
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiverse secondary C(sp3)–H bond functionalization via site-selective trifluoroacetoxylation of aliphatic amines†
Yongzhen
Tang
,
Yuman
Qin
,
Dongmei
Meng
,
Chaoqun
Li
,
Junfa
Wei
 and
Mingyu
Yang
*
and
Mingyu
Yang
*
Key Laboratory of Applied Surface and Colloid Chemistry of MOE, School of Chemistry and Chemical Engineering, Shaanxi Normal University, 620 West Chang'an Ave, Xi'an, 710119, China. E-mail: yangmy05@snnu.edu.cn; yangmy05@gmail.com
First published on 28th June 2018
Abstract
We describe a coinage-metal-catalyzed site-selective oxidation of secondary C(sp3)–H bonds for aliphatic amine substrates. Broad amine scope, good functional compatibility and late-stage diversification are demonstrated with this method. The steric demand of the β-substituents controlled diastereoselectivities under this catalytic system. The site selectivity favors secondary C(sp3)–H bonds over tertiary ones underscoring the unique synthetic potential of this method.
Introduction
Strategies that enable rapid construction and derivatization of late-stage intermediates are highly desirable in modern drug discovery.1 The development of such approaches has the potential of streaming access to search for lead compounds via testing a family of analogues that are derived from a “parent” molecule. During the past decade, direct C–H bond functionalization has become a straightforward tool for derivatizing complex synthetic intermediates. It can convert inert C–H bonds into new functional groups (FGs), selectively and efficiently.2 However, the utility of C–H functionalization for late-stage modification is challenging due to the highly limited range of FGs that can be introduced. Moreover, substrate and/or reaction-condition-controlled protocols generally require de novo synthesis or intensive optimization of reaction conditions, hence limiting rapid derivatization. Therefore, the design of a valuable method by which a latent FG can be initially installed through selective C–H bond functionalization and then easily branched out to other FGs by simple transformation is particularly valuable in modern organic synthesis.Direct C(sp2)–H bond halogenations,3 borylation,4 and silylation5 have served as seminal examples of diverse functionalization and transformations. For unactivated C(sp3)–H bonds, due to the selectivity and inert characteristics of the FGs, diversification of a functionalized product presents a greater challenge.6 Oxygen-containing FGs, such as OH, sulfonyloxy groups (for example, OMs and OTs) and trifluoroacetoxyl groups (OTFA), are generally regarded as common transformation precursors for derivatizations.7 Site-selective C(sp3)–H bond oxidation is a straightforward yet valuable synthetic method to install such groups (Scheme 1, path a).8 However, direct C–H bond oxidation usually suffers from poor selectivity when multiple electronically and sterically similar C–H bonds are present in one molecule. In efforts to better control the selectivity, directed C–H oxidation has been developed as exemplified by transition-metal-catalyzed C–H oxidation reactions.9 However, the diverse functionalization through a suitable installed group via C(sp3)–H oxidation presents a challenge. A typical example has been reported by Dong and co-workers who developed a directed primary C–H bond sulfonyloxylation, followed by diverse functionalization (path b).10 Although this approach shows promising potential, it remains challenging to conduct methylene and remote C(sp3)–H functionalization.11,12 Considering the ubiquity of amino groups, particularly in common FGs in natural products and pharmaceuticals, herein, we describe a coinage-metal-catalyzed site-selective secondary C–H trifluoroacetoxylation of aliphatic amines and late-stage diversification (path c).
