
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibody–nucleotide conjugate as a substrate for DNA polymerases†
J.
Balintová
,
M.
Welter
and
A.
Marx
 *
*
Department of Chemistry, University of Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany. E-mail: andreas.marx@uni-konstanz.de
First published on 24th July 2018
Abstract
Here we report on the development of an antibody-modified nucleotide and its sequence-selective incorporation into nascent DNA catalysed by DNA polymerases. Although the modification of the nucleotide is several orders of magnitude larger than the natural dNTP substrate and even exceeds the size of the DNA polymerase, it is well accepted by the enzyme. Moreover, the recognition of the antibody is not abolished by the conjugation but can be recognized by a secondary antibody that is conjugated to a signal-generating enzyme (i.e., horse radish peroxidase). This product can thus be exploited for a colorimetric read-out of nucleotide incorporation by the naked eye that allows detection of DNA as low as 10 amol. In future, assays like the one described herein might allow nucleic acid diagnostics at single nucleotide resolution without any laboratory equipment.
Introduction
DNA is one of the most important and widely studied molecules due to its high importance in living organisms. Therefore, many approaches to analyse, amplify, and modify DNA are of prime scientific importance. In order to be suitable in the many biotechnological applications that require recognition by a DNA polymerase including sequencing, structure characterization and immobilization, natural nucleotide analogues are substituted with analogues that are modified at the C5 position of pyrimidines and the C7 position of 7-deazapurines.1 During DNA synthesis, DNA polymerases are able to recognize these modified nucleoside triphosphates (dNTP) as substrates and incorporate the modified nucleotide into DNA.1Larger functionalities are often connected to the nucleobase via linkers that vary in composition, length and flexibility. With this strategy a variety of probes, even very bulky modifications, could be incorporated into DNA.2 Along these lines, it has been shown that DNA polymerases are capable of efficient incorporation of nucleotides modified with entities as large as proteins, such as horseradish peroxidase (HRP), despite this “cargo” being more than 100-fold larger than the natural substrates.2a Incorporation of the HRP-modified nucleotide was harnessed for the sequence specific detection of DNA by the naked eye. However, the capability of HRP-labelled nucleotides in the naked-eye detection assay has been limited by a nonspecific background and somewhat high detection limit of 1 fmol DNA. In order to overcome these limitations, we investigated new detection approaches following the principle of sequence-selective DNA polymerase–catalysed nucleotide incorporation and evaluated antibody–nucleotide conjugates since they are widely used for highly sensitive analytics.3 Thus, we focused on the generation and evaluation of an antibody-modified dNTP. This modified nucleotide should in turn be accepted as a substrate for sequence-specific nucleotide incorporation by a DNA polymerase. Of note, the antibody-modified dNTP exceeds the size of the DNA polymerase and – of course – the size of the natural dNTP substrates significantly (Fig. 1).
 | ||
| Fig. 1 Size comparison to scale of KOD DNA polymerase, the antibody IgG and a modified dCTP bearing an azide linker. | ||
ELISA (enzyme-linked immunosorbent assay) is a technique developed for detection and quantification of analytes such as peptides, proteins, antibodies and hormones.4 We envisioned to use ELISA to convert nucleotide incorporation into a signal that is detectable by the naked eye. Thus, a secondary antibody that was conjugated to HRP was used to recognize the incorporated nucleotide–antibody conjugate and thereby converts a supplemented substrate to a product that builds the signal. Enzyme-conjugated antibodies (e.g., antibody-HRP conjugates) offer flexibility in detection because of the variety of substrates available for chromogenic, chemifluorescent and chemiluminescent imaging.5
Indeed, we found that DNA polymerases are capable of using dNTP–antibody conjugates as substrate for sequence-selective nucleotide incorporation into the nascent primer strand despite the unprecedented bulk of the modification. On this basis, we developed a system that enables the naked-eye detection of DNA at single-nucleotide resolution with high sensitivity and a DNA detection limit as low as 10 amol.
