
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
(Salen)Mn(III)-catalyzed chemoselective acylazidation of olefins†
Liang
Zhang
,
Shuya
Liu
,
Zhiguo
Zhao
,
Hongmei
Su
,
Jingcheng
Hao
 and
Yao
Wang
*
and
Yao
Wang
*
School of Chemistry and Chemical Engineering, Key Laboratory of the Colloid and Interface Chemistry, Shandong University, 27 Shanda Nanlu, Jinan 250100, Shandong, China. E-mail: yaowang@sdu.edu.cn
First published on 19th June 2018
Abstract
We describe a (salen)Mn(III)-catalyzed three-component reaction of aldehydes, olefins, and sodium azide for the installation of two useful groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N3) into the double bond. Traditionally, (salen)Mn(III) in conjunction with iodosobenzene is a classical catalysis system for epoxidation of olefins. Owing to the highly competitive oxygenation approaches, it is a true challenge to establish a distinct strategy for the exploration of new olefin transformations based on this (salen)Mn(III) catalysis system. Herein, the key to this (salen)Mn(III)-catalyzed acylazidation of olefins was the rational application of the distinct reactivity of oxomanganese(V) species which is capable of abstracting a hydrogen atom from a substrate C–H bond. This chemoselective reaction occurred in a precisely designed reaction sequence and tolerates complex molecular structures.
O and N3) into the double bond. Traditionally, (salen)Mn(III) in conjunction with iodosobenzene is a classical catalysis system for epoxidation of olefins. Owing to the highly competitive oxygenation approaches, it is a true challenge to establish a distinct strategy for the exploration of new olefin transformations based on this (salen)Mn(III) catalysis system. Herein, the key to this (salen)Mn(III)-catalyzed acylazidation of olefins was the rational application of the distinct reactivity of oxomanganese(V) species which is capable of abstracting a hydrogen atom from a substrate C–H bond. This chemoselective reaction occurred in a precisely designed reaction sequence and tolerates complex molecular structures.
Introduction
Catalytic transformation of olefins is one of the fundamental approaches for the generation of molecular complexity and diversity. A range of elegant and powerful strategies have been established for the installation of a diverse array of useful functional groups into the double bonds, which has essentially advanced the art and practical use of organic synthesis.1 Amongst various catalysts for olefin transformations, manganese Schiff-base complexes have been well-established as powerful catalysts for epoxidation of olefins in the last three decades.2–5 It has been a general notion that the oxomanganese(V) species in situ generated through oxidation of the (salen)Mn(III) complex is an efficient oxygen-transfer species for epoxidation of various olefins.6,7 Owing to the fast and strong oxygenative background reactions,5–7 it remains a substantial challenge to develop a distinct strategy from a fresh perspective that enables the discovery of previously unknown olefin transformations based on this oxidative (salen)Mn(III) catalysis system (Fig. 1A).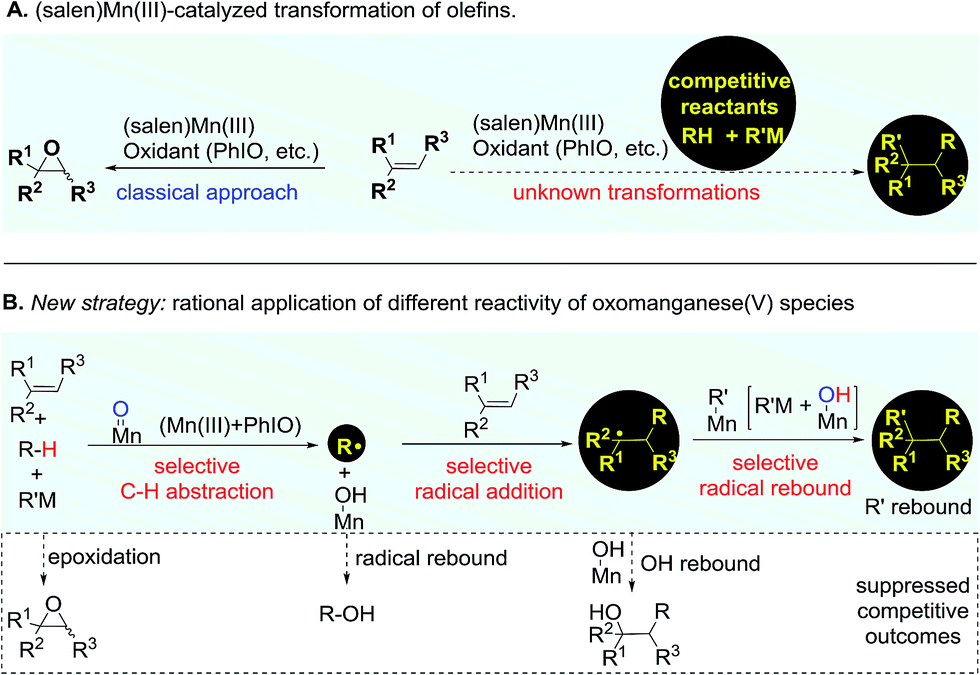 | ||
| Fig. 1 (Salen)Mn(III)-catalyzed competitive transformation of olefins. | ||