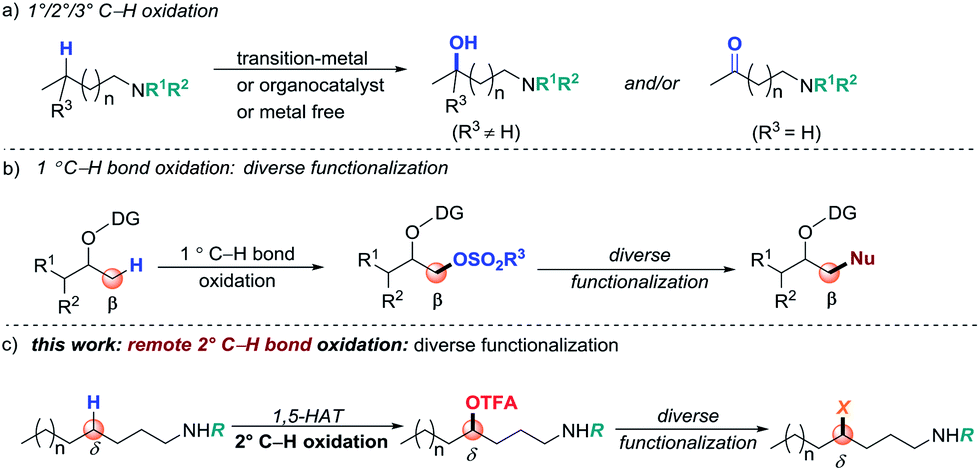 | ||
| Scheme 1 Strategies for directing C–H bond functionalization and transformation. | ||
Results and discussion
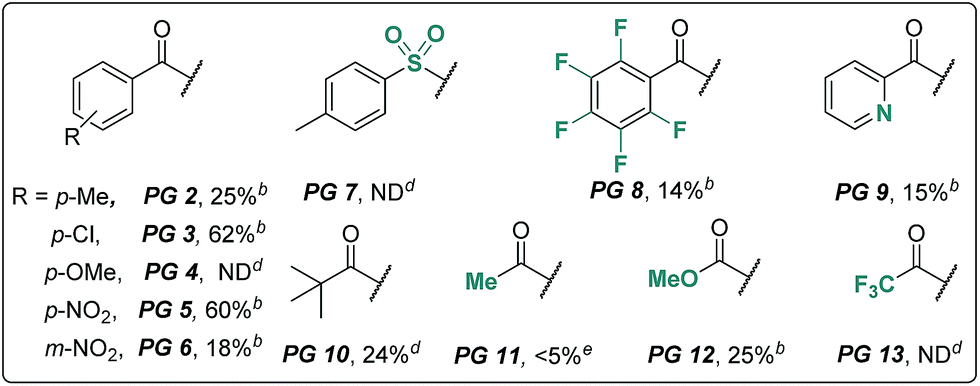
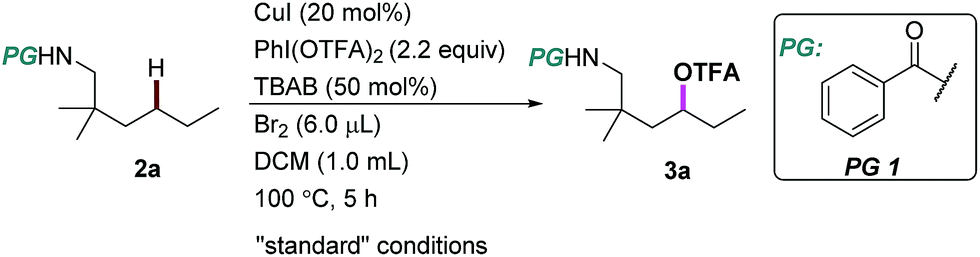
We began our investigation using an aliphatic amide 2a as the model substrate. After examining a range of catalysts, additives, and protecting groups (PGs), we found that the reaction gave exclusively the δ trifluoroacetoxylation (–OTFA) product 3a in 72% isolated yield with 2.2 equiv. of PhI(OTFA)2 as the oxidant, the benzoyl group (Bz) as the PG, and 20 mol% of CuI, 50 mol% of tetrabutylammonium bromide (TBAB) and 1.2 equivalent of Br2 as additives. The selectivity of the reaction was very good; 22% of starting material 2a was recovered and no other byproduct was observed. The influence of the PGs is obvious; e.g., the benzoyl substituent with electron-withdrawing groups is more efficient than the one with electron-donating groups (PG2–PG4). Electron deficient sulphonamide (PG7), which is energetically accessible, using the traditional 1,5-HAT-based approach failed to afford the desired product.10 Notably, pentafluorobenzoyl13 and picolinoyl groups14 have been proved to be powerful directing groups in transition-metal-catalyzed C–H bond activation reactions, affording the product in low yields (PG8–PG9). Finally, alkyl-, or alkoxyl-carbonyl PGs led to a complete or significant loss of reactivity (PG10–PG13).The influence of other reaction parameters was explored (Table 1, entries 2–15). Without a copper catalyst, the yield decreased significantly (Table 1, entry 2), even upon extension of the reaction time to 24 hours (Table 1, entry 3). Copper catalysts other than copper iodide (CuI) also provided the desired product (Table 1, entries 4–7), but product yields were low. Interestingly, silver salt was also effective in this reaction, albeit in relatively low efficiency. The additives Br2 and tetrabutylammonium bromide (TBAB) are necessary to improve the product yield (Table 1, entries 8–10). Compared with other solvents, DCM proved to be the best choice in this oxidation reaction (Table 1, entries 11–13). The reaction can be performed almost equally well at higher temperature (120 °C, Table 1, entry 14). At lower temperature (80 °C) the yield was slightly decreased (Table 1, entry 15).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Variations from “standard” conditions | Yield (%) (rsm)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction was conducted on a 0.1 mmol scale. b rsm: recovered starting material; yields were determined by 1H NMR analysis versus 1,1,2,2-tetrachloroethane as the internal standard. c Reaction time is 24 h. d ND: not detected. Determined by crude 1H NMR. e Detected by crude 1H NMR. f TBAB3: tetrabutylammonium tribromide. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | None | 72 (22) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Without CuI | 25 (66) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Without CuI | 39 (45)c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | CuBr instead of CuI | 33 (50) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CuBr2 instead of CuI | 31 (58) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Cu(OAc)2 instead of CuI | 52 (22) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Cu(OTf)2 instead of CuI | 20 (68) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Without Br2 | 31 (64) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Without TBAB | 6 (89) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | TBAB3f instead of TBAB | 38 (39) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | DCE instead of DCM | 31 (42) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | CHCl3 instead of DCM | 25 (54) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | PhCl instead of DCM | 10 (57) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 120 °C instead of 100 °C | 69 (22) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 80 °C instead of 100 °C | 59 (24) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Subsequently, a range of aliphatic amides with different carbon chains was examined under standard conditions (Table 2). Substituents such as ethyl (3a), methyl (3b) and n-hexyl (3c) groups at the β-position were all well tolerated. Even with much longer carbon chains in 3d or 3e, the reaction occurred only at the δ-position. Exclusive δ-selectivity was observed in the case of 3f bearing a tertiary C–H bond near the reaction center. To our satisfaction, we found that O-containing FGs are well tolerated (3g–3j). Remarkably, the excellent leaving group –OTs (4-tolylsulfonoxyl) was compatible. A gem-dialkyl group on the linear aliphatic amide substrates at the β-position is necessary to improve the product yield. Both a dialkyl group and a six-membered ring are much better than mono-alkyl groups (3k–3p). Interestingly, the primary C–H bond (3k, 3l) and the secondary C–H bond on a six-membered ring (3o, 3p) remained untouched. Significantly, the substituents at the β position of amides play an important role in increasing diastereoselectivities (3q–3s). With a bulky isopropyl group, the diastereomer ratio increased to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (3s). The diastereoselectivities, which are dependent on the steric demand of the β-substituents, may suggest the involvement of a cyclic intermediate during the reaction pathway. The tertiary C–H bond did not afford any desired product (3u). Finally, Bz-protected 2°- and 3°-amine substrates were tested, respectively (3v, 3w).
