
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying site-dependent reactivity in oxidation reactions on single Pt particles†
Shahar
Dery
ab,
Suhong
Kim
 c,
David
Haddad
ab,
Albano
Cossaro
d,
Alberto
Verdini
d,
Luca
Floreano
d,
F. Dean
Toste
*c and
Elad
Gross
*ab
c,
David
Haddad
ab,
Albano
Cossaro
d,
Alberto
Verdini
d,
Luca
Floreano
d,
F. Dean
Toste
*c and
Elad
Gross
*ab
aInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: elad.gross@mail.huji.ac.il
bThe Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 91904, Israel
cDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@berkeley.edu
dCNR-IOM, Laboratorio Nazionale TASC, Basovizza SS-14, Trieste 34012, Italy
First published on 3rd July 2018
Abstract
Catalytic nanoparticles are heterogeneous in their nature and even within the simplest particle various surface sites exist and influence the catalytic reactivity. Thus, detailed chemical information at the nanoscale is essential for understanding how surface properties and reaction conditions direct the reactivity of different surface sites of catalytic nanoparticles. In this work, hydroxyl-functionalized N-heterocyclic carbene molecules (NHCs) were anchored to the surface of Pt particles and utilized as chemical markers to detect reactivity variations between different surface sites under liquid and gas phase oxidizing conditions. Differences in the chemical reactivity of surface-anchored NHCs were identified using synchrotron-radiation-based infrared nanospectroscopy with a spatial resolution of 20 nanometers. By conducting IR nanospectroscopy measurements, along with complementary spatially averaged IR and X-ray spectroscopy measurements, we identified that enhanced reactivity occurred on the particles' periphery under both gas and liquid phase oxidizing conditions. Under gas phase reaction conditions the NHCs' hydroxyl functional groups underwent preferential oxidization to the acid along the perimeter of the particle. Exposure of the sample to harsher, liquid phase oxidizing conditions induced modification of the NHCs, which was mostly identified at the particle's periphery. Analysis of X-ray absorption spectroscopy measurements revealed that exposure of the sample to oxidizing conditions induced aromatization of the NHCs, presumably due to oxidative dehydrogenation reaction, along with reorientation of the NHCs from perpendicular to parallel to the Pt surface. These results, based on single particle measurements, demonstrate the high reactivity of surface sites that are located at the nanoparticle's periphery and the influence of reaction conditions on site-dependent reactivity.
Introduction
Molecular level understanding of catalytic processes is essential for the development of highly efficient and selective catalysts.1–4 However, heterogeneities in the size, structure and composition of solid catalytic particles makes it difficult to directly monitor and identify the influence of various physicochemical parameters on the catalytic reactivity and selectivity.5–7 Thus, non-disruptive, detailed chemical information at the nanoscale is required for understanding how surface properties direct the reactivity of catalytic particles.8–10Conventional spectroscopic techniques (such as infrared, ultraviolet-visible and Raman spectroscopy) for molecular analysis of catalytic reactions provide critical molecular level insight into the catalytic process, but are typically limited in their spatial resolution to the micrometer scale.11–15 Near-field microscopy approaches, such as tip-enhanced Raman spectroscopy (TERS) and infrared nanospectroscopy, can overcome such diffraction limitations by spatially confining light–matter interactions.16–26 These two complementary apertureless approaches overcome the diffraction limit of light to detect the vibrational spectrum of molecules on surfaces with a spatial resolution of ∼20 nm. TERS and IR nanospectroscopy measurements provide the technical capabilities to identify chemical processes at the nanoscale. However, the specific detection of reactants-into-products transformations on the surface of a single nanoparticle is not trivial even while using apertureless methods, since other competing molecular processes, including adsorption, diffusion and desorption, simultaneously occurs on the catalyst's surface.5,11,27
High spatial resolution detection of chemical processes on surfaces can be achieved by using surface-anchored molecules as indicators for chemical reactivity.18,21,22 For example, chemically active NO2- and NH2-functionalized thiols were anchored to Ag and Au surfaces and chemical changes in the thiols functional groups, following exposure of the sample to various reaction conditions, were probed by TERS measurements.18,21 We have recently demonstrated a different approach for high spatial resolution mapping of chemical reactions on catalytic particles.23 In this approach, N-heterocyclic carbene molecules (NHCs) with chemically-active functional groups were anchored to the surface of transition metal particles. The high stability, chemical versatility and ease of preparation of NHCs make them optimal chemical indicators for surface-induced reactivity. The surface-induced reactivity of NHCs was mapped using high spatial resolution synchrotron-based infrared nanospectroscopy (SINS) measurements.23 Using this approach, it was observed that the periphery of the particles, which contain higher density of metal atoms with low coordination numbers, in comparison to the flat regions on top of the particles, were more active in catalyzing oxidation and reduction of the chemically active groups in surface-anchored NHCs.
