
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-bite diboranes for the μ(1,2) complexation of hydrazine and cyanide†
Chang-Hong
Chen
 and
François P.
Gabbaï
*
and
François P.
Gabbaï
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA. E-mail: francois@tamu.edu
First published on 29th May 2018
Abstract
As part of our interest in the chemistry of polydentate Lewis acids as hosts for diatomic molecules, we have investigated the synthesis and coordination chemistry of bidentate boranes that feature a large boron–boron separation. In this paper, we describe the synthesis of a new example of such a diborane, namely 1,8-bis(dimesitylboryl)triptycene (2) and compare its properties to those of the recently reported 1,8-bis(dimesitylboryl)biphenylene (1). These comparative studies reveal that these two diboranes feature some important differences. As indicated by cyclic voltammetry, 1 is more electron deficient than 2; it also adopts a more compact and rigid structure with a boron–boron separation (4.566(5) Å) shorter by ∼1 Å than that in 2 (5.559(4) Å). These differences appear to dictate the coordination behaviour of these two compounds. While 2 remains inert toward hydrazine, we observed that 1 forms a very stable μ(1,2) hydrazine complex which can also be obtained by phase transfer upon layering a solution of 1 with a dilute aqueous hydrazine solution. The stability of this complex is further reflected by its lack of reaction with benzaldehyde at room temperature. We have also investigated the behaviour of 1 and 2 toward anions. In MeOH/CHCl3 (1/1 vol) both compounds selectively bind cyanide to form the corresponding μ(1,2) chelate complexes with a B–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–B bridge at their cores. Competition experiments in protic media show that the anionic cyanide complex formed by 1 is the most stable, with no evidence of decomplexation even in the presence of (C6F5)3B.
N–B bridge at their cores. Competition experiments in protic media show that the anionic cyanide complex formed by 1 is the most stable, with no evidence of decomplexation even in the presence of (C6F5)3B.
Introduction
The chemistry of main group-based polydentate Lewis acids1 has drawn considerable attention over the past decades, leading to applications in anion sensing,2 anion transport,3 small molecule activation and catalysis.4 Diboranes featuring the rigid 1,8-naphthalenediyl5 or ortho-phenylene6 backbones are the most studied examples of such systems. Owing to the short spacing of the two boron centres, these diboranes are well-suited for the chelation of monoatomic anions such as hydride,5a,5b,6a fluoride5a,5e,5g,6a and chloride5c,6b or polyatomic anions amenable to μ(1,1) ligation such as azide.5g,6c The strength of the diborane host-anionic guest interaction in these system has led to the development of application in selective anion sensing5a–c,e,g,6a as well as catalysis.4g,5e,6b,c Although no structural evidence has been obtained for the ditopic complexation of neutral molecules by diboranes of type A and B, it has been demonstrated recently that 9,10-dihydroanthracenes of type C are able to engage 1,2-diazines in complex formation7 and catalyse their Diels Alder chemistry.6g,8 The ability of C to complex 1,2-diazines is reminiscent of the adduct formed by 2,2′-diborabiphenyl and pyridazine.9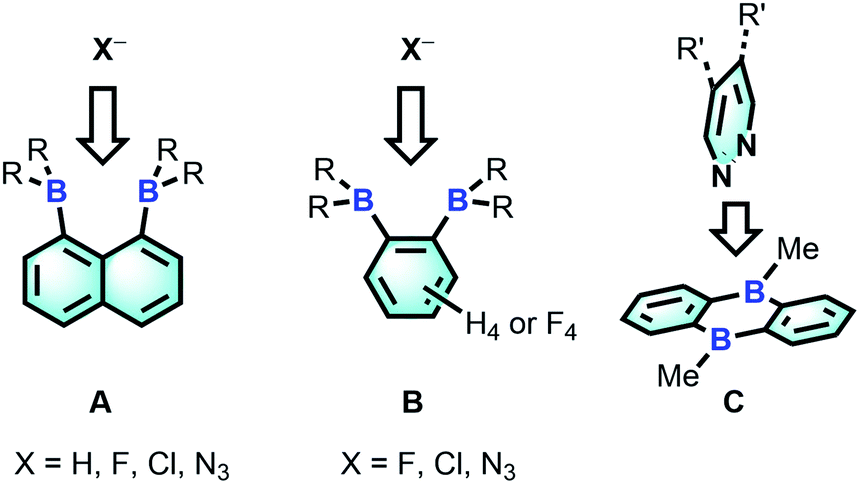
Bearing in mind that the selectivity of these diboranes for their respective guests is dictated by the size match that exists between the host and the guest, we have recently become interested in diboranes with an increased separation between the Lewis acidic centres.10 It occurred to us that such systems may display a different selectivity and may become well adapted to the μ(1,2) chelation of diatomic molecules.11 We also speculated that the rigidity of the targeted diboranes may inform the molecular recognition properties of these bidentate Lewis acidic hosts.
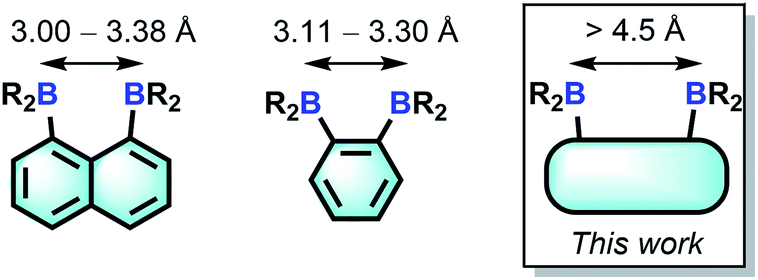
In this article, we report a series of results concerning the synthesis and properties of bidentate diboranes in which the two boron atoms are separated by more than 4.5 Å. We also show that this increased separation allows for the selective μ(1,2) complexation of hydrazine and cyanide, two diatomic molecules that are known for their high toxicity.
Results and discussion
Synthesis, characterization and properties of the diboranes
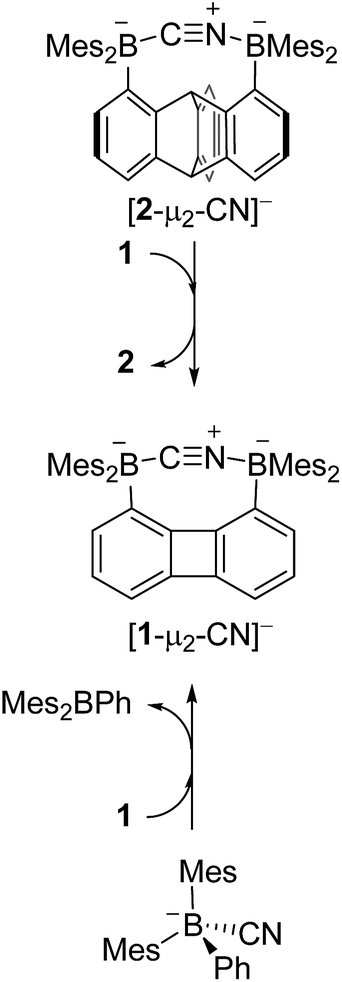
We have recently described the synthesis of 1,8-bis(dimesitylboryl)biphenylene (1),12 a diborane in which the two boron atoms are separated by 4.566(5) Å. To assess the influence of the backbone over the properties of such large-bite diboranes, we have now decided to prepare its 1,8-triptycenediyl analogue (2). By analogy with the approach we employed to access 1, diborane 2 was obtained via the dilithiation of 1,8-dibromotriptycene, followed by metathesis with Mes2BF (Scheme 1).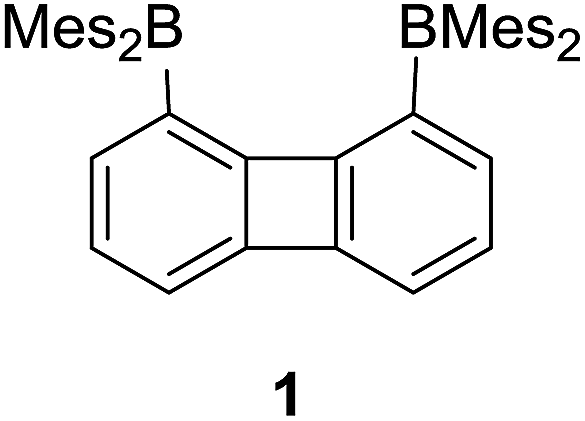