:1 (3s). The diastereoselectivities, which are dependent on the steric demand of the β-substituents, may suggest the involvement of a cyclic intermediate during the reaction pathway. The tertiary C–H bond did not afford any desired product (3u). Finally, Bz-protected 2°- and 3°-amine substrates were tested, respectively (3v, 3w).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Unless otherwise noted, the reaction conditions were as follows: 2a (0.1 mmol), TBAB (0.5 equiv.), Br2 (1.2 equiv.), DCM 1.0 mL, rt, 15 min; then CuI (20 mol%), PhI(OTFA)2 (2.2 equiv.), 100 °C, 5 h. b 8 h. c PhI(OTFA)2 (2.5 equiv.), 8 h. d AgOAc (20 mol%) instead of CuI (20 mol%), PhI(OTFA)2 (2.5 equiv.), 8 h. e PhI(OTFA)2 (3.0 equiv.), 8 h. f BRSM: based on recovered starting materials; isolated BRSM yield in parentheses. g ND: not detected. Determined by crude 1H NMR. h dr: determined by crude 1H NMR. i CuI (20 mol%) instead of AgOAc (20 mol%). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

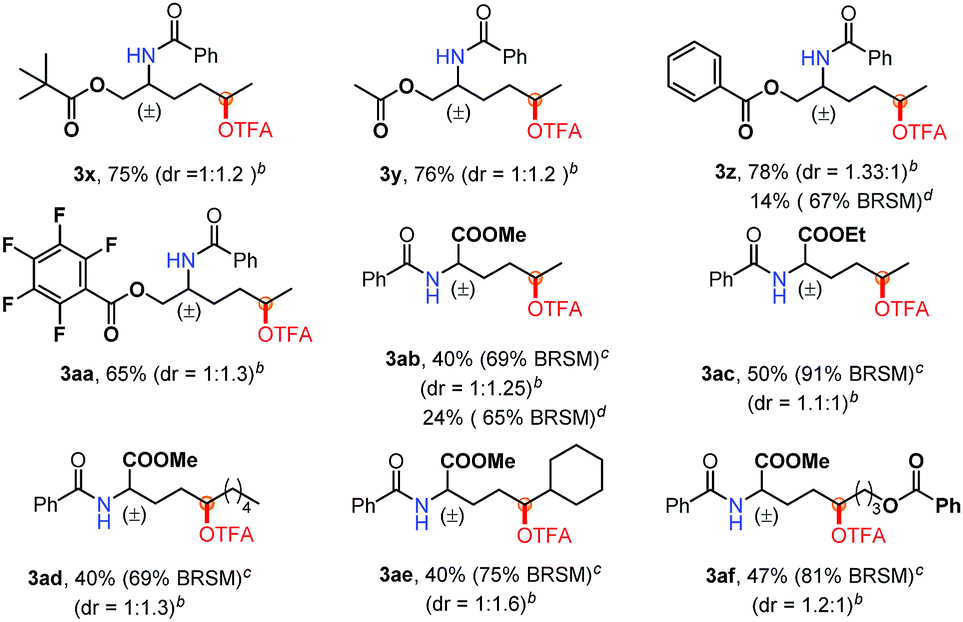
Furthermore, to demonstrate the power of this method, a variety of amino acid derivatives were subjected to this oxidation reaction (Table 3). Notably, the reaction of protected amino alcohols (3x–3aa) proceeded smoothly when using AgOAc as the catalyst instead of CuI (3z, 3ab). The biologically relevant functionalities, such as OPiv, OAc and OBz, as well as pentafluorobenzyl groups are well tolerated. Such functionalized amino alcohols have great potential in the synthesis of biological compounds. Moreover, we investigated various amino acid esters to probe their synthetic utility. The electron-deficient substitutes [CO2Me (3ab–3af) and CO2Et (3ac)] are compatible with this oxidation reaction. In particular, the amino acid ester 2ae gave only a δ position oxidation product (3ae), regardless of the presence of a tertiary C–H bond at the ε position. The mass balance of these reactions [the yield of the product and rsm] (Table 3) indicates that indiscriminate oxidation did not occur.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Unless otherwise noted, the reaction conditions were as follows: 2a (0.1 mmol), TBAB (0.5 equiv.), Br2 (1.2 equiv.), DCM 1.0 mL, rt, 15 min; then AgOAc (20 mol%), PhI(OTFA)2 (2.5 equiv.), 100 °C, 8 h. b dr was determined by crude 1H NMR. c Isolated yield BRSM in parentheses. d Using CuI (20 mol%) as the catalyst, determined by crude 1H NMR. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

To further understand the reactivity of this catalytic system, the benzoyl protected aliphatic amine 2a was subjected to a variety of reported Hofmann–Löffler–Freytag (HLF) type reaction conditions.15 However, neither a halogenation product nor an intramolecular amination product was observed under these conditions, thus resulting in the recovery of the starting material.16
To gain insight into the reaction mechanism, radical capture experiments were carried out to uncover evidence for the presence of any radical intermediate. By adding TEMPO and 2,6-di-tert-butylphenol to the reaction mixture, independently, the reaction was completely inhibited, indicating that a radical process could be involved (see the ESI†). Furthermore, to determine the role of Br2 and TBAB in the transformation, control experiments between Br2 and TBAB were carried out (Scheme 2, eqn (1) and (2)). When TBAB was omitted, the desired product was obtained in very low yield (6%) (Scheme 2, eqn (1)), while in the absence of Br2, the desired product could be obtained in 31% yield. The combination of Br2 and TBAB was necessary for improving the efficiency. This result indicated that N–Br bond formation may not be the only pathway in the initial amidinyl radical generation. Finally, a competition reaction between 2° and 3° C–H bonds was carried out. Only trifluoroacetoxylation of the 2° C–H bond was observed (Scheme 2, eqn (3)). The steric hindrance effect at the δ-position may have inhibited this transformation.