Recent works suggested that variations in the reaction conditions, for example the phase of the reaction, can result in changes to the mechanism and kinetics of oxidation reactions on Pt nanoparticles.28,29 Moreover, it is reasonable to posit that changes in the reaction conditions will differentially impact the various surface sites. However, support for this hypothesis is limited due to the paucity of nanoscale analysis of how modifications in reaction conditions influence the site-dependent reactivity of catalysts.30
Therefore, the aim of this study is to identify, by conducting high spatial resolution single-particle measurements, the ways by which exposure to liquid and gas phase oxidizing conditions modify the chemical reactivity of different surface sites. To achieve this goal we have used OH-functionalized NHCs as indicators for surface-induced reactivity. The NHCs were anchored to the surface of Pt particles so that their functional groups reside in close proximity to catalytically active surface sites. As a consequence, the hydroxyl group can be oxidized upon exposure of the sample to oxidizing conditions (Fig. 1a).
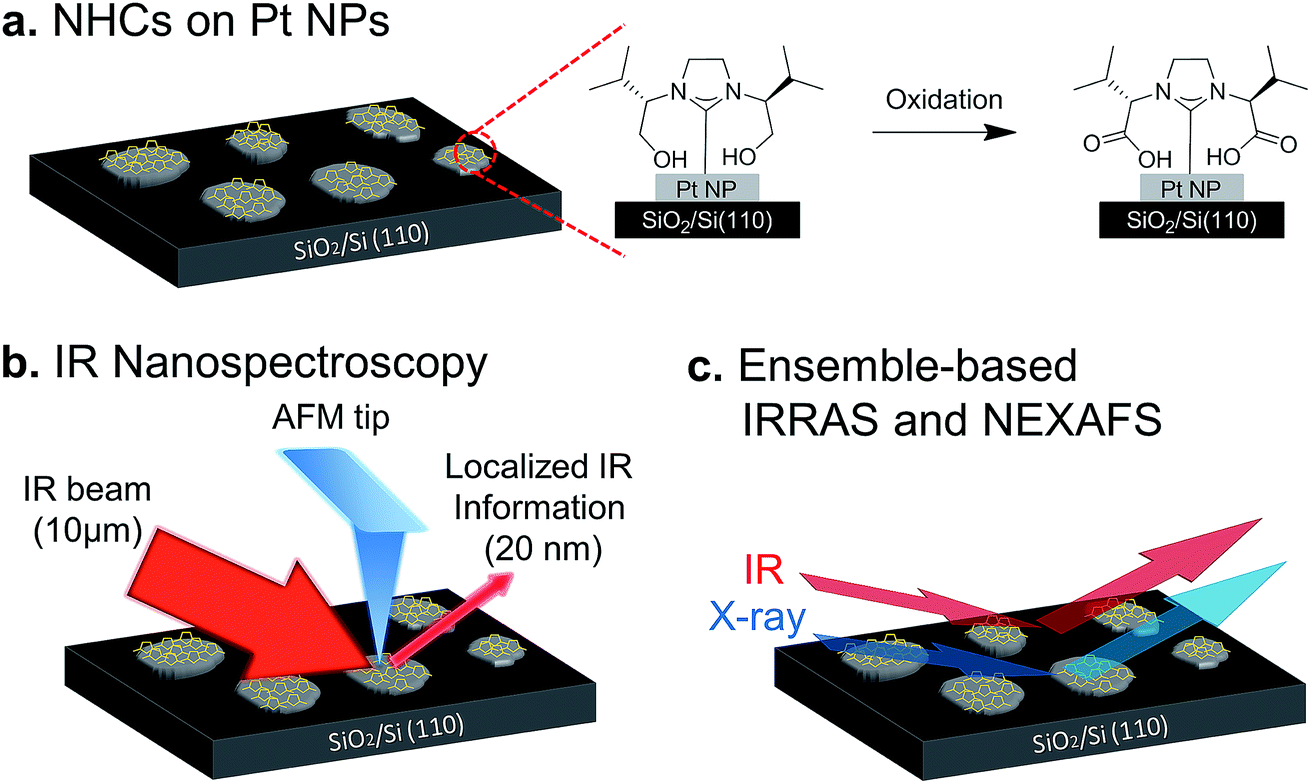 | ||
| Fig. 1 Schematic representation of the experimental approach: (a) OH-functionalized NHCs (marked with orange pentagons) were anchored to the surfaces of Pt particles (grey circles). The particles were supported on a SiO2/Si(110) wafer. Under oxidizing conditions the catalytically active Pt particles oxidize the hydroxyl group of the anchored NHCs. (b) High spatial resolution Synchrotron Infrared Nanospectroscopy (SINS) measurements were performed to identify site-dependent reactivity on single particles. In this setup, an AFM tip acts as an optical antenna by localizing the diffraction-limited IR beam (which has a focal spot of about 10 μm) and inducing an IR scattering signal with a tip-limited spot size of ∼20 nm. (c) Chemical and structural characterization of surface-anchored NHCs was achieved by conducting ensemble-based Near Edge X-ray Absorption Fine Structure (NEXAFS), X-ray photoelectron spectroscopy (XPS) and Infrared Reflection Absorption Spectroscopy (IRRAS) measurements. | ||
High-spatial-resolution SINS measurements were conducted, following exposure of the sample to various oxidizing conditions, to identify site-dependent reactivity on single particles (Fig. 1b). Complementary, surface-sensitive infrared reflection absorption spectroscopy (IRRAS), X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS) measurements were performed to identify how exposure of the sample to gas- or liquid-phase oxidizing conditions impacted the chemical and structural properties of NHCs (Fig. 1c).