The reactive oxomanganese(V) species can readily abstract a hydrogen atom even from inert C–H bonds such as those in alkanes to generate a substrate-derived radical and a hydroxomanganese(IV) intermediate under mild reaction conditions.8,9 This distinct reactivity of oxomanganese(V) species provides a basis and opens up new opportunities for the development of a useful strategy from a fresh perspective enabling the transformation of olefins. As outlined in Fig. 1B, we envisioned that the addition of a carefully selected reactant bearing a weak C–H bond to the oxidative (salen)Mn(III) catalysis system would lead to a preferential C–H abstraction, thus shutting down the highly competitive epoxidation pathway. Considering the fast radical rebound pathway,10,11 it is essential that the in situ generated radical is capable of immediately reacting with the ‘spectator’ olefin to start the precisely designed reaction sequence towards the desirable outcome. Finally, a selective radical rebound reaction would finish the whole reaction sequence. A standing challenge for implementing the designed strategy has been the suppression of these consecutively competitive approaches.12,13
In contrast to alkanes, aldehydes have a weaker (O)C–H bond which can be feasibly cleaved to generate acyl radicals.14,15 Hydroacylation of olefins using aldehydes as an acyl source represents a simple and efficient method for the preparation of ketones.15–18 Under substantially different reaction pathways, aldehydes have been applied in acylation of highly electron-deficient olefins19–22 or unactivated olefins.23–28 Despite the important achievements in radical acylation of olefins, there are considerable limitations that remain to be resolved with regard to the scope of aldehydes and olefins.19–22 At room temperature, acyl radicals are generally reactive towards the addition of highly electron-deficient olefins while they show very poor or no reactivity towards less electron-deficient olefins. Aldehydes are generally restricted to a specific class to avoid the decarbonylation problem and to enable the desirable acyl radical addition of olefins. Furthermore, for an intermolecular hydroacylation approach, the installation of a functional group instead of a hydrogen atom through trapping of the acylation intermediate remains a largely elusive problem. We envisioned that the rationally designed strategy could provide a promising solution to these synthetic problems. Herein, we present our findings on the establishment and application of this strategy in the development of a chemoselective acylazidation of olefins.
Results and discussion
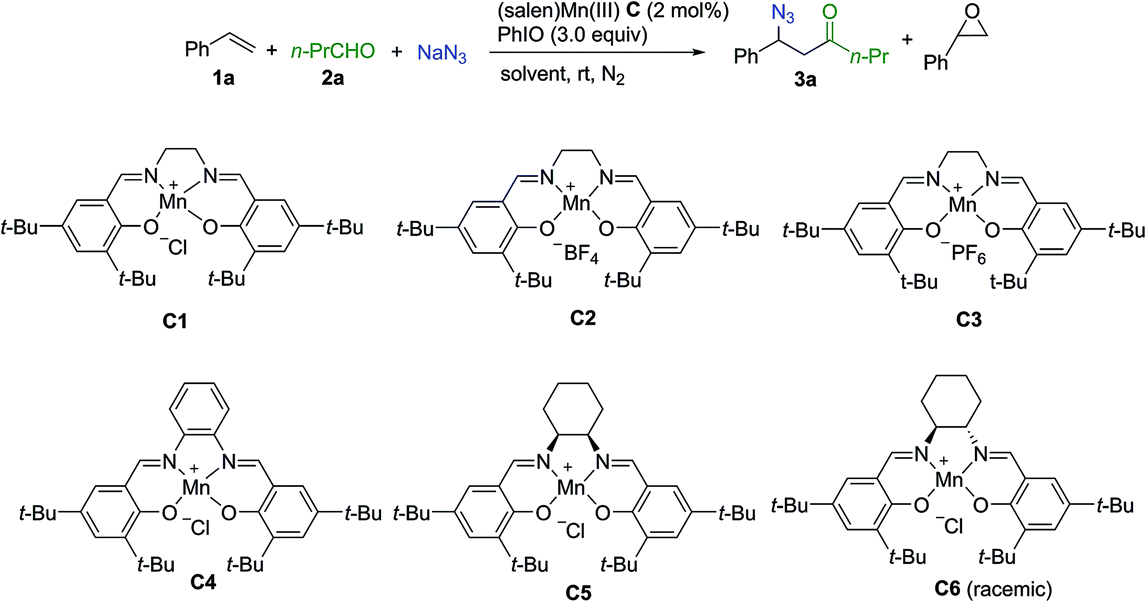
Extensive investigation of numerous possible combinations of competitive reactants was carried out. Encouragingly, we were able to identify that the addition of a combination of aldehyde and sodium azide to the (salen)Mn(III) catalysis system could suppress oxygenation approaches, resulting in the chemoselective formation of an acylazidation product. Organic azides can participate in a range of important reactions including Staudinger ligation,29 1,3-dipolar cycloaddition,30 and the aza-Wittig reaction,31 making them highly attractive targets for organic synthesis.32 A range of elegant methods have been developed for azidation of olefins33 