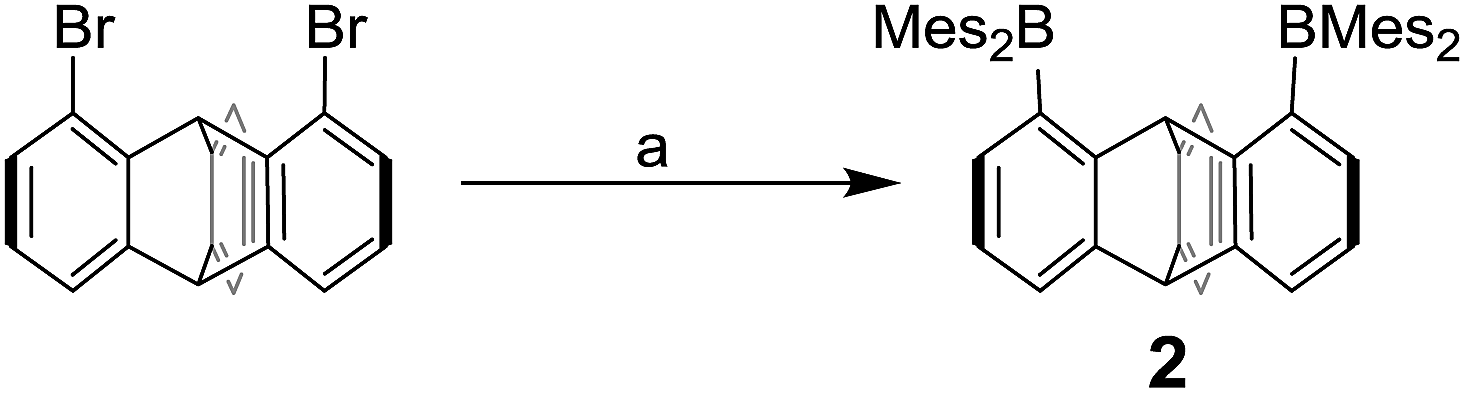 | ||
| Scheme 1 Synthesis of diborane 2. (a) 2.3 equiv. nBuLi, 2.2 equiv. Mes2BF, THF, −78 °C. | ||
Compound 2 is a colourless air-stable solid which has been isolated in moderate yield. The 1H NMR signals of the triptycene backbone are consistent with a symmetrical structure. The detection of six sharp signals arising from the methyl groups and the four meta-H signals of the mesityl groups suggest that the molecule retains mirror symmetry in solution. The 11B NMR spectrum shows a broad peak at 73.0 ppm which falls within the typical range expected for triarylboranes. The cyclic voltammogram (CV) of compound 2 shows two quasi-reversible reduction waves at −2.62 and −3.00 V vs. Fc/Fc+, suggesting that the molecule can be reduced by two electrons (Fig. 1c). Compared to the biphenylene derivative 1 for which the two reduction waves are observed at E1/2 = −2.23 and −2.74 V (Fig. 1c),12 the first reduction of 2 is shifted toward cathodic potentials by 390 mV. This observation indicates that the biphenylene backbone is substantially more electron withdrawing. This conclusion is in agreement with two previous studies dealing with related biphenylene boron derivatives.12,13 Also, the shorter separation observed between the first and second reduction wave in 2 (ΔE1/2 = 0.51 V for 1vs. ΔE1/2 = 0.38 V for 2) signals a decreased electronic communication between the boron atoms, an effect that we assign to the absence of extended conjugation in the triptycene backbone. The importance of electronic communication in 1 is further supported by the fact that the ΔE1/2 measured for 1 (0.51 V) is comparable to the largest value measured for naphthalene-based diboranes (ΔE1/2 range = 0.3–0.52 V), a class of compounds in which the two boron atoms are separated by only 3.002–3.385 Å.5e,14
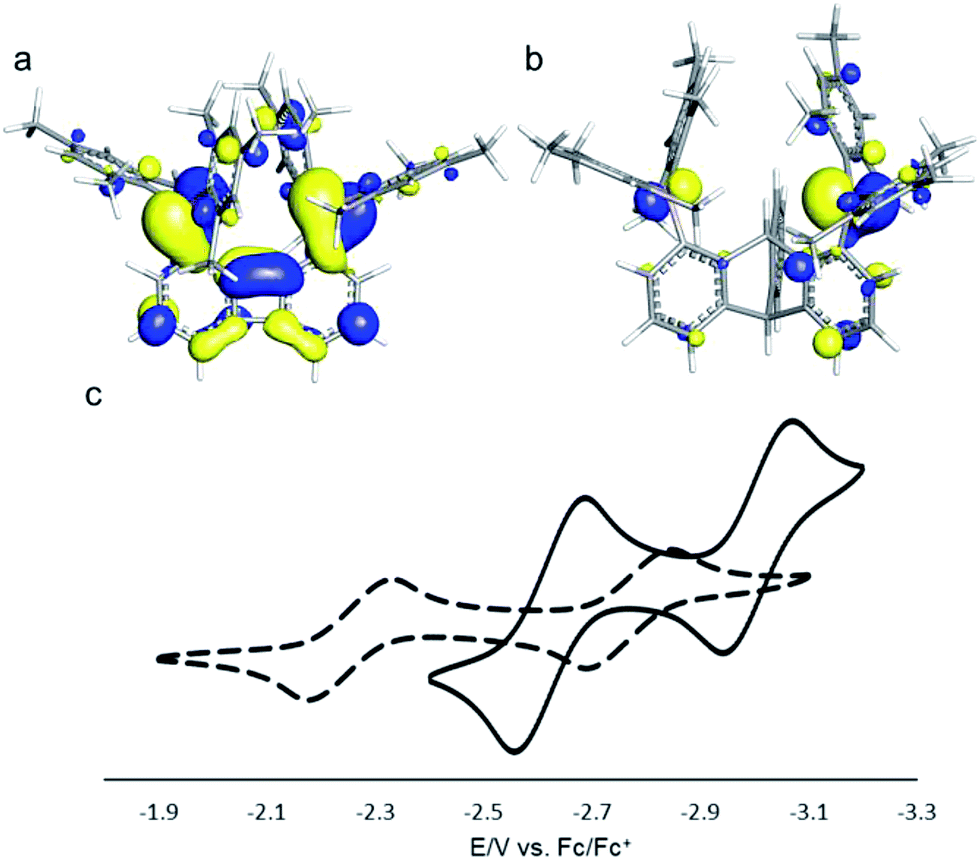 | ||
| Fig. 1 (a) LUMO of 1 (isovalue = 0.03). (b) LUMO of 2 (isovalue = 0.05). (c) Cyclic voltammograms of 1 (dash line, E1/2 = −2.23 and −2.74 V) and 2 (solid line, E1/2 = −2.62 and −3.00 V) in THF. Scan rate = 100 mV s−1. | ||
Computational studies show that the lowest unoccupied molecular orbital (LUMO) of 1 (Fig. 1a) is dominated by the two boron pπ orbitals which are in conjugation with the biphenylene π system. The makeup of this LUMO underscores the effective electronic communication that exists between the two boron centres. In the case of 2, which lacks mirror symmetry because of steric repulsions between the mesityl substituents, the LUMO bears a larger contribution from the pπ orbital of one boron atom. The LUMO + 1, which lies only 0.14 eV above the LUMO, shows a large contribution from the opposite boron atom (see ESI†). More importantly, π-conjugation between the two boron atoms in 2 is interrupted by the sp3 triptycene bridgehead carbon atoms. These features corroborate the conclusion that the two boron atoms of 1 are in closer electronic communication than in 2, as confirmed from the CV measurements.
Single crystals of compound 2 could be obtained by layering a solution of 2 in CH2Cl2 with MeOH. The solid-state structure of 2 shows that the boron atoms adopt a trigonal planar geometry as indicated by the sum of the Caryl–B–Caryl angles (∑∠C–B1–C = 359.7°, ∑∠C–B2–C = 359.4°) (Fig. 2).15 The B1–B2 separation of 5.559(4) Å in 2 is notably larger than that in 1 (4.566(5) Å)12 and 1,8-bis(diphenylboryl)naphthalene (3.002(2) Å).14a In CHCl3/MeOH (1/1 vol), the UV-vis spectra of 1 and 2 feature a low-energy absorption band at 424 nm for 1 (Fig. 3) and 316 nm for 2 (Fig. 4). Time-dependent density functional theory (TD-DFT) calculations and Natural Transition Orbitals (NTO) analysis show the dominant HOMO–LUMO character of these transitions as observed for most triaryl boranes (see ESI†).16 Both 1 and 2 are weakly fluorescent (QY = 0.05 for 1 and 0.04 for 2) at a wavelength of 528 nm and 383 nm, respectively. Given that the boron orbitals are involved in the LUMO of both derivatives, it can be anticipated that the coordination of Lewis bases to the tricoordinate boron centres will dramatically affect these spectral features.16e,17
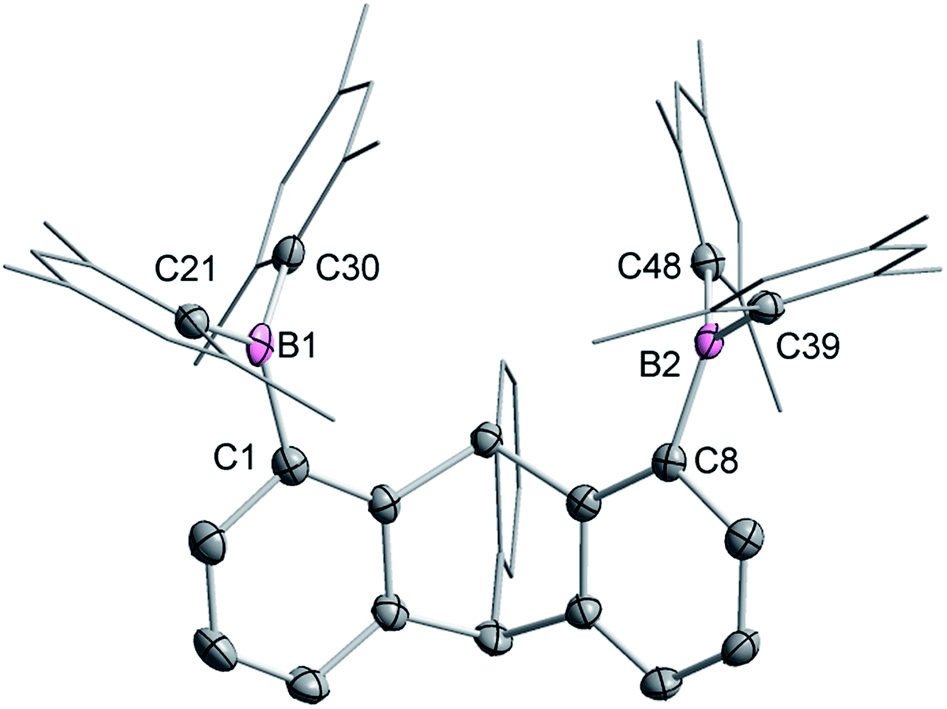 | ||
| Fig. 2 Solid-state structure of 2. Thermal ellipsoids are drawn at the 50% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: B1–C1 1.564(4), B1–C21 1.518(3), B1–C30, 1.571(4), B2–C8 1.572(4), B2–C39 1.577(4), B2–C48 1.570(4); C1–B1–C21 117.5(2), C1–B1–C30 119.1(2), C21–B1–C30 123.1(2), C8–B2–C39 117.6(2), C8–B2–C48 121.1(2), C39–B2–C48 120.7(2). | ||
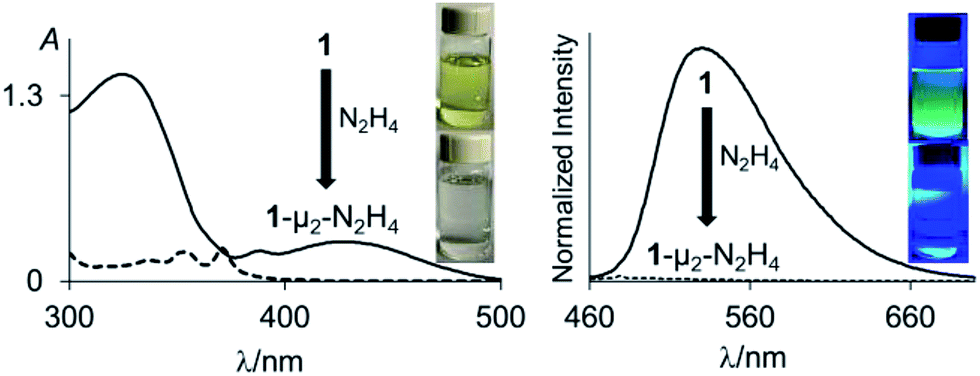 | ||
| Fig. 3 UV-vis absorption (left) and emission (right) spectra of 1 (solid line) and 1-μ2-N2H4 (dash line) (4.52 × 10−5 M, λex = 370 nm) in THF. The pictures of the solutions were taken at the same concentration. The images showing the fluorescence emission were illuminated with a hand-held UV lamp. | ||
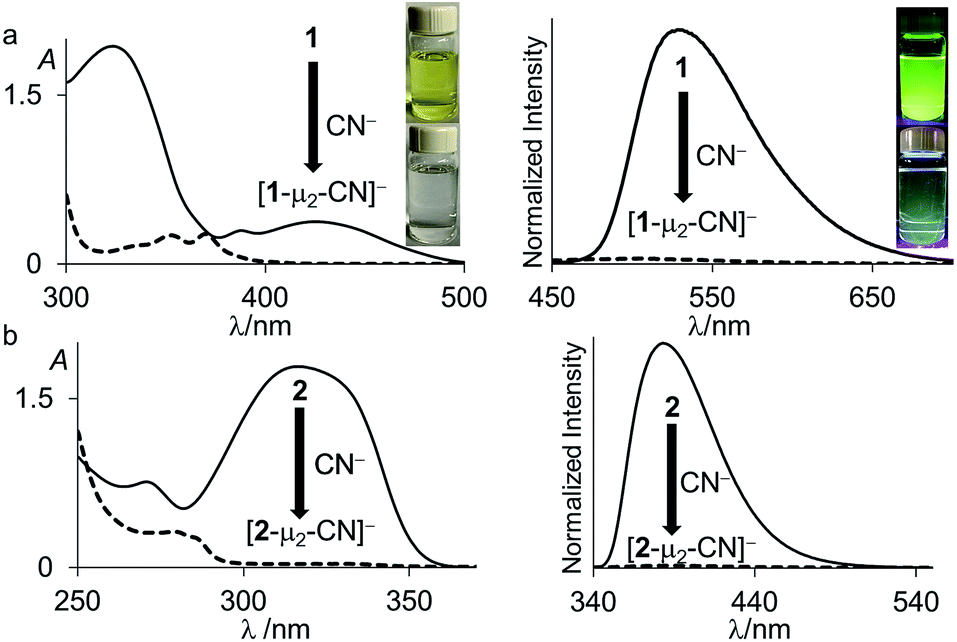 | ||