It should be noted that NHCs were initially developed as ligands in organometallic complexes.31 In the last decade it was widely demonstrated that these molecules can be efficiently anchored to metal surfaces.32–35 The structure and stability of surface anchored NHCs were studied by several surface-sensitive techniques.36–38 However, in depth analysis of the chemical and structural properties of chemically-active NHCs, which were recently utilized as indicators for surface induced reactivity,23,25 remains rare.
In this work, we integrated spatially localized (single-particle) and averaged measurements to identify structure-reactivity correlation on single Pt particles under gas and liquid phase oxidizing conditions. In both cases, higher reactivity was detected on the particles' perimeter. Under gas phase reaction conditions the enhanced reactivity on the particle's periphery led to oxidation of the hydroxyl functional groups into acid. Exposure of the sample to harsher, liquid phase oxidizing conditions induced decarboxylation, which was followed by oxidation, in NHCs that were anchored to the particle's perimeter. The chemical changes in the NHCs' properties were coupled with changes in the NHCs' orientation from perpendicular to parallel to the Pt surface.
Methods and instruments
Sample preparation
Pt particles were prepared by evaporation of metallic Pt film on SiO2/Si(110) surface. This process was followed by annealing of the Pt-coated surface to 823 K for 5 hours, which was conducted under N2 environment to prevent coating of the metallic particles with a SiO2 layer. NHCs were prepared and activated in a glove box according to a recently published protocol.23 The freshly prepared carbene solution was transferred into a vial and covered the Pt-coated Si surface. After 48 h, the Si wafer was removed from the glove box and washed three times with THF (5 ml) and distilled water (5 ml) intermittently. The sample was then flushed with N2 for 5 min and stored in a glove box.SINS measurements
Synchrotron infrared light (provided by the Advanced Light Source, Lawrence Berkeley National Laboratory) was focused onto the apex of an oscillating Pt–Si AFM tip (Nanosensors, PtSi-NCH) at frequency ω in a modified commercial AFM (Innova, Bruker). Because the near-field scattered signal depends nonlinearly on the distance between the tip and the sample, the tip oscillation induces higher harmonics (nω) in the near-field scattered signal. As a result, the near-field signal can be differentiated from the far-field background by detecting the high harmonic frequency 2ω with a lock-in amplifier. A modified commercial FTIR spectrometer (Nicolet 6700, Thermo Scientific) was used to collect the infrared nanospectroscopy signal. Following AFM topography imaging of the surface, infrared nanospectroscopy point measurements were conducted at selected locations of the particles' surface. The Fourier transform of the interferogram provides a complex-valued near-field spectrum. The real (Re(ν), where ν is the wavenumber) and imaginary (Im(ν)) spectra can be represented as spectral amplitude (A(ν)) and phase (φ(ν)). The near-field spectra were reported in the form of a normalized scattering phase (φ(ν) = φsample(ν) − φreference(ν)), using the bare Si surface as a reference point.SINS measurements were performed before and after exposure of the sample to liquid or gas phase oxidizing conditions. All SINS measurements were conducted at room temperature under nitrogen atmosphere. IR nanospectroscopy measurements were conducted on different sites on the surface of at least 10 particles under each reaction conditions. In these measurements at least 8 out of 10 particles, on average, showed a similar trend in their site-dependent reactivity.
In all the performed IR nanospectroscopy measurements it was verified that the AFM tip is positioned either on the center or along the profile of the metallic particles. Therefore, the IR nanospectroscopy signal was mostly induced by molecules that were anchored on the various metal sites with only a minor, if any, contribution from molecules that reside on the metal/metal–oxide interface.