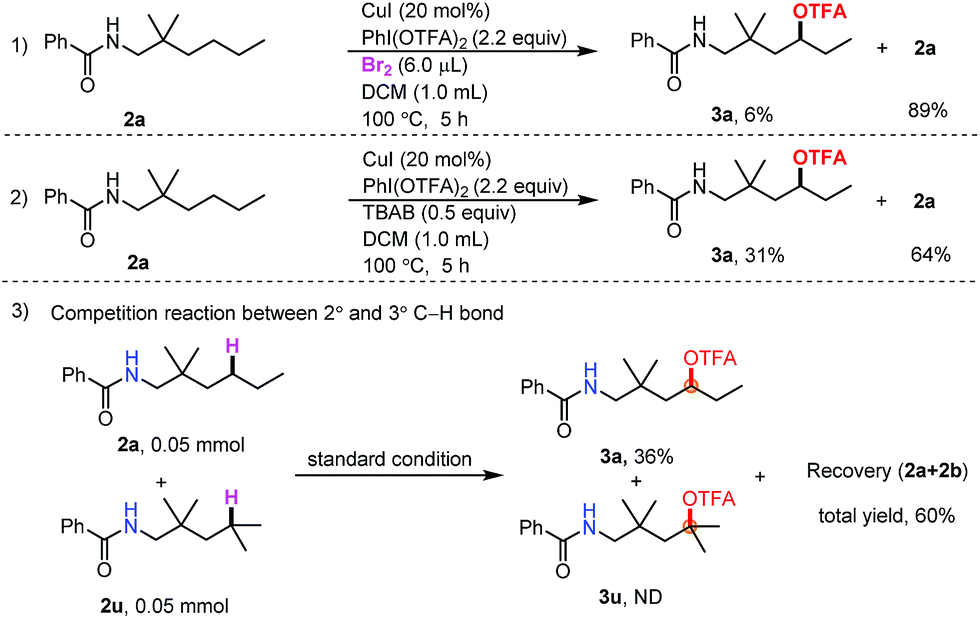 | ||
| Scheme 2 Controlled experiments. | ||
Based on these results, we propose that this transformation proceeds through a single-electron-oxidation process (Scheme 3). With the assistance of PhI(OTFA)2 and the copper catalyst, the N–Br intermediate I (path a) or Cu(III)–amidine intermediate III (path b) can be generated.17 Subsequent homolysis of the N–Br or N–Cu bond afforded amidinyl radical II. This was followed by a 1,5-H radical shift to give the corresponding C-radical V, which resulted in the selective C–H bond trifluoroacetoxylation at the δ position. For the next step, two possible pathways could be followed: one is the oxidative addition of a carbon radical by a Cu(II) catalyst to generate Cu(III)–amide intermediate IV, and after ligand exchange and reductive elimination to afford the trifluoroacetoxylation product 3a (path c). The other is the oxidation of the carbon radical to a carbocation (VI) which is captured by OTFA−, to directly give the product (path d). Although we didn't observe the formation of any δ-brominated product (VII) even with decreased PIFA or shorter reaction time, the possibility that the product is formed through a substitution reaction of intermediate VII cannot be ruled out.