| Fig. 4 UV-vis absorption (left) and emission (right) spectra of (a) 1 (solid line) and [1-μ2-CN]− (dash line) (6.91 × 10−5 M, λex = 370 nm) and (b) 2 (solid line) and [2-μ2-CN]− (dash line) (6.22 × 10−5 M, λex = 280 nm) in CHCl3/MeOH (1/1 vol). The pictures of the solutions were taken at the same concentration. The images showing the fluorescence emission were illuminated with a hand-held UV lamp. | ||
Reactions of the diboranes with neutral diatomic molecules
With 1 and 2 at our disposal, we became eager to investigate the behaviour of these two compounds in the presence of simple diatomic molecules including hydrogen peroxide, hydroxylamine and hydrazine. While complexes containing doubly coordinated peroxide11a,b and hydrazine18 molecules have generated much interest in the context of energy related research, we also note that coordinated hydrazine has been identified as a possible intermediate in nitrogen fixation reactions.10c,19 Because of its acute toxicity, there are also numerous ongoing efforts aimed at developing selective receptors for molecular recognition and sensing applications.20Inspired by the recent discovery that perfluorinated boranes can doubly complex the peroxide and superoxide anions,21 we first tested the reaction of 1 and 2 with 30% H2O2(aq). Unfortunately, we observed decomposition, illustrating the vulnerability of these non-perfluorinated diboranes under oxidative conditions. We also tested the reaction of 1 and 2 with 50% NH2OH(aq), which also resulted in decomposition of the hosts. By contrast, reaction of diborane 1 with hydrazine monohydrate (N2H4·H2O) in THF proceeded smoothly to afford the corresponding hydrazine complex 1-μ2-N2H4 as an air- and moisture-stable off-white solid (Scheme 2). This reaction results in a distinct loss of the yellow colour indicating coordination of the hydrazine to each boron atom. The same effect is responsible for the observed quenching of the green fluorescence (Fig. 3). In stark contrast, diborane 2 does not complex hydrazine under the same condition, even with a large excess of N2H4·H2O. The lack of reactivity of 2 toward hydrazine may be correlated to its lower Lewis acidity as suggested by the CV measurements. It is also possible that the increased spacing between the two boron atoms, the lower rigidity of the backbone22 and the presence of a bridgehead methine group interfere with the ability of 2 to bind hydrazine.
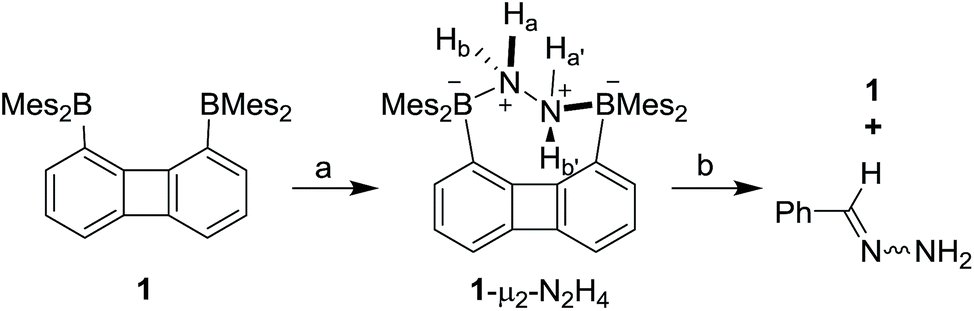 | ||
| Scheme 2 Synthesis of 1-μ2-N2H4 and the reaction of 1-μ2-N2H4 with benzaldehyde. (a) 2.5 equiv. N2H4·H2O, THF, rt. (b) 1 equiv. benzaldehyde, CDCl3, 60 °C. | ||
The 1H NMR spectrum recorded for the resulting hydrazine adduct 1-μ2-N2H4 in CD2Cl2 features six methyl-proton and four meta-proton resonances, indicating that the complex adopts C2 symmetry in solution. The most conspicuous evidence for the formation of a B–NH2–NH2–B bridge is the detection of two broad multiplets at 5.15 and 6.29 ppm, indicating that the B–NH2–NH2–B unit gives rise to an AA′BB′ spin system. These spectroscopic features also indicate that the conformation of the hydrazine molecule in the diboron pocket is rigidly locked in solution. The solid-state structure of 1-μ2-N2H4 offers a consistent picture (Fig. 5). This neutral complex crystallizes in the monoclinic C2/c space group with a half molecule in the asymmetric unit.15 The hydrazine nitrogen atoms are coordinated to the boron atoms via a B–N bond length of 1.688(2) Å which is longer than that in H3B–NH2–NH2–BH3 (1.609 Å)23 but comparable to that recently reported by Szymczak for complex D-μ2-N2H4 (1.697(2) Å and 1.698(2) Å) (Scheme 3).10c
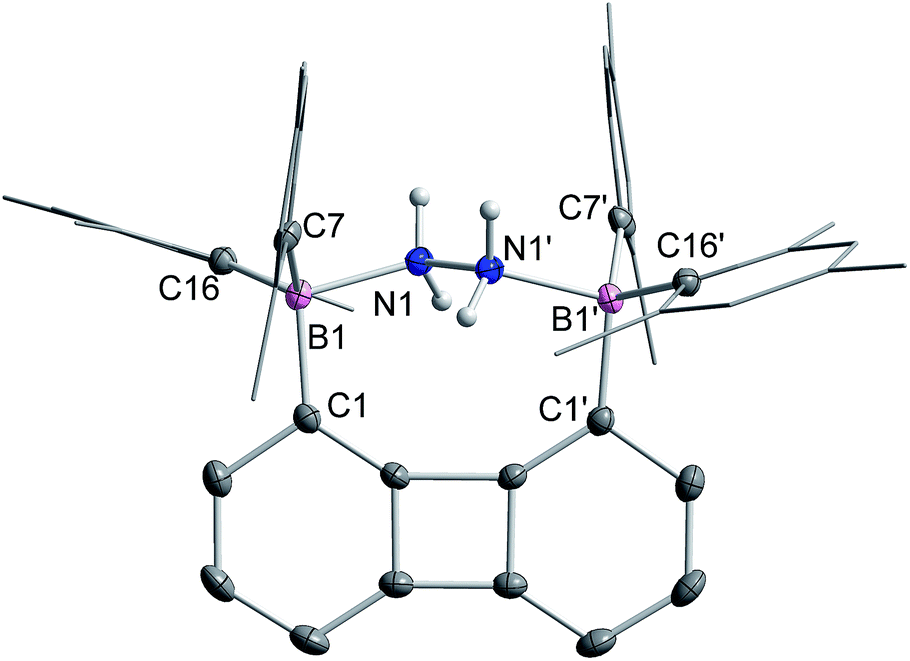 | ||
| Fig. 5 The solid-state structure of 1-μ2-N2H4. Thermal ellipsoids are drawn at the 50% probability level. The hydrogen atoms, except that bound to the nitrogen atoms, are omitted for clarity. Selected bond lengths [Å] and angles [°]: B1–N1 1.688(2), N1–N1′ 1.469(2), B1–C1 1.627(21), B1–C7 1.651(2), B1–C16 1.647(2); B1–N1–N1′ 113.7(1), C7–B1–C16 118.7(1), C1–B1–C7 119.7(1), C1–B1–C16 106.6(1). | ||
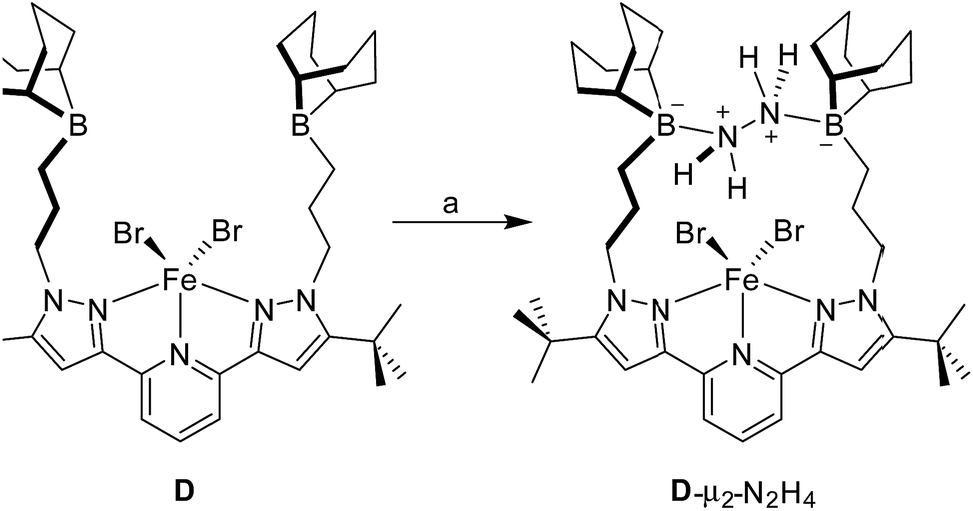 | ||
| Scheme 3 Reaction of iron complex (D) with hydrazine to afford D-μ2-N2H4 according to Szymczak et al. (a) 1 equiv. N2H4, THF, rt. | ||
The N–N bond length of 1.469(2) Å is similar to the N–N bond length determined previously in the literature.24 This observation clearly indicates that diborane 1 is well suited for the chelation of hydrazine molecule. We also note that D can bind two hydrazine molecules, one at each boron atom. This is not the case of diborane 1 which only forms a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, even in the presence of an excess of hydrazine. This difference in behaviour illustrates the selectivity imparted by the rigid preorganization of the two boron atoms in 1.10c
:1 complex, even in the presence of an excess of hydrazine. This difference in behaviour illustrates the selectivity imparted by the rigid preorganization of the two boron atoms in 1.10c