IRRAS measurements
Measurements were conducted at room-temperature under vacuum on reflection–absorption cell (Harrick, Inc.) with FTIR spectrometer (Vertex V70, Bruker). Measurement parameters: 1024 scans with a resolution of 4 cm−1 using mercury cadmium telluride (MCT) detector.NEXAFS and XPS measurements
X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption experiments (NEXAFS) measurements were performed at the ALOISA beamline of the ELETTRA synchrotron facility in Trieste (Italy).39 X-ray absorption spectra measurements were taken in partial electron yield using a channeltron detector equipped with a front grid biased at negative voltage (−230 V) to filter out the low energy secondary electrons. The NEXAFS spectra at the carbon K-edge were measured with the resolution set to ∼80 meV, keeping the sample at a constant grazing angle of 6°. The orientation of the surface with respect to the photon beam polarization was changed from s-polarization to close to p-polarization by rotating the sample coaxially to the photon beam axis. NEXAFS spectra were reported in the form of a normalized absorption amplitude (I(E) = I/I(reference)), using NEXAFS measured on bare Pt particles (not coated with NHCs) as a reference. X-ray photoelectron spectra of C1s, N1s and Pt4f were acquired. The binding energies of N1s XP-spectra were calibrated according to the binding energy of the C–C peak position (285.5 eV) in the C1s spectra. Analysis of the XPS peaks and their fitting was performed using CasaXPS software.Results and discussion
Pt particles with an average size of 100 ± 30 nm were prepared by annealing (823 K, 5 h, 1 atm N2) of a 20 nm Pt film that was deposited on SiO2/Si(110) wafer (Fig. S1†). OH-functionalized NHCs were anchored on the supported Pt particles according to a recently published protocol.23 No detectable changes in the particles' morphology were observed following the attachment of NHCs on their surface (Fig. S1†).IR nanospectroscopy measurements were performed to identify the NHCs' reactivity on Pt particles following exposure of the sample to gas-phase (1 atm O2, 50 °C, 10 h) and liquid-phase (0.1% w/w H2O2, rt, 10 h) oxidizing conditions. No additional chemical changes were obtained in the properties of surface-anchored NHCs while conducting longer-duration (beyond 10 hours) exposure time, ensuring that the surface anchored molecules reached their local chemical equilibrium.
AFM measurements mapped the topography of NHC-coated Pt particles (left panels, Fig. 2). Subsequently, single point SINS measurements were conducted at the center of the particles, yielding the corresponding IR spectra (right panels, Fig. 2). The location of each SINS measurement is marked by a colored circle in the AFM image.
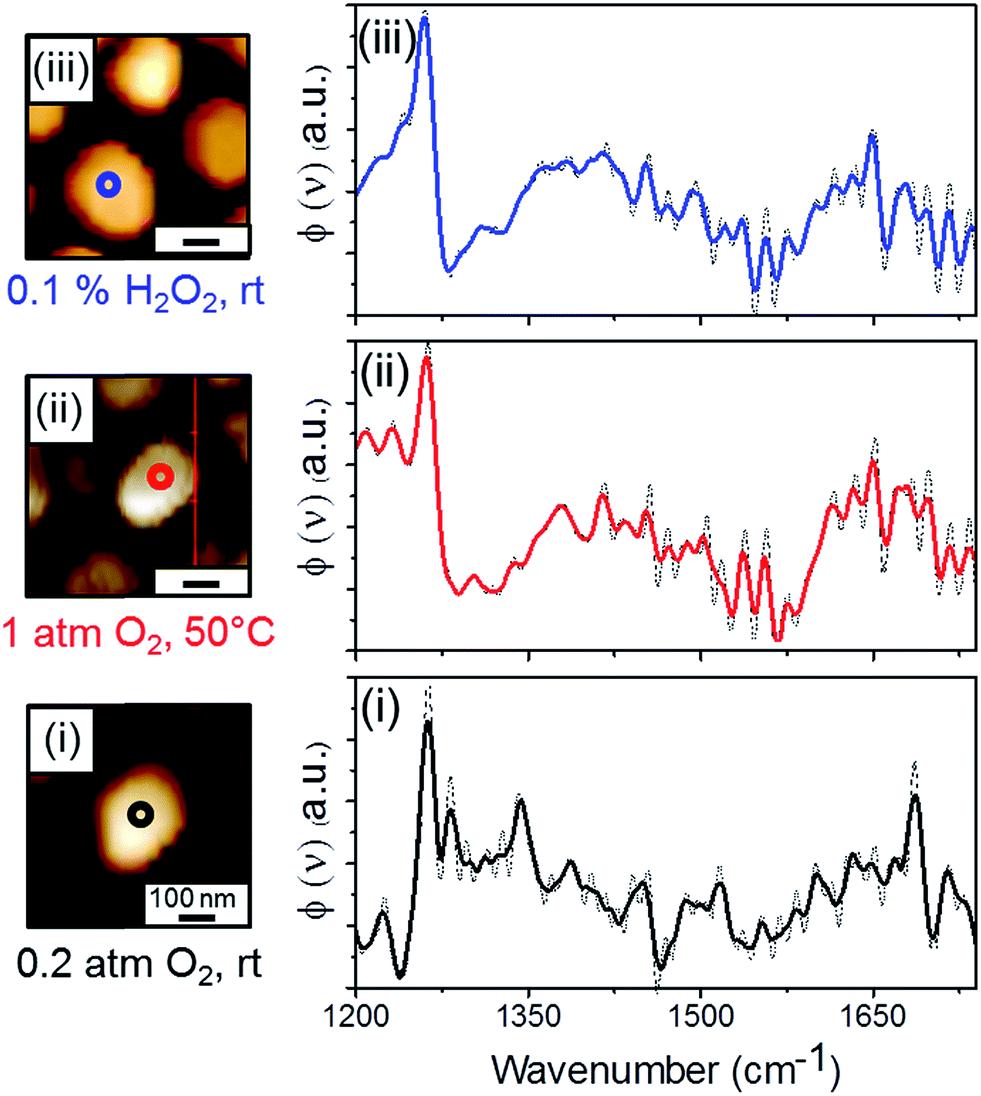 | ||