Results and discussion
Synthesis of functionalized dNTP
The most commonly employed method for conjugation of monoclonal antibodies to other molecules is the use of heterobifunctional cross-linkers to introduce a linker between the antibody and the molecule of interest.6 Labelling of amines is a widely used method to tag antibodies. To obtain the desired antibody-labelled dNTP, the direct bioconjugation of a functionalized nucleotide (dCC16N3TP) to an antibody (mouse IgG1-Apoliprotein A1 monoclonal antibody, pAb) was developed herein (Scheme 1). | ||
| Scheme 1 (a) Conjugation of azide-modified nucleotide to antibody; (b) structure of antibody-modified nucleotide (dCpAbTP). NHS: N-hydoxysuccinimide, DBCO: dibenzocyclooctyne. | ||
Our approach of the bioconjugation process was based on strain-promoted alkyne–azide cycloaddition (SPAAC),7 in which the antibody is activated with a dibenzocyclooctyne (DBCO) moiety and subsequently covalently linked with an azide-modified dNTP (Scheme 1, ESI-Schemes 1 and 2†). First, the azide-modified nucleotide (dCC16N3TP) was synthesized by using HATU-promoted coupling of 5-(aminopentynyl)-2′-deoxycytidine triphosphate8 with 16-azidohexadecanoic acid in 35% yield (ESI-Scheme 1†).6f For DBCO functionalization of the herein used pAb, DBCO-PEG4-NHS was employed to react with amines of the antibody's lysines.6f DBCO-PEG4-NHS is poorly soluble in buffer and it requires solubilisation in an organic co-solvent such as DMSO. The presence of the organic solvent helps to prevent the labelling reagent to precipitate and improves efficacy of the conjugation reaction. However, we found that the co-solvent should not exceed 10% of the reaction mixture in order to retain activity and prevent aggregation. After incubation of pAb with DBCO-PEG4-NHS (5 eq.) for 2 hours we obtained 3.1 DBCO molecules per pAb after the reaction using the molar extinction coefficient of DBCO (for experimental details see ESI†). To obtain the antibody-labelled nucleotide (dCpAbTP), the DBCO-functionalized antibody was conjugated to the azide-labelled nucleotide (dCC16N3TP, 10 eq.) by incubation in PBS buffer (pH 7.4) at 4 °C for 16 h.
Processing of antibody labelled dNTP by DNA polymerases
In order to validate whether DNA polymerases are capable of incorporating the antibody-labelled dNTP (dCpAbTP) into DNA, single-nucleotide incorporation experiments were performed by employment of the antibody-labelled nucleotide (dCpAbTP). We used a primer extension (PEx) assay with a 5′-32P labelled primer, a template in the sequence context of the B-type Raf kinase (BRAF) T1796A point mutation (see ESI for sequences Table 1†) and different DNA polymerases [KOD, KlenTaq (KTq) and 9°N DNA polymerases (DNA pol), also see ESI-Fig. 1†]. The insertion of modified nucleotides into DNA was subsequently analysed by denaturing polyacrylamide gel electrophoresis (PAGE) and autoradiography. We found that the employment of antibody-labelled dNTP (dCpAbTP) in these reactions resulted in a drastically slower migrating product when a G was present at the incorporation site in the template sequence (Fig. 2, BRAF-G and ESI-Fig. 1†). Little if any incorporation was observed opposite the other nucleobases A, C and T (Fig. 2, BRAF-A, C and T). Thus, the incorporation is selective towards its cognate nucleobase pair despite the modifications of the nucleotide.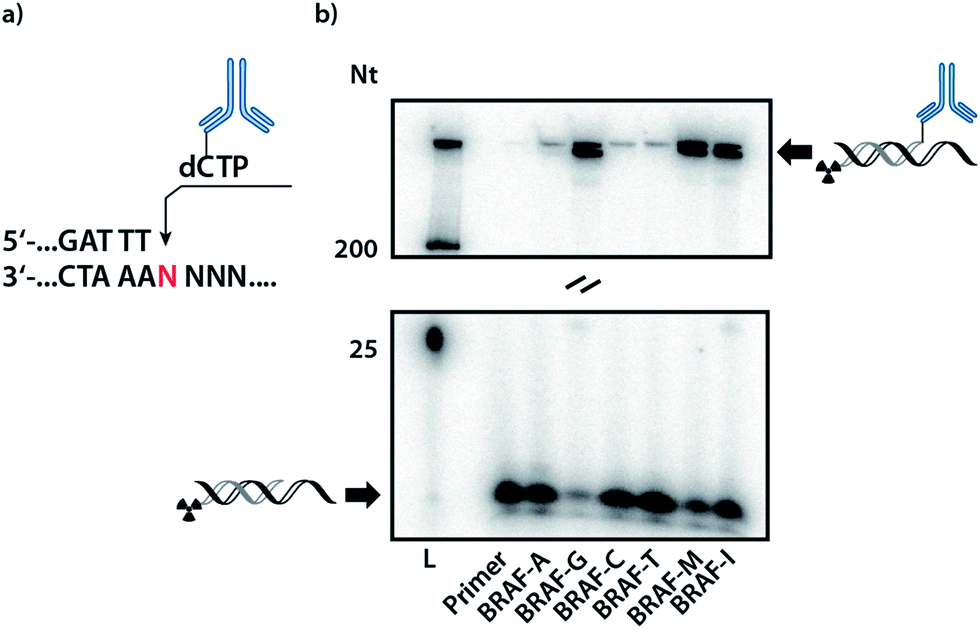 | ||
| Fig. 2 PAGE of PEX single and multiple incorporation of the modified nucleotide (dCpAbTP). (a) Partial sequence of the primer and the template at the incorporation site; (b) autoradiography of a primer extension reaction employing the conjugate (dCpAbTP) with KOD DNA polymerase on templates containing the BRAF T1796A point mutation site with mismatch pairings and multiple incorporation sites (see sequence table). Samples were collected after 5 min. L: Marker. | ||
For all incorporations we noted that the bands observed are split into two separate bands. This might be caused by the mutli-labeled conjugate being incorporated into a second primer strand, thus increasing the retardation in the gel.