and C–H azidation.34 To optimize the reaction conditions, styrene was used as a model substrate while n-butylaldehyde and sodium azide were employed as a combination of competitors. As shown in Table 1, initially, several control experiments were conducted. No product was observed in the absence of either the (salen)Mn(III) catalyst or PhIO (entries 1–2). In the absence of n-butylaldehyde and sodium azide, as previously reported by Kochi and co-workers,12 a fast consumption of styrene was observed (<1 h, >95% conversion of styrene, 30% epoxide, entry 3). The acylazidation product was formed in 57% yield catalyzed by C1 in CH3CN (entry 4). Solvent optimization showed that a mixed solvent of EtOAc and H2O is an optimal choice, which afforded the desired product in 75% yield (entries 5–9). The evaluation of different (salen)Mn(III) complexes revealed that catalyst C1 is a promising candidate (entries 10–14). Upon using 1 mol% of catalyst C1, the reaction yield was decreased (entry 15). Further optimization of the amount of aldehyde and iodosobenzene did not improve the reaction yield (entries 16–18). The optimized reaction conditions did not provide an observable quantity of the epoxidation product. In contrast, 41% epoxide was obtained in the absence of n-butylaldehyde and sodium azide (entry 19).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Epoxideb (%) | Yieldb (3a%) |
| a Unless otherwise noted, all the reactions were carried out with 1a (0.3 mmol), 2a (1.5 mmol), NaN3 (1.2 mmol), PhIO (0.9 mmol) and C (2 mol%) in 3.0 mL solvent as indicated at room temperature. b Isolated yield. c Without PhIO. d Without 2a and NaN3. e 6.4 mL mixed solvent was used (H2O/organic solvent = 1/0.6, entries 8–19). f 1 mol% of C was used. g 0.9 mmol of 2a was used. h 2.0 equiv. of PhIO were used. i 4.0 equiv. of PhIO were used. n.r. = no reaction. | |||||
| 1 | — | CH3CN | 12 | n.r. | n.r. |
| 2c | C1 | CH3CN | 12 | n.r. | n.r. |
| 3d | C1 | CH3CN | <1 | 30 | — |
| 4 | C1 | CH3CN | 6 | <5 | 57 |
| 5 | C1 | EtOAc | 6 | 6 | 45 |
| 6 | C1 | DCM | 6 | <5 | 27 |
| 7 | C1 | Toluene | 6 | <5 | 39 |
| 8e | C1 | H2O/CH3CN | 3 | <5 | 52 |
| 9 | C1 | H2O/EtOAc | 3 | <5 | 75 |
| 10 | C2 | H2O/EtOAc | 3 | <5 | 65 |
| 11 | C3 | H2O/EtOAc | 3 | <5 | 68 |
| 12 | C4 | H2O/EtOAc | 5 | 7 | 11 |
| 13 | C5 | H2O/EtOAc | 9 | <5 | 65 |
| 14 | C6 | H2O/EtOAc | 5 | <5 | 55 |
| 15f | C1 | H2O/EtOAc | 6 | <5 | 58 |
| 16g | C1 | H2O/EtOAc | 6 | <5 | 54 |
| 17h | C1 | H2O/EtOAc | 6 | <5 | 49 |
| 18i | C1 | H2O/EtOAc | 6 | <5 | 51 |
| 19d | C1 | H2O/EtOAc | 4 | 41 | — |
The substrate scope was investigated next. As shown in Scheme 1, remarkably, all the representative aldehydes, including linear- and branched-aliphatic aldehydes and aromatic aldehydes, could be tolerated and the desired products were obtained in reasonable yields (3a–d). Aromatic rings of olefins bearing electron-donating or electron-withdrawing groups were tolerated. Meanwhile, substituent groups could be incorporated in diversified positions, delivering ortho-, meta-, and para-substituted products. Furthermore, multi-substituted aromatic rings were amenable to this transformation (3m–o). The employment of several α-substituted styrene derivatives for the synthesis of a range of tertiary azides was successful (3p–s). Both