| Fig. 2 AFM topography images (left panels) and single point SINS measurements (right panels) of Si-supported Pt particles that were coated with OH-functionalized NHCs. IR nanospectroscopy measurements were conducted at the center of single Pt particles (the position of each IR measurement is marked by colored circles) after NHCs' deposition (i) and following exposure of the sample to gas phase (1 atm O2, 50 °C, 10 h) (ii) and liquid phase (0.1 w/w% H2O2, rt, 10 h) (iii) oxidizing conditions. The dash and color lines represent the as-received and smoothed spectra, respectively. | ||
SINS measurement was performed following exposure of the sample to air, with the AFM tip located above the center of the NHC-coated Pt particle (marked with a black circle in Fig. 2, AFM image (i)). An infrared spectrum with few vibrational peaks was obtained (Fig. 2, spectrum (i)). The main peak at 1250 cm−1 was assigned to C–OH vibration. A minor peak was detected at ∼1700 cm−1 that can be correlated to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch. However, its relatively small weight compared to the dominant C–OH vibration indicates that the majority of the NHCs' hydroxyl groups were not oxidized after their exposure to air.
O stretch. However, its relatively small weight compared to the dominant C–OH vibration indicates that the majority of the NHCs' hydroxyl groups were not oxidized after their exposure to air.
The infrared spectrum collected after exposure of the sample to gas phase oxidizing conditions (measurement position is marked with a red circle in Fig. 2, AFM image (ii)) exhibited a peak at 1250 cm−1 and a broad contribution at 1600–1700 cm−1, with integrated intensity larger than that region in spectrum (i) (Fig. 2, spectrum (ii)). The detection of an IR contribution at ∼1650 cm−1 and the fact that the intensity of the C–OH vibration peak was unchanged after exposure of the sample to gas phase oxidizing conditions indicate that the OH-functionalized molecules that reacted under these conditions were oxidized into acid, with no detectable formation of aldehyde. The higher noise level and the features at 1400–1600 cm−1 can be related to atmospheric water lines.
The NHC-coated Pt particles were also exposed to liquid phase oxidizing conditions, where H2O2 was used as an oxidizing agent (0.1% w/w H2O2, rt, 10 h). Single point SINS measurement was performed at the center of a single Pt particle (measurement position is marked with a blue circle in Fig. 2, AFM image (iii)). Two main peaks, at 1250 cm−1 and at 1600–1700 cm−1, were obtained in the SINS spectrum and assigned to C–OH and CO vibrations, respectively (Fig. 2, spectrum (iii)).
Interestingly, although H2O2 is a much more reactive oxidizing agent than O2, the acquired SINS spectrum was similar to the one obtained after exposure of the sample to gas phase oxidizing conditions. The high similarity of the two SINS spectra (Fig. 2, spectrum (ii) and (iii)) reveals that exposure of the sample to liquid phase oxidizing conditions did not noticeably change the NHCs' chemical properties or induced NHCs' deformation, in comparison to the results obtained following exposure of the sample to gas phase oxidizing conditions.