To further establish the high affinity that 1 displays for hydrazine, we have studied the complexation reaction under dilute conditions. We observed that diborane 1 is capable of capturing hydrazine from dilute aqueous solutions under biphasic conditions. Quantitative formation of 1-μ2-N2H4 was observed by 1H NMR spectrometry when a solution of 1 in CH2Cl2 (1 mL, [1] = 7.7 mM) was stirred for 24 hours with an aqueous solution (2.5 mL) containing 1 weight% of N2H4. Complex 1-μ2-N2H4 is remarkably stable. It can be stored in air and shows no evidence of decomposition for months. It is also resistant to reactions with aldehydes. For example, 1-μ2-N2H4 failed to react with benzaldehyde in CDCl3 at room temperature over the course of 20 h. However, upon heating to 60 °C, a clean, yet slow reaction is observed leading to the quantitative formation of the free diborane 1 and benzaldehyde hydrazone after 20 h as confirmed by 1H NMR spectroscopy (Scheme 2).25
Reaction of the diboranes with the cyanide anion
Next in our survey of the properties of these diboranes, we decided to focus on the case of anions and especially the cyanide anion which is known for its acute toxicity and which has often been considered as a target in molecular recognition assays,26 including using boron-based hosts.17b,27Both diboranes quickly react with KCN in a CH2Cl2/MeOH (1/1 vol) mixture containing dibenzo-18-crown-6. As expected, this reaction is accompanied by a quenching of both the absorption and emission band (Fig. 4). In the case of 1, these changes result in a distinct loss of both the yellow colour and the green fluorescence of the starting diborane (Fig. 4a). Workup of these reactions afforded the corresponding cyanide complexes [1-μ2-CN]− and [2-μ2-CN]− as air-stable [K(dibenzo-18-crown-6)]+ salts (Scheme 4). The 11B NMR signals detected for these anionic complexes (−15.8 ppm for [1-μ2-CN]− and −11.7 ppm for [2-μ2-CN]−) are consistent with the existence of four-coordinate boron atoms. These signals are somewhat broad, possibly indicating the presence of two closely spaced and overlapping resonances corresponding to the NCN-bound and CCN-bound boron atoms, respectively. The presence of the cyanide anion in these complexes is confirmed by IR spectroscopy which shows that, in both cases, the cyanide stretching frequency (νCN = 2229 cm−1 for 1 and 2184 cm−1 for 2) is higher than that of KCN (νCN = 2158 cm−1). The higher energy of these vibrations is typical of cyanoborates. This effect can be rationalized by invoking a stabilization of the π molecular orbitals of the cyanide anion.28 The absence of π-backbonding can also be invoked as a cause for this effect. The greater νCN observed for [1-μ2-CN]− can be corroborated to the CV results and again suggest that the biphenylene backbone is more electron withdrawing than the triptycene backbone. The methyl region of the 1H NMR spectrum recorded for both [1-μ2-CN]− and [2-μ2-CN]− as [K(dibenzo-18-crown-6)]+ salts in CDCl3 shows a level of complexity that is not consistent with equivalence of the two boryl moieties. We interpret these features as an evidence that the two boryl moieties are differentiated by their ligation to the C or N terminus of the cyanide anion.
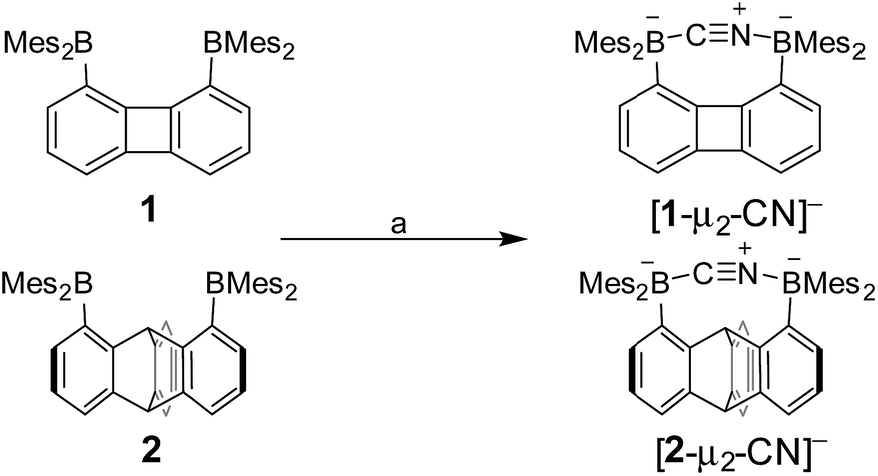 | ||
| Scheme 4 Synthesis of [1-μ2-CN]− and [2-μ2-CN]− as [K(dibenzo-18-crown-6)]+ salts. (a) 1 equiv. KCN, 1 equiv. dibenzo-18-crown-6, CH2Cl2/MeOH (1/1 vol), rt. | ||
Interestingly, diborane 1 and 2 appear to be highly selective for cyanide since no interaction with HCO3−, HSO4−, H2PO4−, CH3COO−, Cl−, Br−, I−, and N3− is observed in CHCl3/MeOH (1/1 vol) by UV-vis spectrometry. Boranes are known to also display a large affinity for the fluoride anion. However, under these conditions, neither 1 nor 2 show any evidence of binding with fluoride. We propose that this selectivity is assisted by the specific architecture of the compounds that are well adapted to cyanide complexation (vide infra). However, considering that this selectivity could be biased by the protic nature of the medium which solvates the fluoride anion more effectively than the cyanide anion, we also tested the reaction with fluoride in dry CDCl3. When 1 and 2 were combined with tetrabutylammonium difluorotriphenylsilicate (TBAT), no changes were observed in the 1H NMR spectra of the diboranes indicating the absence of any interaction. When tetrabutylammonium fluoride trihydrate (TBAF·3H2O) was used in dry CDCl3, 1 underwent hydrolysis as previously described while 2 remained unperturbed.12 Under the same conditions, both 1 and 2 quickly react with tetrabutylammonium cyanide (TBACN) to give the corresponding cyanide complexes. These results show that these diboranes are also selective for cyanide in organic solvents. Such selectivity is not unprecedented in the chemistry of boron-based Lewis acids.17b,27 Lastly, these diboranes do not react with LiHBEt3 in dry CDCl3.