trans- and cis-stilbene could be used as substrates to afford the same product with similar diastereoselectivities in 50% and 54% yield, respectively (3t). Under standard conditions using cis-stilbene as a substrate, acylazidation product 3t was obtained in 30% yield along with 17% epoxide. Interestingly, reducing the catalyst loading to 1 mol% was effective in bringing about a vigorous competition between the reactions of acylazidation and epoxidation of stilbene since the acylazidation approach overwhelmingly dominated the reaction pathways. A heterocyclic substrate could also be tolerated in this reaction (3u).
 | ||
| Scheme 1 Substrate scopea,b. aUnless otherwise noted, all the reactions were carried out with 1a (0.3 mmol), 2a (1.5 mmol), NaN3 (1.2 mmol) and C (2 mol%) in 6.4 mL H2O/EtOAc (1/0.6) at room temperature. bThe diastereoselectivity was determined by 1H NMR analysis of the crude reaction mixture and yields refer to the isolated major product. c1 mol% of catalyst C1 and 6.0 equiv. of PhIO were used (compound 3t). dThe diastereoselectivities of 4b, 5a and 5b refer to the ratio of the two major isomers. | ||
Next, the use of electron-deficient α,β-unsaturated amides as substrates was studied. All the α,β-unsaturated amides smoothly afforded the desired products with moderate to good diastereoselectivity in reasonable yields (Scheme 1). Aromatic groups with different electrical properties such as electron-withdrawing or electron-donating groups were tolerated, as were ortho-, meta-, and para- substituted aromatic rings. The structure of 3aa was confirmed by single-crystal X-ray diffraction.35 This methodology could be applied to more complex contexts. Estrone derivatives could be applied in this transformation. It is of significance to incorporate the azido group into peptide derivatives for the synthesis of a wide range of complex molecules with promising biological activities.36,37 This methodology provides a concise pathway for the preparation of azidopeptide derivatives (Scheme 1).
As shown in Fig. 2, a series of control experiments were carried out to probe the mechanism (see the ESI†). Considering the significant work done by Jacobsen and co-workers who established that azide ring-opening of epoxides can be efficiently catalyzed by metal–salen complexes,38,39 the replacement of styrene with styrene oxide did not generate product 3a either under standard reaction conditions or without PhIO. This experiment can exclude the possibility that 3a was generated from complex transformation of styrene oxide. Furthermore, established work revealed that α,β-unsaturated ketones could be generated through copper-catalyzed oxidative coupling of alkenes with aldehydes.40 Meanwhile, Jacobsen and co-workers discovered that a Lewis acid was able to catalyze conjugate addition of azide to α,β-unsaturated ketones.41,42 However, no α,β-unsaturated ketone ((E)-1-phenylhex-1-en-3-one) was observed under standard reaction conditions using 1a and 2a as the starting materials. The reaction between a synthetic α,β-unsaturated ketone ((E)-1-phenylhex-1-en-3-one) and sodium azide catalyzed by (salen)Mn(III) C1 with (or without) PhIO failed to deliver the conjugate product 3a, which excludes the possibility of a Lewis acid-catalyzed conjugate addition pathway.
