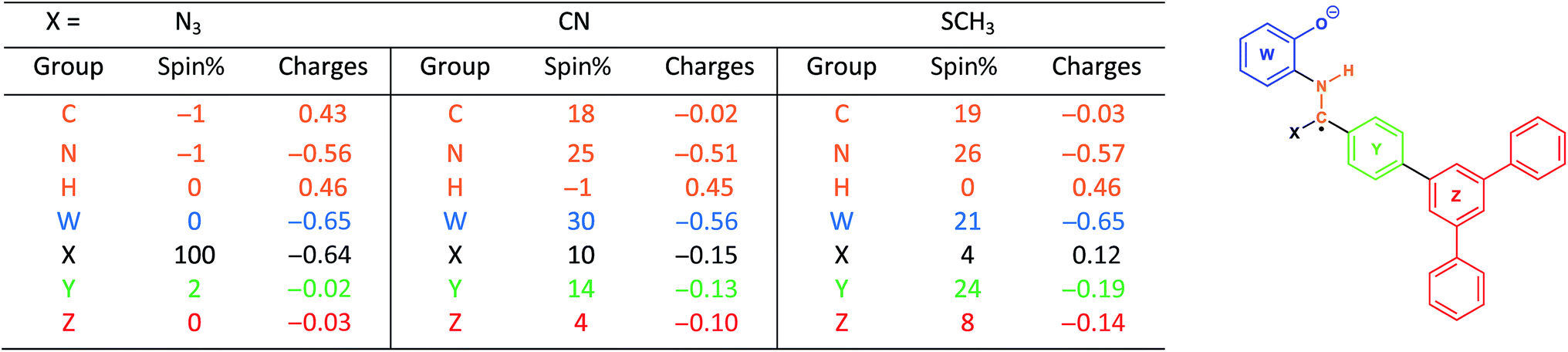
|
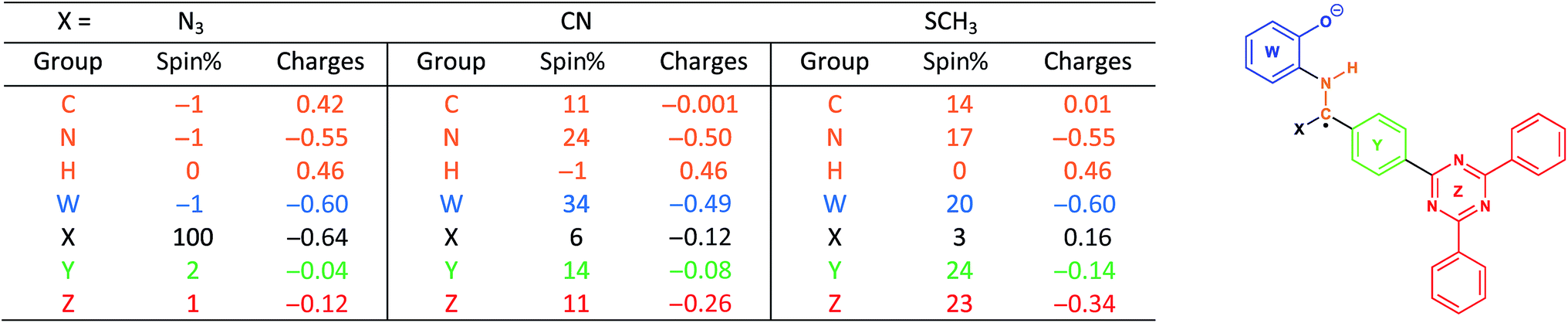
|
Based on the experimental and computational data collected, we propose the following mechanism for the cyclization of the BBO-linkage during the nucleation process (Scheme 2). The aminophenol and formyl precursors initially undergo an imine condensation reaction to produce (A) followed by subsequent proton transfer to form the protonated imine intermediate (B). Then, cyanide attacks the imine α-carbon of (B) to generate the sp3 hybridized intermediate (C). Afterwards, triplet state oxygen moves in to abstract the hydrogen atom at the sp3 hybridized α-carbon of (C) to produce intermediate (D) and generate a hydroperoxyl radical species. Later, this hydroperoxyl radical species returns to abstract a hydrogen atom from the β-nitrogen of (D) to eliminate hydrogen peroxide and produce intermediate (E). From here, an intramolecular cyclization followed by elimination of the cyano group produces the BBO-linkage. Although the proposed mechanism does shed more light on the role of cyanide during the cyclization process, it is unclear if radical–radical interactions49 between the adjacent layers of BBO-COFs occur or assist with the nucleation and growth process.