The structures of the cyanide complexes as [K(dibenzo-18-crown-6)]+ salts have been investigated by single crystal X-ray diffraction which confirms the μ(1,2) chelation of the cyanide anions (Fig. 6).15 Both structures were solved on the basis of a model in which the cyanide anion is disordered over two overlapping head-to-tail positions, which as indicated by the refinements, contribute almost equally to the observed structures. In both structures, the boron atoms are distinctly pyramidal as indicated by the sum of the Caryl–B–Caryl angles which fall in the 341.7(4)–343.2(4)° range for [1-μ2-CN]− and 342.0(5)–344.6(5)° for [2-μ2-CN]−. The accuracy of the crystallographic measurements does not allow for a comparison of the B–CCN and B–NCN bond distances which fall in the 1.54(4)–1.62(3) Å for [1-μ2-CN]− and 1.63(5)–1.67(8) Å for [2-μ2-CN]−. The small difference in the B1–B2 separations in [1-μ2-CN]− (4.187 (4) Å) and 1 (4.566(5) Å) reflects the rigidity of the molecule. Compared to 1, the large variation of the B1–B2 separation on going from 2 (B1–B2 5.559(4) Å) to [2-μ2-CN]− (B1–B2 4.321(5) Å) speaks to the flexibility of the triptycene backbone and its pincer ability toward the cyanide anion. The behaviour of 1 and 2 and their ability to coordinate to both ends of the cyanide anion is reminiscent of the behaviour of dicopper(II) complexes (E) which also form μ(1,2) cyanide complexes as described by Krämer (Scheme 5).29 This bonding mode differs from that observed for stibonium (F) and sulfonium boranes (G) in which the cyanide anion interact primarily with the boron atom, with a weak side on contact to the adjacent stibonium or sulfonium cation (Scheme 5).27d,30 Finally, it is important to point out that while [1-μ2-CN]− and [2-μ2-CN]− are the first bimolecular μ(1,2) complexes formed by a diborane and cyanide, their cores are reminiscent of that found in termolecular complexes of general formula [Ar3B–CN–BAr3]− (Ar = Ph or C6F5).31
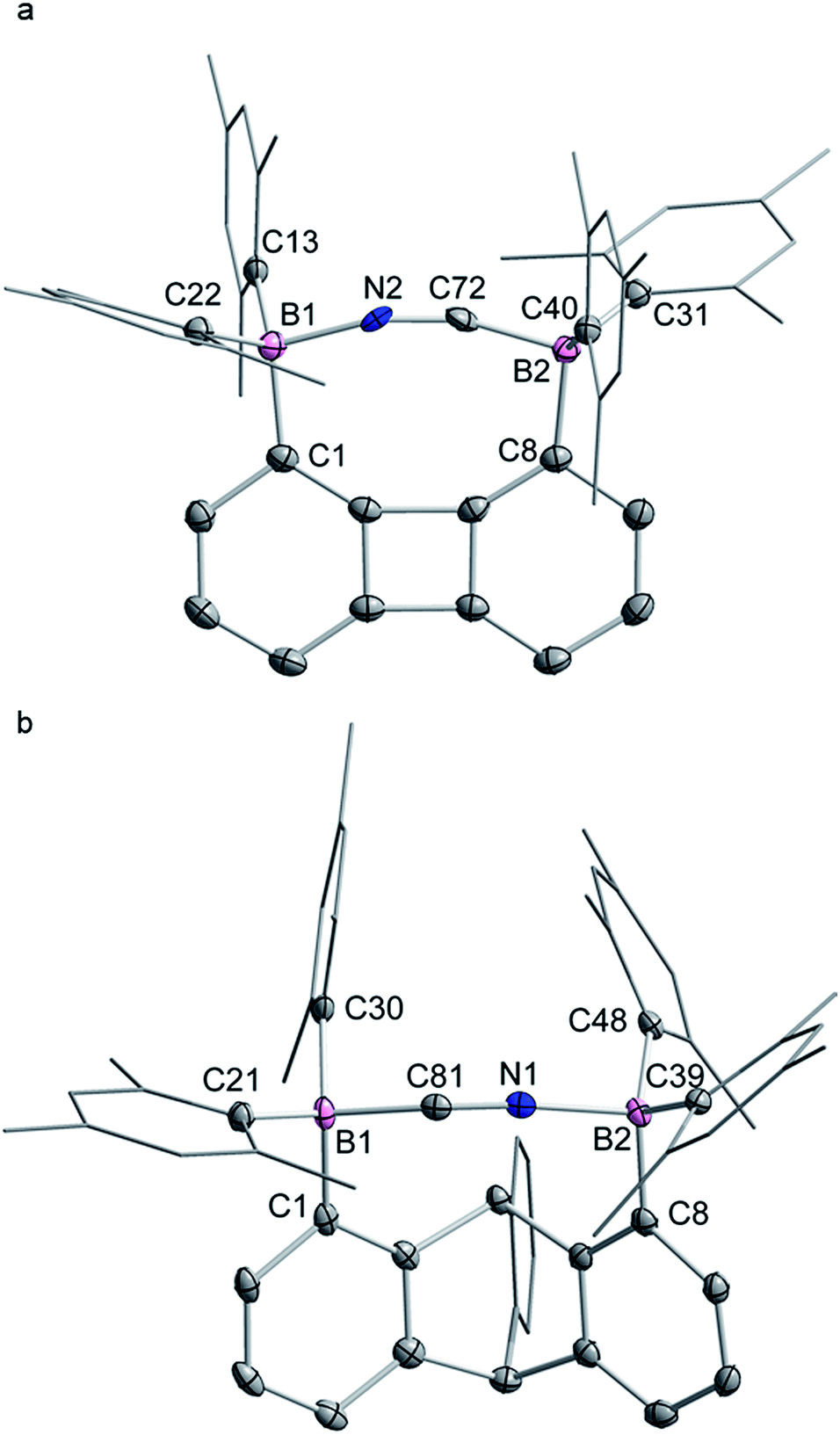 | ||
| Fig. 6 Solid-state structures of the cyanide complexes [K(dibenzo-18-crown-6)][1-μ2-CN]-(CH2Cl2) (a) and [K(dibenzo-18-crown-6)][2-μ2-CN]-(CH2Cl2)2 (b). Thermal ellipsoids are drawn at the 50% probability level. The [K(dibenzo-18-crown-6)]+, the solvate molecules and the hydrogen atoms are omitted for clarity. The cyanide anions shown correspond to that with the higher positional occupancy. Selected bond lengths [Å] and angles [°] for [1-μ2-CN]−: B1–C1 1.635(4), B1–C13 1.649(4), B1–C22 1.639(4), B1–N2 1.58(2), B2–C8 1.641(4), B2–C31 1.643(4), B2–C40 1.657(3), B2–C72 1.54(4) C72–N2 1.21(4); C1–B1–C13 121.1(2), C13–B1–C22 113.8(2), C1–B1–C22 108.3(2), C1–B1–N2 100.9(7), C13–B1–N2 98.7(8), C22–B1–N2 112.9(7), B1–N2–C72 157.0(19), C8–B2–C31 108.4(2), C8–B2–C40 118.0(2), C31–B2–C40 115.3(2), C8–B2–C72 99.8(8), C31–B2–C72 115.8(9), C40–B2–C72 98.5(9), B2–C72–N2 163(2); for [2-μ2-CN]−: C1–B1 1.657(5), C21–B1 1.644(5), C30–B1 1.665(5), C81–B1 1.66(4), C81–N1 1.16(8), C8–B2 1.632(5), C39–B2 1.646(5), C48–B2 1.649(5), N1–B2 1.64(5); C1–B1–C21 109.1(3), C1–B1–C30 120.2(3), C21–B1–C30 112.7(3), C1–B1–C81 102.6(11), C21–B1–C81 111.4(16), C30–B1–C81 99.8(15), B1–C81–N1 158(6), C8–B2–C39 113.5(3), C8–B2–C48 113.1(3), C39–B2–C48 118.0(3), C8–B2–N1 99.8(15), C39–B2–N1 110.6(18), C48–B2–N1 99.1(13), B2–N1–C81 168(6). | ||
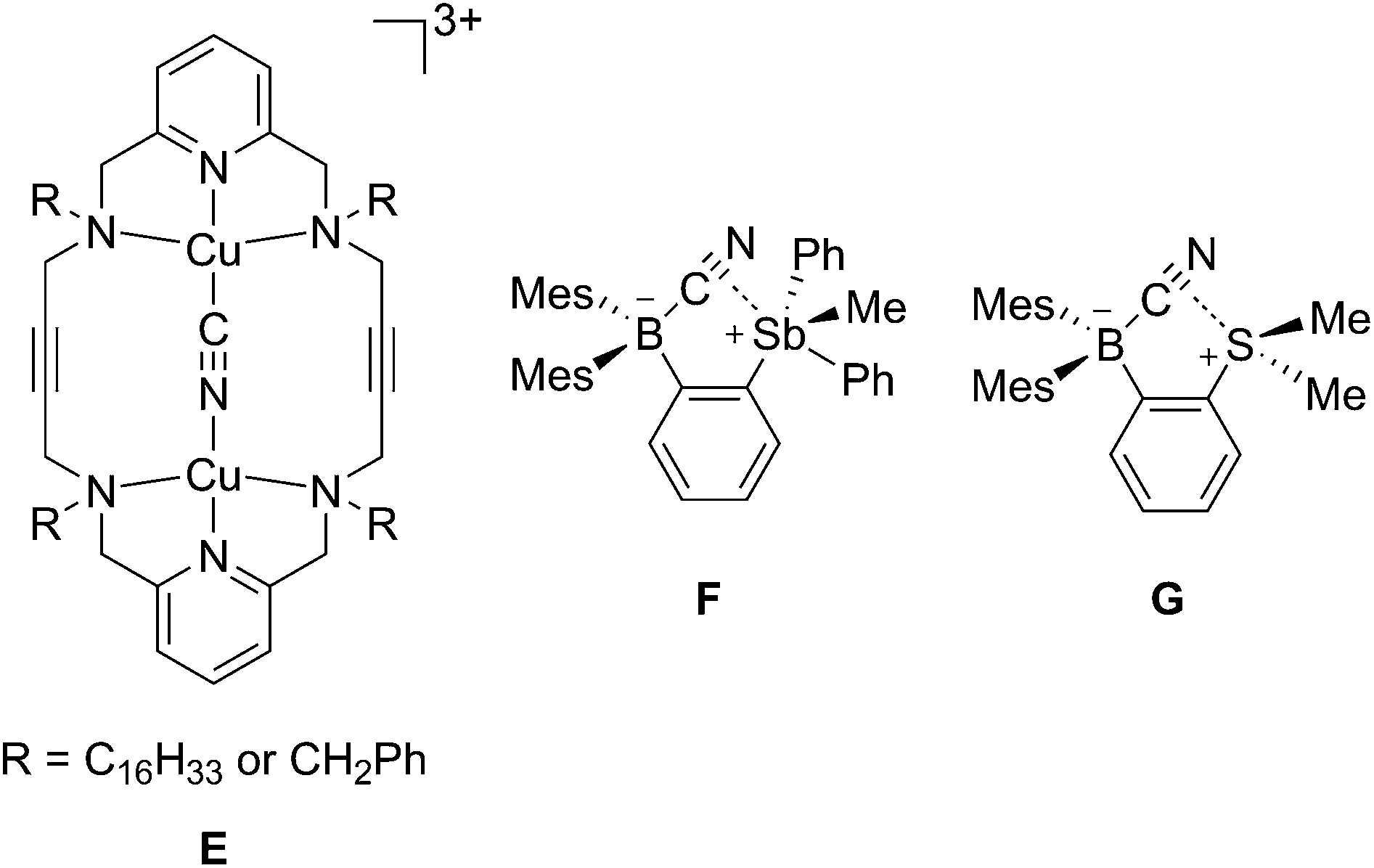 | ||
| Scheme 5 Structure of the cyanide complexes of ditopic hosts E–G. | ||
Since the CV data of 1 and 2 and the IR data of [1-μ2-CN]− and [2-μ2-CN]− suggest that 1 is a stronger Lewis acid than 2, we became eager to experimentally confirm that 1 would indeed outcompete 2 with regard to cyanide binding. In accordance with this prediction, we observed, using 1H NMR spectroscopy, the quantitative transfer of the cyanide anion from 2 to 1 when [nBu4N][2-μ2-CN] was mixed with an equimolar quantity of 1 in CDCl3/CD3OD (1/1 vol) at 60 °C over the course of 12 h (Scheme 6). This observation confirms the higher cyanide ion affinity of 1 in protic solution, which is consistent with the electrochemical and IR measurements described above. Furthermore, a competition experiment was also performed with the monofunctional model borane Mes2BPh (3). In these tests, the cyanide anion was transferred from [3-CN]− to 1 quantitatively in CDCl3/CD3OD (1/1 vol) at 60 °C (Scheme 6). However, no transfer was observed between [3-CN]− and 2. The absence of a reaction points to the weaker cyanide affinity of 2 when compared to 1. The reversibility of the cyanide binding was also investigated by allowing [nBu4N][1-μ2-CN] and [nBu4N][2-μ2-CN] to react with one equivalent of (C6F5)3B in CDCl3/CD3OD (1/1 vol).32 Under these conditions, [2-μ2-CN]− was readily converted into 2 while [1-μ2-CN]− remained untouched, again supporting the superior Lewis acidity of 1.
 | ||
| Scheme 6 Competition reactions of [nBu4N][2-μ2-CN] and 1 (top) and [nBu4N][3-CN] and 1 (bottom). The [nBu4N]+ cations are not shown for clarity. | ||
Conclusions