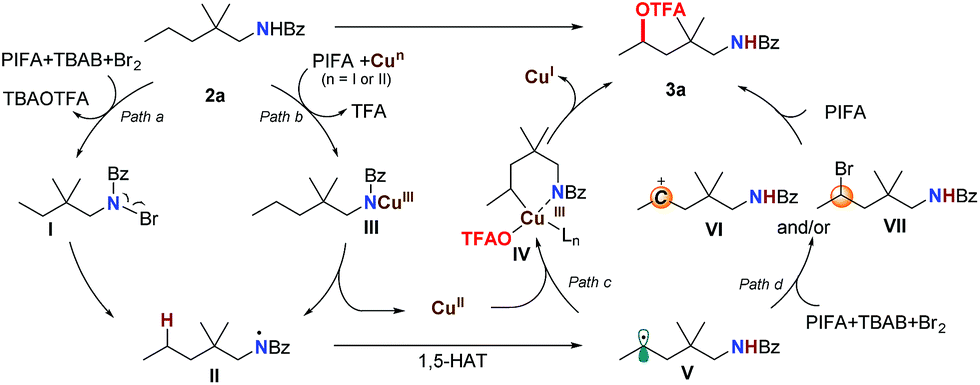 | ||
| Scheme 3 Proposed mechanism. | ||
Finally, the relatively large scale experiment and application of complex molecules with potential bioactivity, as well as late-stage functionalization of these molecules were demonstrated (Scheme 4). The commercially available lithocholic acid derivative 2ag was converted to 3ag in 63% yield, which serves as a common intermediate for diversification. Intramolecular cyclization of 3ag gave the pyrrolidine derivative 4ag. The subsequent SN2 reactions afforded, rapidly, a variety of 3ag derivatives through the formation of C–N (5ag), C–O (6ag), and C–Cl (7ag) bonds. Hydrolysis of 3ag afforded the alcohol 6ag, followed by oxidation to give the ketone 8ag.
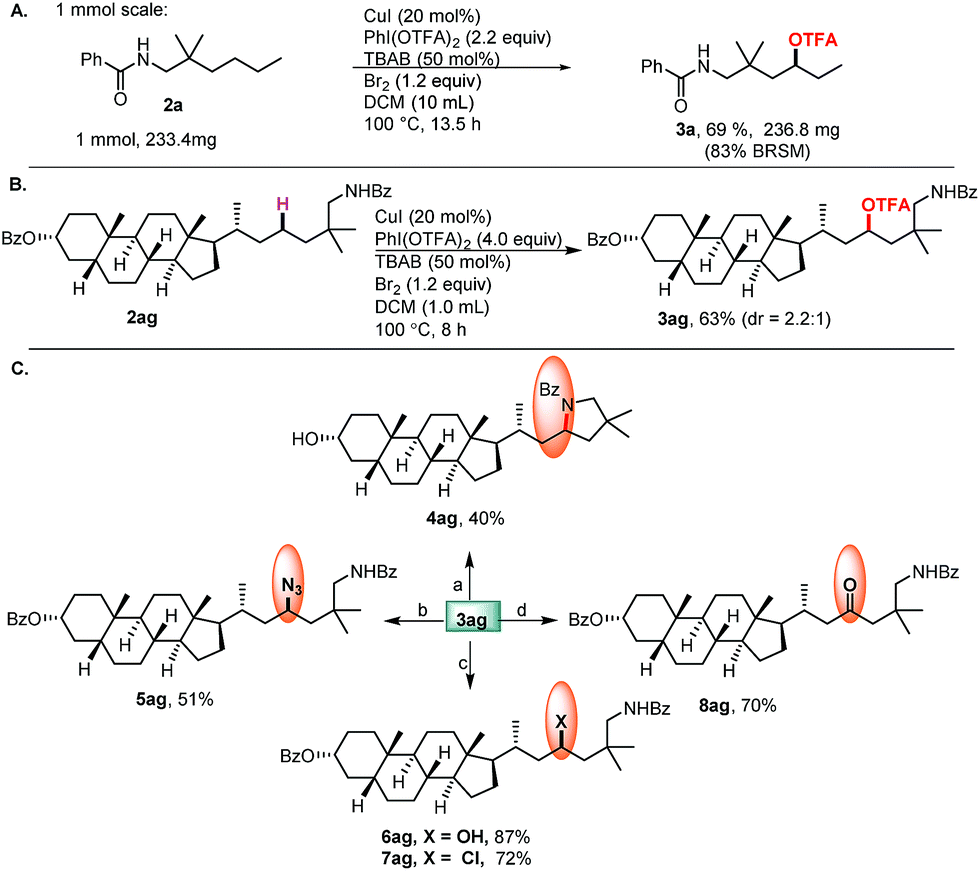 | ||
| Scheme 4 (A) 1 mmol scale experiment; (B) site-selective oxidation of complex molecules; (C) late-stage functionalization of steroidal compounds.a aReagents and conditions: (a) t-BuOK (3.6 equiv.), THF, −78 °C, 30 min, then rt, 15 h, 40%, dr = 1:0.9; (b) NaN3 (2.0 equiv.), DMF, 90 °C, 16 h, 51%, dr = 2:3; (c) K2CO3 (4.0 equiv.), DMF, 100 °C, 5 h, 87%, dr = 2.2:1; SOCl2 (2.0 equiv.), DCM, 100 °C, 1.5 h, 72%, dr = 1:2; (d) K2CO3 (4.0 equiv.), DMF, 100 °C, 5 h; PCC (1.2 equiv.), DCM, rt, 2 h, 70%. | ||
Conclusions
In summary, we have reported a diverse secondary C(sp3)–H functionalization of aliphatic amines. Both secondary C–H bond oxidation and late-stage diversification were achieved through a simple trifluoroacetoxylation intermediate. Broad amine scope, good functional compatibility, and exceptional site selectivity for secondary C(sp3)–H bonds over tertiary ones underscore the unique synthetic potential of this method. Furthermore, mechanistic studies and efforts to expand the scope of this transformation are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (21572124 and 21602128), Fundamental Research Funds for the Central Universities (GK201802008), and start-up funds from Shaanxi Normal University, Shaanxi Provincial Natural Science Foundation (2016ZDJC-03, 2018JM2010). We thank Professor M. Sawamura (Hokkaido University) for assistance in the manuscript revision. We also thank Prof. Ning Jiao (Perking University), Prof. Da-Gang, Yu (Sichuan University), Prof. Bijie, Li (Tsinghua University), Prof. Yu-Ming, Zhao and Prof. Tao Wang (Shannxi Normal University) for valuable discussion and suggestions.Notes and references
- (a) L. A. Thompson and J. A. Ellman, Chem. Rev., 1996, 96, 555 CrossRef PubMed; (b) M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007 CrossRef PubMed; (c) K. C. Nicolaou, Chem. Biol., 2014, 21, 1039 CrossRef PubMed.
- For recent selected reviews on direct C–H functionalization, see: (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef PubMed; (b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef PubMed; (d) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC. Direct C–H functionalization, see: (e) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef PubMed; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed; (g) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef PubMed; (i) G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef PubMed.
- (a) T.-S. Mei, D.-H. Wang and J.-Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef PubMed; (b) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef PubMed.
- (a) T. Ishiyama, J. Takagi, K. Ishida and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 390 CrossRef PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef PubMed; (c) S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058 CrossRef PubMed; (d) T. Ishiyama, K. Sato, Y. Nishio and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 5346 CrossRef PubMed.
- (a) T. Lee, T. W. Wilson, R. Berg, P. Ryberg and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 6742 CrossRef PubMed; (b) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef PubMed; (c) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592 CrossRef PubMed.
- (a) T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306 CrossRef PubMed; (b) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847 CrossRef PubMed; (c) V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389 CrossRef PubMed.