 | ||
| Fig. 2 Probing mechanism. | ||
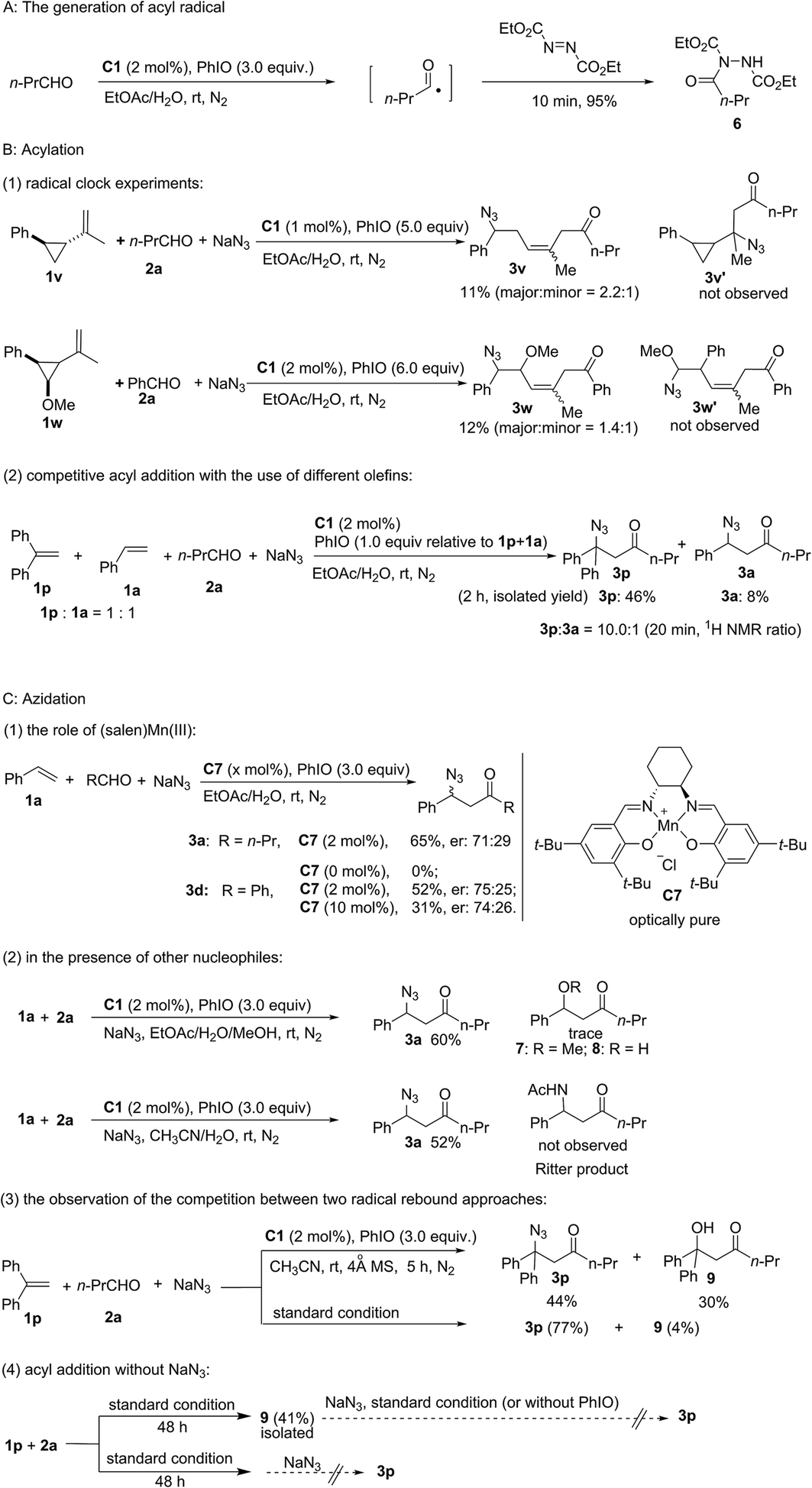
The generation of an acyl radical in the presence of Mn(III) and PhIO was observed by using diethyl azodicarboxylate as a trapping agent.43 Furthermore, using radical clock substrate 1v afforded the ring-opening product 3v. Newcomb-type cyclopropane substrate 1w was employed to differentiate between a radical approach and a cationic pathway.44,45 Acyl addition to the olefin 1w occurs with subsequent cleavage of the benzylic cyclopropyl bond rather than the α-methoxy cyclopropyl bond. Furthermore, an intermolecular competitive reaction between substrates 1p and 1a was carried out. Despite greater steric hindrance, compound 3p was obtained as a major product and a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (20 min, 3p:3a) was observed upon using a 1:1 mixture of 1p and 1a. This observation can be attributed to the competitive generation of a more stable dibenzylic radical. All these results clearly point to an acyl radical addition pathway.