In summary, we describe two “large-bite” diboranes that are ideally suited for the selective μ(1,2) complexation of hydrazine and cyanide, two diatomic molecules that are known for their high toxicity. The high selectivity displayed for these two specific molecules highlights the defining role played by the backbone. The most potent binder is the biphenylene-based diborane which complexes both hydrazine and cyanide while the triptycene derivative only binds cyanide. A similar picture emerges from competition experiments which show that 1 is a better molecular recognition unit for cyanide than 2. Our results and analyses indicate that the enhanced properties of 1 arise from the electron withdrawing nature of the biphenylene backbone. We also propose that the rigidity of this diborane is a favourable factor that helps sequester hydrazine and the cyanide anion in the diboron pocket. This binding appears to be irreversible in the case of the cyanide anion. In the case of the hydrazine complex, we observe that slow release can be triggered upon elevation of the temperature and in the presence of a reaction partner such as benzaldehyde. This last feature shows that such bidentate hosts could be used for the slow release of reactive compounds.Experimental
Synthesis of 2
n BuLi (2.65 M, 1 mL, 2.65 mmol) was slowly added to a solution of 1,8-dibromotriptycene (500 mg, 1.2 mmol) in dry THF (8 mL) under N2 at −78 °C. After 30 min of stirring at low temperature, a solution of Mes2BF (715 mg, 2.6 mmol) in THF (5 mL) was slowly transferred into the reaction flask using a cannula. The resulting solution was stirred for an additional 12 h at room temperature. The solution was then treated with saturated NH4Cl(aq) (1 mL) and the solvent was removed under vacuum. The resulting white solid was then dissolved in CH2Cl2 (5 mL). The resulting solution was filtered and brought to dryness under vacuum. The off-white solid was then washed with MeOH (15 mL) twice and dried under vacuum to afford diborane 2 as a white powder in 60% yield. 1H NMR (399.50 MHz, 25 °C, CDCl3): δ 7.49 (d, 2H, 3JH–H = 6.6 Hz, triptycene-CH), 7.23 (d, 1H, 3JH–H = 7.0 Hz, triptycene-CH), 6.97 (pseudo t, 2H, 3JH–H = 7.4 Hz, triptycene-CH), 6.90–6.86 (m, 3H, triptycene-CH), 6.82 (s, 2H, mes-CH), 6.77 (s, 2H, mes-CH), 6.73 (s, 2H, mes-CH), 6.61 (pseudo t, 1H, 3JH–H = 7.4 Hz, triptycene-CH), 6.35 (s, 2H, mes-CH), 5.40 (s, 1H, triptycene-CH), 5.30 (d, 1H, 3JH–H = 7.4 Hz, triptycene-CH), 5.16 (s, 1H, triptycene-CH), 2.34 (s, 6H, –CH3), 2.30 (s, 6H, –CH3), 2.17 (s, 6H, –CH3), 2.13 (s, 6H, –CH3), 1.79 (s, 6H, –CH3), 0.60 (s, 6H, –CH3) ppm. 13C NMR (100.46 MHz, 25 °C, CDCl3): δ 150.86, 145.50, 145.23, 144.42, 144.27, 143.12, 141.52, 141.17, 140.90, 140.77, 139.89, 138.58, 138.57, 131.21, 129.92, 129.90, 128.47, 128.14, 127.90, 126.58, 126.17, 125.05, 124.16, 123.50, 121.60, 55.45 (triptycene-CH), 54.26 (triptycene-CH), 25.20 (–CH3), 24.96 (–CH3), 22.37 (–CH3), 22.03 (–CH3), 21.39 (–CH3), 21.25 (–CH3) ppm. 11B (128.16 MHz, 25 °C, CDCl3): δ 73.0 (br) ppm. Elemental analysis calculated (%) for C56H56B2: C, 89.60; H, 7.52; found C, 89.62; H, 7.47.Synthesis of 1-μ2-N2H4
A solution of N2H4·H2O (20 mg, 0.4 mmol) in THF (2 mL) was combined with a solution of diborane 1 (100 mg, 0.154 mmol) in THF (5 mL) at room temperature. The resulting solution was stirred for an additional hour and brought to dryness under vacuum. The resulting solid was washed twice with MeOH (5 mL) to afford 1-μ2-N2H4 as a off-white powder in 90% yield. 1H NMR (499.48 MHz, 25 °C, CD2Cl2): δ 6.85 (s, 2H, mes-CH), 6.80 (s, 2H, mes-CH), 6.68 (s, 2H, mes-CH), 6.57 (s, 2H, mes-CH), 6.55–6.50 (m, 6H, biphenylene-CH), 6.31–6.28 (m, 2H, –NH2–), 5.17–5.13 (m, 2H, –NH2–), 2.25 (s, 6H, –CH3), 2.24 (s, 6H, –CH3), 2.15 (s, 6H, –CH3), 1.80 (s, 6H, –CH3), 1.74 (s, 6H, –CH3), 1.67 (s, 6H, –CH3) ppm. 13C NMR (125.61 MHz, 25 °C, CD2Cl2): δ 151.26, 149.80, 143.94, 141.61, 139.50, 137.63, 137.42, 136.01, 135.58, 132.50, 131.24, 130.65, 130.60, 128.00, 115.17, 26.77(–CH3), 25.72(–CH3), 23.61(–CH3), 23.21(–CH3), 21.07(–CH3), 20.87(–CH3) ppm. 11B (128.16 MHz, 25 °C, CDCl3): not observed. Elemental analysis calculated (%) for C48H54B2N2·0.93 × CHCl3: C, 74.24; H, 6.99; found C, 74.22; H, 7.03. This EA result indicates the crystal sample obtain from CHCl3/MeOH contains interstitial solvent molecules. This view is consistent with the crystallographic measurements which show the presence of disordered interstitial solvents equivalent to an electron count of 59 as indicated by application of the SQUEEZE protocol.33 This electron count corresponds almost exactly to the number of electrons of a chloroform molecule (58). The EA results suggest partial loss of the interstitial chloroform molecule.Synthesis of [K(dibenzo-18-crown-6)][1-μ2-CN]
A solution of KCN (10 mg, 0.15 mmol) in MeOH (2 mL) was slowly added at room temperature into a CH2Cl2 solution (10 mL) containing 1 (100 mg, 0.15 mmol) and dibenzo-18-crown-6 (56 mg, 0.15 mmol). The resulting colourless solution was stirred for an additional 1 h at room temperature. The solvent was removed under vacuum and the resulting solid was washed by Et2O (10 mL) twice to afford [K(dibenzo-18-crown-6)][1-μ2-CN] as a white powder in 90% yield. 1H NMR (499.53 MHz, 25 °C, CDCl3): δ 7.00–6.97 (m, 4H, –C6H4–), 6.84–6.79 (m, 4H, –C6H4–), 6.66 (t, 2H, 3J = 8.78 Hz, biphenylene-CH), 6.50 (br, 8H, mes-CH), 6.20–6.16 (m, 2H, biphenylene-CH), 6.10 (d, 3J = 6.34 Hz, biphenylene-CH), 4.03–4.01 (m, 8H, –CH2–), 3.76–3.74 (m, 8H, –CH2–), 2.13 (br, 12H, –CH3), 1.84 (br, 24H, –CH3) ppm. 1H NMR (399.46 MHz, −50 °C, CDCl3): δ 6.95–6.94 (m, 4H, –C6H4–), 6.77–6.74 (m, 4H, –C6H4–), 6.67 (t, 2H, 3J = 9.34 Hz, biphenylene-CH), 6.58–6.52 (m, 6H, mes-CH), 6.41 (s, 1H, mes-CH), 6.39 (s, 1H, mes-CH), 6.19–6.12 (m, 4H, biphenylene-CH), 3.94 (br, 8H, –CH2–), 3.37 (br, 8H, –CH2–), 2.18 (s, 6H, –CH3), 2.14 (s, 3H, –CH3), 2.11 (s, 3H, –CH3), 2.05 (s, 6H, –CH3), 1.84–1.80 (m, 12H, –CH3), 1.70 (s, 6H, –CH3) ppm. 13C NMR (125.62 MHz, 25 °C, CDCl3): δ 158.33, 157.36, 150.05, 146.14, 137.01, 136.28, 132.48, 132.37, 128.55, 124.36, 122.25, 112.02, 111.77, 80.29, 68.97 (–CH2–), 66.59 (–CH2–), 24.44 (–CH3), 20.82 (–CH3) ppm. 11B (128.16 MHz, 25 °C, CDCl3): δ −15.8 ppm. IR νCN = 2229 cm−1. Elemental analysis calculated (%) for C69H74B2KO6·0.55 × CH2Cl2: C, 75.48; H, 6.84; found C, 75.49; H, 6.78. These EA results indicate partial loss of the interstitial solvent molecules.Synthesis of [K(dibenzo-18-crown-6)][2-μ2-CN]