- (a) O. Mitsunobu, Comprehensive Organic Synthesis. Selectivity, Strategy and Efficiency in Modern Organic Chemistry, ed. B. M. Trost and I. Fleming, Oxford, Pergamon, 1991, vol. 6, pp. 1–33 Search PubMedFor OTFA as a leaving group, see: (b) S. V. Pronin, C. A. Reiher and R. A. Shenvi, Nature, 2013, 501, 195 CrossRef PubMed; (c) C. A. Reiher and R. A. Shenvi, J. Am. Chem. Soc., 2017, 139, 3647 CrossRef PubMed.
- For selected reviews on C–H oxidation, see: (a) M. Costas, K. Chen and L. Que Jr, Coord. Chem. Rev., 2000, 200–202, 517 CrossRef; (b) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef PubMed; (c) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899 RSC. For selected transition-metal catalyzed C–H oxidation, see: (d) C. Kim, K. Chen, J. Kim and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 5964 CrossRef; (e) B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391 CrossRef PubMed; (f) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef PubMed; (g) Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 13776 CrossRef PubMed; (h) J. B. C. Mack, J. D. Gipson, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 9503 CrossRef PubMed.
- For selected directing group assisted C–H bond oxidation, see: (a) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef PubMed; (b) D.-H. Wang, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef PubMed; (c) B.-J. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 6586 CrossRef PubMed; (d) R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef PubMed.
- Y. Xu, G. Yan, Z. Ren and G. Dong, Nat. Chem., 2015, 7, 829 CrossRef PubMed.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef PubMed; (c) D. Meng, Y. Tang, J. Wei, X. Shi and M. Yang, Chem. Commun., 2017, 53, 5744 RSC; (d) L. R. Reddy, B. V. S. Reddy and E. J. Corey, Org. Lett., 2006, 8, 2819 CrossRef PubMed; (e) N. R. Paz, D. Rodíguez-Sosa, H. Valdés, R. Marticorena, D. Melián, M. B. Copano, C. C. González and A. J. Herrera, Org. Lett., 2015, 17, 2370 CrossRef PubMed; (f) R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994 CrossRef PubMed.
- (a) G. C. Choi, Q. Zhu, D. Miller, C. Gu and R. R. Knowels, Nature, 2016, 539, 268 CrossRef PubMed; (b) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef PubMed; (c) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692 CrossRef PubMed; (d) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771 CrossRef PubMed; (e) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2017, 57, 296 CrossRef PubMed; (f) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340 CrossRef PubMed; (g) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857 CrossRef PubMed. For oxyradicals, see: (h) L. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 128, 1904 CrossRef; (i) A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612 CrossRef PubMed; (j) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (k) X. Hu, J. Chen and W. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960 CrossRef PubMed; (l) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (m) D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897 CrossRef PubMed; (n) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569 CrossRef PubMed.
- T. M. Figg, M. Wasa, J.-Q. Yu and D. G. Musaev, J. Am. Chem. Soc., 2013, 135, 14206 CrossRef PubMed.
- S.-Y. Zhang, G. He, Y. Zhao, K. Wright, A. William, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef PubMed.
- (a) C. Betancor, J. I. Concepcion, R. Hernandez, J. A. Salazar and E. Suárez, J. Org. Chem., 1983, 48, 4430 CrossRef; (b) C. G. Francisco, A. J. Herrera and E. Suárez, J. Org. Chem., 2003, 68, 1012 CrossRef PubMed; (c) C. Martínez and K. Muńiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef PubMed; (d) C. Q. O'Broin, P. Fernández, C. Martínez and K. Muńiz, Org. Lett., 2016, 18, 436 CrossRef PubMed; (e) E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef PubMed.
- Under Yu reported reaction conditions, a little amount of bromination product was detected from analysis of the crude 1H NMR.
- For the generation of amidinyl radicals with copper and PIFA, see: H. Chen, S. Sanjaya, Y.-F. Wang and S. Chiba, Org. Lett., 2013, 15, 212 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and compound characterization. See DOI: 10.1039/c8sc01788c |
| This journal is © The Royal Society of Chemistry 2018 |