Exposure of the sample to reducing conditions (1 atm H2, 80 °C, 10 h), after its exposure to liquid or gas phase oxidizing conditions, led to a considerable decrease in the CO stretch regions, indicating that the oxidized functional groups of the NHCs can be reduced back to hydroxyl (Fig. S2†).23
One of the advantages in conducting high spatial resolution IR nanospectroscopy measurements is the ability to identify the influence of different surface sites on the reactivity. Herein, site-dependent reactivity was identified by comparing SINS spectra that were measured on the center and the periphery of a single Pt particle, following exposure of the sample to various oxidizing conditions. Differences in the reactivity on various surface sites were identified by subtracting the spectrum that was measured at the particle's perimeter (φ(ν)side) from the one measured at the particle's center (φ(ν)center) (Fig. 3). An example for the acquired SINS spectra at the side and center of the same particle is shown in Fig. S3.†
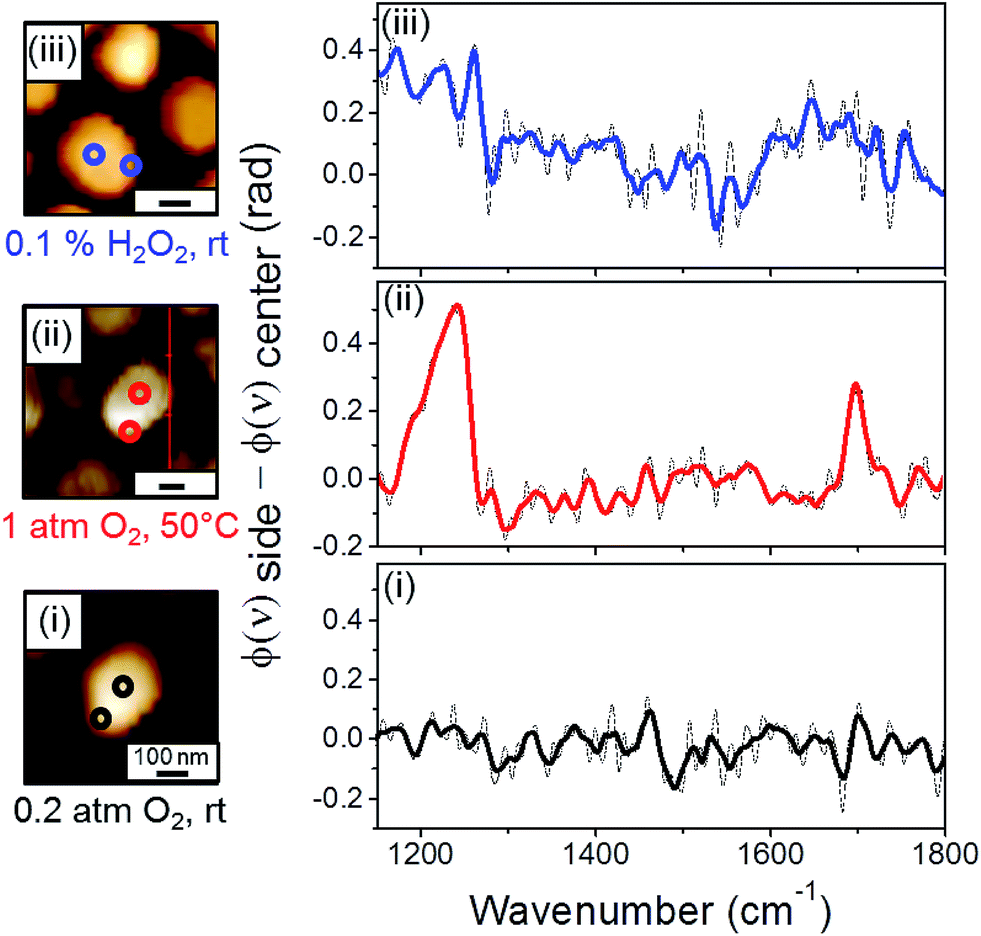 | ||
| Fig. 3 Infrared nanospectroscopy point measurements were conducted on the edge and center of single NHC-coated Pt particles. The location of IR measurements is marked by colored circles in the AFM images (left panels). The differences in near-field spectra that were acquired on the side (φ(ν)side) and center (φ(ν)center) of each particle are reported (right panels). IR nanospectroscopy measurements were conducted following NHCs' deposition (i) and after exposure of the sample to gas phase (1 atm of O2, 50 °C, 10 h) (ii) and liquid phase (0.1% w/w H2O2, rt, 10 h) (iii) oxidizing conditions. The dash and color lines represent the as-received and smoothed spectra, respectively. | ||
SINS difference (φ(ν)side − φ(ν)center) spectrum of NHC-coated Pt particles that were exposed to air did not show any noticeable signal (Fig. 3, spectrum (i)). Thus, almost no difference was detected between the chemical properties of NHCs that were anchored to the side and center of a Pt particle. This result indicates that under ambient conditions different surface sites do not induce detectable changes in the reactivity. Moreover, the similarity in the IR nanospectroscopy signals on various surface sites demonstrates that the local spectroscopic signal is not influenced by the position of the near-field measurement.
However, once the sample was exposed to gas phase oxidizing conditions (1 atm O2, 50 °C, 10 h), clear features were obtained in the difference spectrum (Fig. 3, spectrum (ii)). Two peaks at 1200 and 1700 cm−1 were detected in the difference spectrum and were correlated to C–OH and CO vibrations. This result reveals that exposure of the sample to mild gas phase oxidizing conditions led to preferential oxidation of the hydroxyl functional groups in NHCs that were anchored on the particle's edge. It is hypothesized that the reason for the enhanced reactivity across the periphery of the particle is the high density of low-coordinated metal atoms at the particle's edge.40 Cross section and profile analysis of the supported Pt particles was performed (ESI Fig. S4†). Based on analysis of several probed particles it was identified that the height of the particles was 25 ± 5 nm, while their profile length was 20 ± 5 nm. Such profile length within non-crystalline particles will induce corrugated edges with high density of low-coordinated surface atoms.