:1 (20 min, 3p:3a) was observed upon using a 1:1 mixture of 1p and 1a. This observation can be attributed to the competitive generation of a more stable dibenzylic radical. All these results clearly point to an acyl radical addition pathway.
To probe the azidation process, several control experiments were carried out. Nearly the same moderate enantioselectivity of 3d was obtained in the presence of 2 mol% and 10 mol% of Jacobsen's catalyst C7 while no product was observed without this catalyst. However, an intramolecular reaction using 2-(allyloxy)benzaldehyde as the substrate could proceed in the absence of a catalyst to afford chroman derivatives.46 Considering the potential pathway in which the oxidation of radical intermediate by manganese would lead to the formation of a carbocation, 20 equiv. of methanol was added to the standard reaction system to competitively trap the carbocation. However, neither methoxylated product 7 nor hydroxylated product 8 was observed. Furthermore, no product of the competitive Ritter reaction was detected upon using CH3CN/H2O as the solvent. Furthermore, competition between the two radical rebound approaches was observed and a considerable amount of hydroxylated product 9 (30%) was isolated in dry CH3CN. This experiment suggests that the formation of an acyl radical and a hydroxomanganese(IV) intermediate originated from (O)C–H abstraction by the reactive oxomanganese(V) species. These experiments indicate an azido-rebound pathway. Only 4% of 9 was isolated, which indicates that the rate of azido-rebound is much faster than the rate of hydroxy-rebound under standard reaction conditions. Furthermore, in the absence of NaN3, product 9 was isolated in 41% yield, which clearly revealed that both the generation of the acyl radical and the subsequent radical addition of the olefin can occur in the absence of NaN3. Further control experiments revealed that the acylazidation product 3p is not generated from the transformation of hydroxylated compound 9.
Based on these obtained results, a possible mechanism was proposed and is depicted in Fig. 3. Initially, a ligand exchange process resulted in a manganese-bound azide, M1. The remaining (salen)Mn(III) is oxidized by PhIO, generating a reactive oxomanganese(V) intermediate, M2. The oxomanganese(V) species is capable of selectively abstracting a hydrogen from the aldehyde to form intermediate M3 and an acyl radical. The subsequent acyl radical addition of a manganese-activated alkene would generate a pending radical intermediate. Finally, azido-rebound via complex M4 releases the final product and (salen)Mn(III) catalyst.
 | ||
| Fig. 3 Plausible mechanism. | ||
Conclusions
In summary, enabled by the rational application of the distinct reactivity of oxomanganese(V) species, a new strategy was established for the transformation of olefins. A unique finding was that (salen)Mn(III) in conjunction with iodosobenzene can induce an intermolecular acylazidation of olefins using aldehydes and sodium azide as competitive reactants. Both carbonyl and azido groups were incorporated into simple olefinic compounds as well as complex targets. This transformation enables electron-deficient, neutral and even electron-rich alkenes to participate in an acyl radical addition process at room temperature. The representative aldehydes, including linear- and branched-aliphatic aldehydes and aromatic aldehydes, could be used as effective reactants. Potentially, the strategy described herein could open the doors to new catalytic reactivity that has been unexplored.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21772113, 21302075, and 11501454), the Key Research and Development Plan of Shandong Province (2017GGX70109), and the Fundamental Research Funds of Shandong University (2017JC004). We thank Prof. Di Sun for assistance with the X-ray crystal structure analysis.Notes and references
- Comprehensive Organic Synthesis II, P. Knochel and G. A. Molander, Elsevier, Amsterdam, The Netherlands, 2014 Search PubMed.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef.
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef.
- F. Teixeira and M. N. D. S. Cordeiro, Catalysts, 2017, 7 DOI:10.3390/catal7010002.
- T. Katsuki, Chem. Soc. Rev., 2004, 33, 437–444 RSC.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef PubMed.
- C. T. Dalton, K. M. Ryan, V. M. Wall, C. Bousquet and D. G. Gilheany, Top. Catal., 1998, 5, 75–91 CrossRef.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef PubMed.