A solution of KCN (10 mg, 0.15 mmol) in MeOH (2 mL) was slowly added at room temperature into a CH2Cl2 solution (10 mL) containing 2 (116 mg, 0.15 mmol) and dibenzo-18-crown-6 (56 mg, 0.15 mmol). The resulting colourless solution was stirred for an additional 1 h at room temperature. The solvent was removed under vacuum and the resulting solid was washed by Et2O (10 mL) twice to afford [K(dibenzo-18-crown-6)][2-μ2-CN] as a white powder in 88% yield. 1H NMR (499.49 MHz, 25 °C, CD2Cl2): δ 7.32 (d, 1H, 3JH–H = 7.3 Hz, triptycene-CH), 7.09 (d, 2H, 3JH–H = 7.3 Hz, triptycene-CH), 7.04–6.98 (m, 5H), 6.93–6.88 (m, 6H), 6.84 (d, 1H, 3JH–H = 7.3 Hz, triptycene-CH), 6.66 (s, 2H, mes-CH), 6.58 (pseudo t, 2H, 3JH–H = 7.3 Hz, triptycene-CH), 6.53 (s, 1H, mes-CH), 6.52 (s, 2H, mes-CH), 6.48 (s, 1H, mes-CH), 6.41 (d, 1H, 3JH–H = 7.8 Hz, triptycene-CH), 6.37 (d, 1H, 3JH–H = 7.8 Hz, triptycene-CH), 6.31 (s, 1H, mes-CH), 6.27 (s, 1H, mes-CH), 5.74 (s, 1H, triptycene-CH), 5.26 (s, 1H, triptycene-CH), 4.18–4.16 (m, 8H, –CH2–), 3.93–3.91 (m, 8H, –CH2–), 2.25 (s, 3H, –CH3), 2.23 (s, 3H, –CH3), 2.19 (s, 6H, –CH3), 2.14 (s, 3H, –CH3), 2.02 (s, 3H, –CH3), 2.00 (s, 3H, –CH3), 1.95 (s, 3H, –CH3), 1.63 (s, 3H, –CH3), 1.62 (s, 3H, –CH3), 1.18 (s, 3H, –CH3), 1.13 (s, 3H, –CH3) ppm. 13C NMR (125.61 MHz, 25 °C, CD2Cl2): δ 151.30, 150.72, 147.52, 146.65, 146.59, 144.42, 144.27, 143.19, 142.76, 142.53, 142.21, 141.89, 141.66, 140.88, 133.17, 130.01, 132.75, 132.63, 131.47, 131.34, 129.29, 129.12, 129.02, 128.90, 128.73, 128.60, 128.15, 126.69, 124.49, 123.46, 122.85, 122.57, 122.48, 121.56, 120.50, 120.40, 111.95, 69.85 (–CH2–), 67.21 (–CH2–), 56.15 (triptycene-CH), 51.86 (triptycene-CH), 26.87 (–CH3), 25.97 (–CH3), 25.91 (–CH3), 25.77 (–CH3), 25.55 (–CH3), 25.48 (–CH3), 24.79 (–CH3), 24.48 (–CH3), 21.24 (–CH3), 20.86 (–CH3) ppm. 11B (128.16 MHz, 25 °C, CDCl3): δ −11.7 ppm. IR νCN = 2184 cm−1. Elemental analysis calculated (%) for C77H80B2KNO6·0.43 × CH2Cl2: C, 76.69; H, 6.72; found C, 76.70; H, 6.71. These EA results indicate partial loss of the interstitial solvent molecules.Computational details
Density functional theory (DFT) structural optimizations of 1 and 2 were carried with the Gaussian 09 program. In all cases, the structures were optimized using the B3LYP functional and the following mixed basis set: C/H, 6-31g; B, 6-31g+(d′). For all optimized structures, frequency calculations were carried out to confirm the absence of imaginary frequencies. The molecular orbitals were visualized and plotted using the Jimp2 program. The TD-DFT and NTO calculations were carried out using the MPW1PW91 functional and the above-mentioned basis sets.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this research (Grant 56871-ND3). We also acknowledge support from the Welch Foundation (Grant A-1423), Texas A&M University (Arthur E. Martell Chair of Chemistry), and the Laboratory for Molecular Simulation at Texas A&M University (software and computational resources) is gratefully acknowledged.References
- (a) T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef PubMed; (b) E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC; (c) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef PubMed; (d) Anion Coordination Chemistry, ed., K. Bowman-James, A. Bianchi and E. Garcia-Espana, Wiley-VCH Verlag GmbH & Co. KGaA, 2012 Search PubMed; (e) H. Zhao, L. A. Leamer and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC; (f) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef PubMed; (g) P. A. Gale, E. N. W. Howe, X. Wu and M. J. Spooner, Coord. Chem. Rev., 2018 DOI:10.1016/j.ccr.2018.02.005.
- (a) M. H. Lee and F. P. Gabbaï, Inorg. Chem., 2007, 46, 8132–8138 CrossRef PubMed; (b) J. K. Day, C. Bresner, N. D. Coombs, I. A. Fallis, L.-L. Ooi and S. Aldridge, Inorg. Chem., 2008, 47, 793–804 CrossRef PubMed; (c) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef PubMed; (d) P. Chen, A. S. Marshall, S.-H. Chi, X. Yin, J. W. Perry and F. Jäkle, Chem.–Eur. J., 2015, 21, 18237–18247 CrossRef PubMed; (e) M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 CrossRef PubMed.
- (a) M. E. Jung and H. Xia, Tetrahedron Lett., 1988, 29, 297–300 CrossRef; (b) M. Rothmaier and W. Simon, Anal. Chim. Acta, 1993, 271, 135–141 CrossRef; (c) I. H. A. Badr, M. Diaz, M. F. Hawthorne and L. G. Bachas, Anal. Chem., 1999, 71, 1371–1377 CrossRef PubMed; (d) N. Chaniotakis, K. Jurkschat, D. Mueller, K. Perdikaki and G. Reeske, Eur. J. Inorg. Chem., 2004, 2283–2288 CrossRef; (e) Z. Yan, Z. Zhou, Y. Wu, I. A. Tikhonova and V. B. Shur, Anal. Lett., 2005, 38, 377–388 CrossRef; (f) A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed; (g) S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef PubMed.