Next to the mismatch templates, we employed templates encoding for multiple incorporation in an either consecutive (Fig. 2, BRAF-M) or iterative fashion (BRAF-I). As the shifted bands of a monoincorporation are only barely migrating in an 9% PAGE gel, we used gradient SDS-PAGE to check for multiple incorporation (see ESI-Fig. 2†). Here we observed the formation of a new band for the consecutive template, indicating the possibility to incorporate more than one conjugate in PEx assays. However, we found no additional band in case of the iterative template. As the analysis of a possible incorporation after the conjugate is however strongly impeded by the highly retarded migration in the gel, we cannot completely rule out, that the primer can be further elongated by natural nucleotides.
In order to evaluate the efficiency of incorporation of the antibody-modified nucleotide (dCpAbTP) in comparison to natural nucleotides, we performed single-nucleotide incorporation experiments in which the antibody-labelled nucleotide directly competed for incorporation with its natural counterpart, as described before.2a The ratio of unmodified dCTP vs. modified nucleotide (dCpAbTP) was determined by PAGE through the significantly different migration caused by the incorporation of the bulky modification. We observed that KOD DNA pol incorporates the antibody-modified nucleotide (dCpAbTP) with approximately 78-fold lower efficiency than the natural nucleotide (ESI-Fig. 3†).
Naked-eye detection assay employing dCpAbTP
We anticipated that incorporation of the antibody-modified nucleotide into a DNA by PEx can be converted into a signal by incubation with a HRP-conjugated secondary antibody and the subsequent addition of a HRP substrate that is in-turn converted into a product enabling a colorimetric read-out (Fig. 3).9 We envisaged that this approach could be used as a new method for the development of a DNA sequence-selective naked-eye detection system. | ||
| Fig. 3 The naked-eye detection assay employing dCpAbTP. | ||
This “sandwich” assay offers major advantages: first, each antibody contains several epitopes that can be bound by the labelled secondary antibody, allowing signal amplification. Secondly, different entities that are conjugated to the secondary antibody allow visualization by different modes (e.g., fluorescent tags or enzymes such as horseradish peroxidase and alkaline phosphatase) using the same primary antibody.4
For our naked-eye detection system, a biotinylated primer strand is first immobilized on streptavidin-coated beads. Due to the template-dependency of the DNA polymerase reaction, a primer that binds sequence-selectively to a target sequence is extended by elongation with an antibody-modified nucleotide. After the removal of the excess of the antibody-modified dNTP, an enzyme-conjugated secondary antibody (goat anti-mouse IgG H + L, HRP) with specificity against the primary antibody was added in order to bind to the antibody-labelled DNA. After the removal of the excess of the secondary antibody and subsequent addition of the substrate (either the chromogenic substrate o-dianisidine or the fluorogenic substrate Amplex Red),10 HRP, which is conjugated to the secondary antibody, catalyses the conversion of the substrate to a detectable product, allowing– in the end – direct visualization of the nucleotide incorporation event (Fig. 4, ESI-Fig. 4†).
 | ||
| Fig. 4 Quantification of the naked-eye detection assay using dCpAbTP by UV/Vis spectroscopy (a and c) and naked eye (b and d) using KOD DNA pol in the presence/absence of the indicated amount of template and dCpAbTP (0.17 μM) using the substrates o-dinanisidine (a and b) and Amplex red (c and d). | ||
The negative control reactions (absence of template and absence of antibody-modified nucleotide dCpAbTP) have been performed to reveal false positive signals caused by deficient washing or unspecific binding of the antibody-modified nucleotides and the secondary antibody (ESI-Fig. 4†). When employing the antibody-modified nucleotide (dCpAbTP), we were able to detect 50 amol of DNA by using the chromogenic substrate o-dianisidine (Fig. 4c) and 10 amol of DNA by using the fluorogenic substrate Amplex red (Fig. 4d).