As previously demonstrated,41 a relatively small increase in the density of low-coordinated surface atoms can induce a major enhancement in the catalytic reactivity. Thus, a local increase in the density of defects on the periphery of the particle, in comparison to their density on the center of the particle, can lead to the enhanced reactivity on these surface sites, which was detected by conducting site-dependent IR nanospectroscopy measurements. In addition, it cannot be excluded that the edges of the particle expose various low index surface planes that are different and more reactive than those located on the flat center of the particle and induce the observed enhancement of the catalytic reactivity on the particle's periphery.
Site-dependent reactivity was also observed after exposure of the sample to liquid phase oxidizing conditions (0.1% w/w H2O2, rt, 10 h) (Fig. 3, spectrum (iii)). Interestingly, the obtained difference spectrum displayed a lineshape distinct from that detected after gas-phase oxidation: the CO contribution was broader and shifted to lower energy (1570–1720 cm−1), while the sharp C–OH peak was smaller and shifted to 1250 cm−1. The chemical interpretation for the observed changes of peak energy and intensity will be discussed later.
The side-center difference spectra (Fig. 3) nicely demonstrate that exposure of the sample to gas- or liquid-phase oxidizing conditions had a different influence on the chemical properties of molecules that were anchored on the perimeter of the particle. These chemical differences were not observed while sampling NHCs that were anchored on the center of the particle (Fig. 2, spectrum (ii) and (iii), respectively).
Exposure of the gas phase oxidized sample to harsher reducing conditions (1 atm H2, 80 °C, 10 h) diminished the chemical differences between NHCs that were anchored to the side and center of the particle (Fig. S5†). This result shows that the signals in the difference spectra (Fig. 3) were induced by variations in the chemical properties of surface-anchored NHCs and not by changes in the density or orientation of NHCs on different surface sites.
Following the results obtained by SINS measurements, complementary spatially averaged IRRAS and XPS measurements were conducted to better identify the reasons for the detected changes in the SINS spectrum at different surface sites (Fig. 4). IRRAS spectra of the as-deposited NHCs featured a dominant peak at 1250 cm−1, which was assigned to C–OH vibration (Fig. 4a, spectrum (i)). N1s XP-spectrum of the as-deposited NHCs was measured (Fig. 4b, spectrum (i)) and its peak area was fit by two Gaussians (Fig. S6†) centered at 401.2 and 403.7 eV, which were assigned to N–C and N–Ox/amide species with a spectral weight of 86% and 14% of the total peak area, respectively (Table 1). The detection of a relatively wide (FWHM = 3 eV) N1s XPS peak of surface anchored NHCs was previously reported.25,32,42 C1s XP-spectrum (Fig. 4c, spectrum (i)) was fit by two Gaussians (Fig. S5†), centered at 285.5 and 286.6 eV, which correspond to C–C and C–O/N species with a spectral weight of 44% and 56%, respectively (Table 1).