- (a) S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef PubMed; (b) M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298–7301 CrossRef PubMed; (c) M. Hirai, J. Cho and F. P. Gabbaï, Chem.–Eur. J., 2016, 22, 6537–6541 CrossRef PubMed; (d) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef PubMed; (e) L. Wang, S. Zhang, Y. Hasegawa, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2017, 53, 5499–5502 RSC; (f) S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC; (g) D. Chen, G. Xu, Q. Zhou, L. W. Chung and W. Tang, J. Am. Chem. Soc., 2017, 139, 9767–9770 CrossRef PubMed.
- (a) H. E. Katz, J. Org. Chem., 1985, 50, 5027–5032 CrossRef; (b) H. E. Katz, J. Am. Chem. Soc., 1985, 107, 1420–1421 CrossRef; (c) H. E. Katz, Organometallics, 1987, 6, 1134–1136 CrossRef; (d) M. Reilly and T. Oh, Tetrahedron Lett., 1994, 35, 7209–7212 CrossRef; (e) M. Melaïmi, S. Sole, C.-W. Chiu, H. Wang and F. P. Gabbaï, Inorg. Chem., 2006, 45, 8136–8143 CrossRef PubMed; (f) C. Jiang, O. Blacque and H. Berke, Chem. Commun., 2009, 5518–5520 RSC; (g) H. Zhao and F. P. Gabbaï, Organometallics, 2012, 31, 2327–2335 CrossRef.
- (a) V. C. Williams, W. E. Piers, W. Clegg, M. R. J. Elsegood, S. Collins and T. B. Marder, J. Am. Chem. Soc., 1999, 121, 3244–3245 CrossRef; (b) S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686–14687 CrossRef PubMed; (c) J. Chai, S. P. Lewis, S. Collins, T. J. J. Sciarone, L. D. Henderson, P. A. Chase, G. J. Irvine, W. E. Piers, M. R. J. Elsegood and W. Clegg, Organometallics, 2007, 26, 5667–5679 CrossRef; (d) L. D. Henderson and W. E. Piers, J. Organomet. Chem., 2007, 692, 4661–4668 CrossRef; (e) M. V. Metz, D. J. Schwartz, C. L. Stern, P. N. Nickias and T. J. Marks, Angew. Chem., Int. Ed., 2000, 39, 1312–1316 CrossRef; (f) V. C. Williams, G. J. Irvine, W. E. Piers, Z. Li, S. Collins, W. Clegg, M. R. J. Elsegood and T. B. Marder, Organometallics, 2000, 19, 1619–1621 CrossRef; (g) S. N. Kessler, M. Neuburger and H. A. Wegner, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef PubMed; (h) E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef PubMed; (i) J. W. Taylor, A. McSkimming, C. F. Guzman and W. H. Harman, J. Am. Chem. Soc., 2017, 139, 11032–11035 CrossRef PubMed.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC.
- (a) S. N. Kessler, M. Neuburger and H. A. Wegner, Eur. J. Org. Chem., 2011, 2011, 3238–3245 CrossRef; (b) L. Schweighauser and H. A. Wegner, Chem.–Eur. J., 2016, 22, 14094–14103 CrossRef PubMed.
- D. J. H. Emslie, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2003, 42, 1252–1255 CrossRef PubMed.
- (a) H. E. Katz, J. Org. Chem., 1989, 54, 2179–2183 CrossRef; (b) H. Wang and F. P. Gabbaï, Organometallics, 2005, 24, 2898–2902 CrossRef; (c) J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 18194–18197 CrossRef PubMed.
- (a) N. Lopez, D. J. Graham, R. McGuire, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 CrossRef PubMed; (b) M. Nava, N. Lopez, P. Müller, G. Wu, D. G. Nocera and C. C. Cummins, J. Am. Chem. Soc., 2015, 137, 14562–14565 CrossRef PubMed; (c) E. W. Dahl, H. T. Dong and N. K. Szymczak, Chem. Commun., 2018, 54, 892–895 RSC.
- C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 521–525 CrossRef PubMed.
- S. Kirschner, J.-M. Mewes, M. Bolte, H.-W. Lerner, A. Dreuw and M. Wagner, Chem.–Eur. J., 2017, 23, 5104–5116 CrossRef PubMed.
- (a) J. D. Hoefelmeyer and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 9054–9055 CrossRef; (b) J. D. Hoefelmeyer, S. Solé and F. P. Gabbaï, Dalton Trans., 2004, 1254–1258 RSC.
- CCDC 1838442 (1-μ2-N2H4), 1827003 (2), 1827004 ([K(dibenzo-18-crown-6)][1-μ2-CN]-(CH2Cl2)) and 1827005 ([K(dibenzo-18-crown-6)][2-μ2-CN]-(CH2Cl2)2) contain the supplementary crystallographic data for this paper.†.
- (a) K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef PubMed; (b) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef; (c) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef PubMed; (d) H. Y. Zhao and F. P. Gabbai, Nat. Chem., 2010, 2, 984–990 CrossRef PubMed; (e) A. L. Brazeau, K. Yuan, S.-B. Ko, I. Wyman and S. Wang, ACS Omega, 2017, 2, 8625–8632 CrossRef.
- (a) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef PubMed; (b) T. Matsumoto, C. R. Wade and F. P. Gabbaï, Organometallics, 2010, 29, 5490–5495 CrossRef; (c) K. C. Song, K. M. Lee, N. V. Nghia, W. Y. Sung, Y. Do and M. H. Lee, Organometallics, 2013, 32, 817–823 CrossRef.
- (a) S. Pylypko, E. Petit, P. G. Yot, F. Salles, M. Cretin, P. Miele and U. B. Demirci, Inorg. Chem., 2015, 54, 4574–4583 CrossRef PubMed; (b) G. Qi, K. Wang, K. Yang and B. Zou, J. Phys. Chem. C, 2016, 120, 21293–21298 CrossRef.
- (a) Y. Yu, W. W. Brennessel and P. L. Holland, Organometallics, 2007, 26, 3217–3226 CrossRef PubMed; (b) Y. Lee, N. P. Mankad and J. C. Peters, Nat. Chem., 2010, 2, 558 CrossRef PubMed; (c) C. T. Saouma, C. C. Lu and J. C. Peters, Inorg. Chem., 2012, 51, 10043–10054 CrossRef PubMed; (d) K. Umehara, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 6754–6757 CrossRef PubMed; (e) Y. Li, Y. Li, B. Wang, Y. Luo, D. Yang, P. Tong, J. Zhao, L. Luo, Y. Zhou, S. Chen, F. Cheng and J. Qu, Nat. Chem., 2013, 5, 320 CrossRef PubMed.
- B. Roy and S. Bandyopadhyay, Anal. Methods, 2018, 10, 1117–1139 RSC.
- (a) J. T. Henthorn and T. Agapie, Angew. Chem., Int. Ed., 2014, 53, 12893–12896 CrossRef PubMed; (b) X. Tao, C. G. Daniliuc, O. Janka, R. Pöttgen, R. Knitsch, M. R. Hansen, H. Eckert, M. Lübbesmeyer, A. Studer, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2017, 56, 16641–16644 CrossRef PubMed.
- C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 CrossRef PubMed.
- S. Mebs, S. Grabowsky, D. Förster, R. Kickbusch, M. Hartl, L. L. Daemen, W. Morgenroth, P. Luger, B. Paulus and D. Lentz, J. Phys. Chem. A, 2010, 114, 10185–10196 CrossRef PubMed.
- K. Kohata, T. Fukuyama and K. Kuchitsu, J. Phys. Chem., 1982, 86, 602–606 CrossRef.
- E. Leusmann, F. Schneck and S. Dehnen, Organometallics, 2015, 34, 3264–3271 CrossRef.
- (a) Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC; (b) Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC; (c) F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC; (d) T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- (a) T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef PubMed; (b) C.-W. Chiu and F. P. Gabbaï, Dalton Trans., 2008, 814–817 RSC; (c) C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60–61 CrossRef PubMed; (d) Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef PubMed; (e) A. E. J. Broomsgrove, D. A. Addy, A. Di Paolo, I. R. Morgan, C. Bresner, V. Chislett, I. A. Fallis, A. L. Thompson, D. Vidovic and S. Aldridge, Inorg. Chem., 2010, 49, 157–173 CrossRef PubMed; (f) C. R. Wade and F. P. Gabbaï, Inorg. Chem., 2010, 49, 714–720 CrossRef PubMed; (g) Y. Kim, H.-S. Huh, M. H. Lee, I. L. Lenov, H. Zhao and F. P. Gabbaï, Chem.–Eur. J., 2011, 17, 2057–2062 CrossRef PubMed.
- D. F. Shriver and J. Posner, J. Am. Chem. Soc., 1966, 88, 1672–1677 CrossRef.
- (a) B. Ahlers, K. Cammann, S. Warzeska and R. Krämer, Angew. Chem., Int. Ed., 1996, 35, 2141–2143 CrossRef; (b) S. Warzeska and R. Krämer, Chem. Commun., 1996, 499–500 RSC.
- C. R. Wade and F. P. Gabbaï, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 1199–1205 Search PubMed.
- (a) C. M. Giandomenico, J. C. Dewan and S. J. Lippard, J. Am. Chem. Soc., 1981, 103, 1407–1412 CrossRef; (b) S. J. Lancaster, D. A. Walker, M. Thornton-Pett and M. Bochmann, Chem. Commun., 1999, 1533–1534 RSC; (c) J. Zhou, S. J. Lancaster, D. A. Walker, S. Beck, M. Thornton-Pett and M. Bochmann, J. Am. Chem. Soc., 2001, 123, 223–237 CrossRef PubMed; (d) N. M. Brunkan, D. M. Brestensky and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 3627–3641 CrossRef PubMed.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra of novel compounds, crystallographic data, absorption and emission spectra, DFT optimization results. CCDC 1838442, 1827003–1827005. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01877d |
| This journal is © The Royal Society of Chemistry 2018 |