Conclusions
In summary, we have reported the first in-depth mechanistic investigation into the cyclization and crystallization of BBO-COFs utilizing different nucleophiles (e.g. NaN3 and NaSCH3). However, we found that NaCN provided BBO-COFs with the highest surface areas and best crystallinity. Our experimental and electron spin population studies also confirm that the nucleophiles do not promote the direct cyclization of the BBO-linkage but instead, initiates an oxidative dehydrogenation mechanistic pathway by producing radical intermediates that are stabilized by a captodative effect. In addition, we discovered that the electron deficient TFPT monomer not only aids in increasing crystallinity of the materials by promoting the formation of D–A stacking layers, but also participates in the delocalization of radicals generated during the oxidative dehydrogenation process. We believe this study provides a fundamental understanding of how BBO-COFs are formed, and an intricate thread towards unravelling the mysteries surrounding the crystallization of COFs. Such investigations are necessary for obtaining high quality COF structures for practical applications.Conflicts of interest
There are no conflicts of interest.Acknowledgements
P. L. M acknowledges the National Science Foundation (NSF) and Georgia Tech Facilitating Academic Careers in Engineering and Science (GT-FACES) for a Career Initiation Grant, and funding from The American Chemical Society Petroleum Research Fund (55562-DNI7) and the Center for Emergent Materials (DMR-1420451) at The Ohio State University. C. M. H. acknowledges financial support from the NSF (CHE-1609889) as well as generous allocations of computational resources from the Ohio Supercomputer Center.Notes and references
- M. Dogru and T. Bein, Chem. Commun., 2014, 50, 5531–5546 RSC.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068–16086 CrossRef.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef PubMed.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef PubMed.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878–2882 CrossRef PubMed.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef PubMed.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef PubMed.
- G. H. V. Bertrand, V. K. Michaelis, T. C. Ong, R. G. Griffin and M. Dincă, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4923–4928 CrossRef PubMed.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. A. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920–2924 CrossRef PubMed.
- S. Duhović and M. Dincă, Chem. Mater., 2016, 28, 2855–2873 Search PubMed.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef PubMed.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef PubMed.
- C. R. Deblase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. Dichtel, J. Am. Chem. Soc., 2014, 135, 16821–16824 CrossRef PubMed.
- C. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef PubMed.
- S. Chandra, D. R. Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074–2080 CrossRef.
- B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2014, 136, 8783–8789 CrossRef PubMed.
- B. J. Smith, N. Hwang, A. D. Chavez, J. L. Novotney and W. R. Dichtel, Chem. Commun., 2015, 51, 7532–7535 RSC.
- H. Li, A. D. Chavez, H. Li, W. R. Dichtel and J.-L. Bredas, J. Am. Chem. Soc., 2017, 139, 16310–16318 CrossRef PubMed.
- C. M. Thompson, G. Occhialini, G. T. McCandless, S. B. Alahakoon, V. Cameron, S. O. Nielsen and R. A. Smaldone, J. Am. Chem. Soc., 2017, 139, 10506–10513 CrossRef PubMed.
- X. Chen, M. Addicoat, S. Irle, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 546–549 CrossRef PubMed.
- L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC.
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef.
- Y.-H. Cho, C.-Y. Lee, D.-C. Ha and C.-H. Cheon, Adv. Synth. Catal., 2012, 354, 2992–2996 CrossRef.
- Y.-H. Cho, C.-Y. Lee and C.-H. Cheon, Tetrahedron Lett., 2013, 69, 6565–6573 CrossRef.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 18, 734–736 RSC.
- H. A. Patel, D. Ko and C. T. Tavuz, Chem. Mater., 2014, 26, 6729–6733 CrossRef.
- M. G. Rabbani, T. Islamoglu and H. M. El-Kaderi, J. Mater. Chem A., 2017, 5, 258–265 RSC.
- B. C. Tlach, A. L. Tomlinson, A. G. Ryno, D. D. Knoble, D. L. Drochner, K. J. Krager and M. Jefferies-EL, J. Org. Chem., 2013, 78, 6570–6581 CrossRef PubMed.
- M. A. Saeed, H. T. M. Le and O. S. Miljanić, Acc. Chem. Res., 2014, 47, 2074–2083 CrossRef PubMed.
- W. Chen, W. An, Y. Wang and A. Yu, J. Org. Chem., 2016, 81, 10857–10862 CrossRef PubMed.
- H. G. Viehe, Z. Janousek and R. Merényi, Acc. Chem. Res., 1985, 18, 148–154 CrossRef.
- H. Tanaka, Prog. Polym. Sci., 2003, 28, 1171–1203 CrossRef.
- T. Stuyver, T. Zeng, Y. Tsuji, S. Fias, P. Geerlings and F. De Proft, J. Phys. Chem. C, 2018, 122, 3194–3200 CrossRef.
- T. Ishi-i, K. Yaguma, T. Thiemann, M. Yashima, K. Ueno and S. Mataka, Chem. Lett., 2004, 33, 1244–1245 CrossRef.
- During this research, Wang and coworkers reported the interesting photocatalytic properties of benzoxazole-linked LZU-191, which is identical to BBO-COF 3. However, LZU-191 was constructed using a different synthetic route, and no in-depth mechanistic formation studies were performed. For more details, please see: P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef PubMed.
- F. Hasse, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 831, 1736–154104 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gompers, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranaginghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Straoverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revission A.03, Gaussian, Inc, Wallingford CT, 2016 Search PubMed.
- Y. Wang, M. Frasconi, W.-G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876–885 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, FT-IR, solid-state 13C NMR, TGA, PXRD, SEM, optimized XYZ coordinates, vibrational frequencies, and full NPA results. See DOI: 10.1039/c8sc01683f |
| This journal is © The Royal Society of Chemistry 2018 |