Conclusions
Here we report on the development of an antibody-modified nucleotide and its sequence-selective incorporation into nascent DNA catalysed by DNA polymerases. The ‘cargo’ of the modified nucleotide is not only several orders of magnitude larger than the natural dNTP substrates but also exceeds the size of the DNA polymerase (see Fig. 1). However, the antibody-conjugated nucleotide is incorporated in a template and sequence specific manner despite its unprecedented size. Moreover, the recognition of the antibody is not abolished by the conjugation but can even be recognized by a secondary antibody that is conjugated to a signal-generating enzyme (i.e., HRP) and thus be exploited for a colorimetric read-out of nucleotide incorporation by the naked eye. The major advantage of this “sandwich” assay is, that the antibody contains several epitopes that can be bound by the labelled secondary antibody, allowing signal amplification and gain of sensitivity. We show that the colorimetric read-out of the nucleotide incorporation is detectable by the naked eye and allows detection of DNA as low as 10 amol. This is an about 100-fold increase in sensitivity compared to the approach in which HRP was directly appended to the nucleotide.2aIn future, assays like the one described herein might allow nucleic acid diagnostics at single nucleotide resolution without any laboratory equipment, like thermocyclers, and thus will be suited for in field analysis and point-of-care testing (POCT).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support by the Alexander von Humboldt-Foundation (stipend to J. B.), the Deutsche Forschungsgemeinschaft (DFG) and Konstanz Research School Chemical Biology.Notes and references
- (a) D. R. Bentley, et al. , Nature, 2008, 456, 53 CrossRef PubMed; (b) T. D. Harris, et al. , Science, 2008, 320, 106 CrossRef PubMed; (c) R. Drmanac, et al. , Science, 2010, 327, 78 CrossRef PubMed; (d) P. R. Langer, A. A. Waldrop and D. C. Ward, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 6633 CrossRef PubMed; (e) S. Ishino and Y. Ishino, Front. Microbiol., 2014, 5 Search PubMed; (f) M. Hocek, J. Org. Chem., 2014, 79, 9914 CrossRef PubMed; (g) A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418 CrossRef PubMed.
- (a) M. Welter, D. Verga and A. Marx, Angew. Chem., Int. Ed., 2016, 55, 10131 CrossRef PubMed; (b) L. H. Thoresen, G.-S. Jiao, W. C. Haaland, M. L. Metzker and K. Burgess, Chem. –Eur. J., 2003, 9, 4603 CrossRef PubMed; (c) D. Cherkasov, T. Biet, E. B-uml, W. Traut and M. Lohoff, Bioconjugate Chem., 2010, 21, 122 CrossRef PubMed; (d) Z. Zhu, J. Chao, H. Yu and A. Waggoner, Nucleic Acids Res., 1994, 22, 3418 CrossRef PubMed; (e) C. J. Lacenere, N. K. Garg, B. M. Stoltz and S. R. Quake, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 9 CrossRef PubMed.
- D. Wild, The Immunoassay Handbook, Elsevier Science, 4th edn, 2013 Search PubMed.
- (a) E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871 CrossRef PubMed; (b) R. M. Lequin, Clin. Biochem., 2005, 51, 2415 Search PubMed; (c) T. Waritani, J. Chang, B. McKinney and K. Terato, MethodsX, 2017, 4, 153 CrossRef PubMed.
- S. Aydin, Peptides, 2015, 72, 4 CrossRef PubMed.
- (a) N. Kotagiri, Z. Li, X. Xu, S. Mondal, A. Nehorai and S. Achilefu, Bioconjugate Chem., 2014, 25, 1272 CrossRef PubMed; (b) A. M. Sochaj, K. W. Świderska and J. Otlewski, Biotechnol. Adv., 2015, 33, 775 CrossRef PubMed; (c) N. J. Alves, N. Mustafaoglu and B. Bilgicer, Bioconjugate Chem., 2014, 25, 1198 CrossRef PubMed; (d) D. Zeng, Y. Guo, A. G. White, Z. Cai, J. Modi, R. Ferdani and C. J. Anderson, Mol. Pharmaceutics, 2014, 11, 3980 CrossRef PubMed; (e) P. L. Ross and J. L. Wolfe, J. Pharm. Sci., 2016, 105, 391 CrossRef PubMed; (f) H. Gong, I. Holcomb, A. Ooi, X. Wang, D. Majonis, M. A. Unger and R. Ramakrishnan, Bioconjugate Chem., 2016, 27, 217 CrossRef PubMed; (g) P. Akkapeddi, S.-A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954 RSC.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- A. Baccaro, A.-L. Steck and A. Marx, Angew. Chem., Int. Ed., 2012, 51, 254 CrossRef PubMed.
- (a) D. Verga, M. Welter, A. L. Steck and A. Marx, Chem. Commun., 2015, 51, 7379 RSC; (b) D. Verga, M. Welter and A. Marx, Bioorg. Med. Chem. Lett., 2016, 26, 841 CrossRef PubMed.
- L. F. Cheow, S. H. Ko, S. J. Kim, K. H. Kang and J. Han, Anal. Chem., 2010, 82, 3383 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01839a |
| This journal is © The Royal Society of Chemistry 2018 |