- A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC.
- M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef.
- J. T. Groves, Models and mechanisms of cytochrome P450 action, in Cytochrome P450: Structure, Mechanisms and Biochemistry, ed. P. R. Ortiz de Montellano, 3rd edn, Plenum Press, New York, 2005 Search PubMed.
- K. Srinivasan, P. Michaud and J. K. Kochi, J. Am. Chem. Soc., 1986, 108, 2309–2320 CrossRef PubMed.
- J. T. Groves, W. J. Kruper and R. C. Haushalter, J. Am. Chem. Soc., 1980, 102, 6375–6377 CrossRef.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, 2007 Search PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and L. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef PubMed.
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef PubMed.
- J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202–2209 RSC.
- X. Lan, N. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef.
- S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 119, 2583–2586 CrossRef.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef PubMed.
- S. A. Moteki, A. Usui, S. Selvakumar, T. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060–11064 CrossRef PubMed.
- L. Lv and Z. Li, Chin. J. Chem., 2017, 35, 303–306 CrossRef.
- L. Xiao, X. Fu, M. Zhou, J. Xie, L. Wang, X. Xu and Q. Zhou, J. Am. Chem. Soc., 2016, 138, 2957–2960 CrossRef PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Chem. Sci., 2015, 6, 174–180 RSC.
- Q. A. Chen, D. K. Kim and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 3772–3775 CrossRef PubMed.
- M. Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef PubMed.
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef PubMed.
- W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRef PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef PubMed.
- F. Palacios, C. Alonso, D. Aparicio, G. Rubiales and J. M. de los Santos, Tetrahedron, 2007, 63, 523–575 CrossRef.
- For selected reviews, see: (a) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef; (b) J. Fu, G. Zanoni, E. A. Anderson and X. Bi, Chem. Soc. Rev., 2017, 46, 7208–7228 RSC; (c) M. Goswami and B. de Bruin, Eur. J. Org. Chem., 2017, 1152–1176 CrossRef PubMed; (d) K. Wu, Y. Liang and N. Jiao, Molecules, 2016, 21, 352–372 CrossRef PubMed; (e) T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef PubMed; (f) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- For selected examples, see: (a) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef PubMed; (b) X. Sun, X. Li, S. Song, Y. Zhu, Y. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059–6066 CrossRef PubMed; (c) T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692–11695 CrossRef PubMed; (d) J. Xu, X. Li, Y. Gao, L. Zhang, W. Chen, H. Fang, G. Tang and Y. Zhao, Chem. Commun., 2015, 51, 11240–11243 RSC; (e) J. Waser, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 8294–8295 CrossRef PubMed; (f) H. Yin, T. Wang and N. Jiao, Org. Lett., 2014, 16, 2302–2305 CrossRef PubMed; (g) D. J. Guerin, T. E. Horstmann and S. J. Miller, Org. Lett., 1999, 1, 1107–1109 CrossRef PubMed.
- For selected examples, see: (a) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751–759 CrossRef; (b) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef PubMed; (c) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300–5303 CrossRef PubMed.
- CCDC 1546115 (3aa) contains the supplementary crystallographic data for this paper.
- J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- L. E. Martínez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef.
- J. F. Larrow, S. E. Schaus and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 7420–7421 CrossRef.
- J. Wang, C. Liu, J. Yuan and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 2256–2259 CrossRef PubMed.
- M. S. Taylor, D. N. Zalatan, A. M. Lerchner and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 1313–1317 CrossRef PubMed.
- G. M. Sammis and E. N. Jacobsen, J. Am. Chem. Soc., 2003, 125, 4442–4443 CrossRef PubMed.
- H.-B. Zhang, Y. Wang, Y. Gu and P.-F. Xu, RSC Adv., 2014, 4, 27796–27799 RSC.
- M.-H. Le Tadic-Biadatti and M. Newcomb, J. Chem. Soc., Perkin Trans. 2, 1996, 1467–1473 RSC.
- M. M. Roubelakis, G. C. Vougioukalakis, Y. S. Angelis and M. Orfanopoulos, Org. Lett., 2006, 8, 39–42 CrossRef PubMed.
- J. Zhao, P. Li, X. Li, C. Xia and F. Li, Chem. Commun., 2016, 52, 3661–3664 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1546115. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01882k |
| This journal is © The Royal Society of Chemistry 2018 |