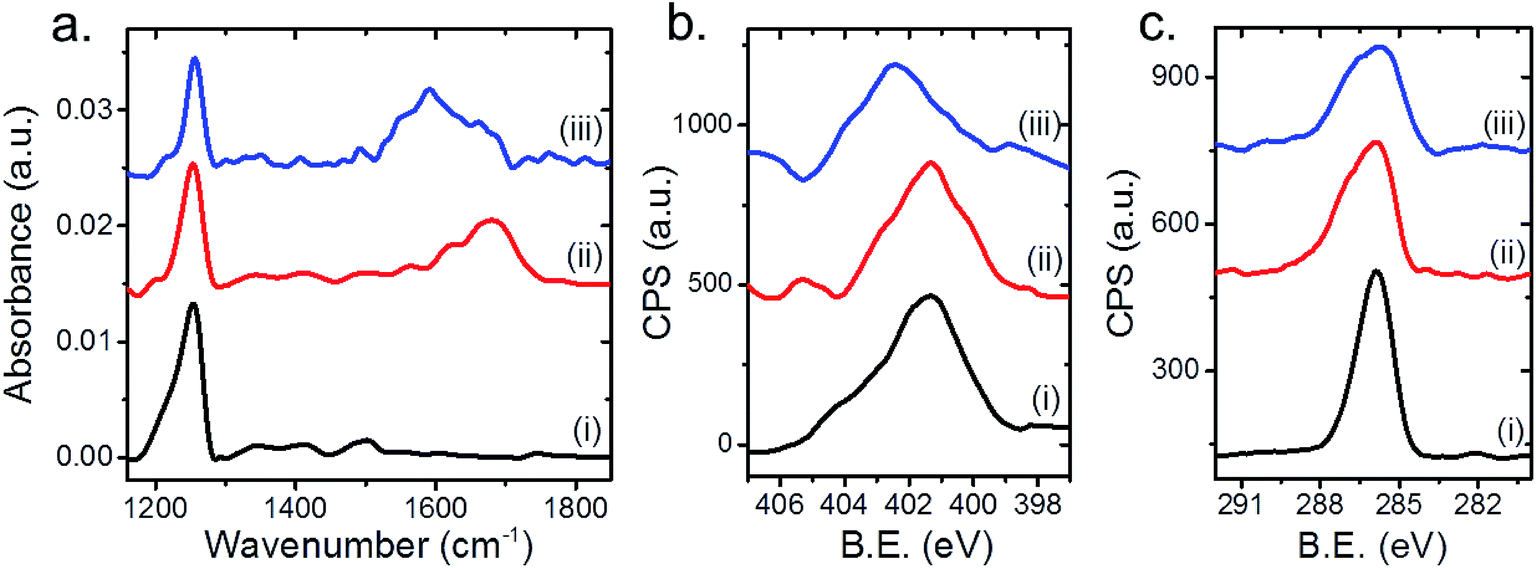 | ||
| Fig. 4 IRRAS (a), N1s XPS (b) and C1s XPS (c) spectra of OH-functionalized NHCs that were anchored to the surface of supported Pt particles. Spectroscopic measurements were conducted before (i) and after exposure of the sample to gas phase (1 atm O2, 80 °C, 10 h) (ii) and liquid phase (0.1% w/w H2O2, rt, 10 h) (iii) oxidizing conditions. | ||
| XPS signal | ||||||
|---|---|---|---|---|---|---|
| Treatment | N1s | C1s | ||||
| NC (398.5 eV) |
N–C (401.2 eV) | Amide/N–Ox (402.7–403.7 eV) | C–C (285.5 eV) | C–O/N (286.6 eV) | COOH (288.4 eV) | |
| Air, rt | — | 86% | 14% | 44% | 56% | — |
| Gas phase oxidation | 5% | 83% | 12% | 32% | 60% | 8% |
| Liquid phase oxidation | 10% | 30% | 60% | 33% | 60% | 7% |
The lack of IR peaks in the CO region and the absence of a COOH feature in the C1s XPS peak indicate that under ambient conditions most of the surface-anchored NHCs were OH-terminated and were not oxidized. The detection of a minor CO vibration signal (∼1700 cm−1) in the IR nanospectroscopy measurement (Fig. 2, spectrum (i)), which was not observed in the IRRAS measurement (Fig. 4a, spectrum (i)), can be correlated to the enhanced sensitivity of IR nanospectroscopy measurements to dipoles that are positioned vertical to the surface, thus making it feasible to detect the contribution of this minor species by near-field IR nanospectroscopy.
The detection of a small fraction (<15%) of high binding energy nitrogen species on the surface, correlated to N–Ox or amide, can be induced by the strong interaction between the NHCs and the Pt surface which led to partial deformation of the surface anchored NHCs.
NEXAFS measurements at the carbon K-edge were performed to identify the geometry of surface anchored NHCs. By changing the orientation of the probed surface with respect to the linearly polarized photon beam, both p- and s-polarized NEXAFS spectra (marked in Fig. 5 with solid and dotted lines, respectively) were acquired. The main features in the p- and s-polarized NEXAFS spectra of the as-deposited OH–NHCs (Fig. 5, spectra (i), solid and dotted lines, respectively) were associated to the C1s → 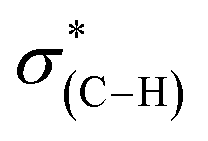 (287.5 eV) and to
(287.5 eV) and to 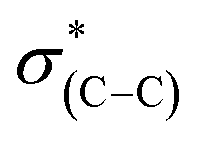 or/and
or/and 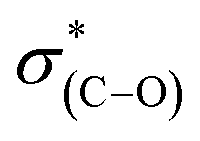 transitions (at 289–293 eV).43–45 In addition, a peak was detected in the s-polarized NEXAFS spectrum at 285.2 eV, which was not detected in the p-polarized spectrum and was assigned to
transitions (at 289–293 eV).43–45 In addition, a peak was detected in the s-polarized NEXAFS spectrum at 285.2 eV, which was not detected in the p-polarized spectrum and was assigned to 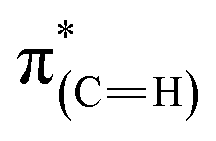 transitions.46,47 This dichroism implies that the imidazoline rings of the as-deposited NHCs display a preferential orientation perpendicular to the surface.
transitions.46,47 This dichroism implies that the imidazoline rings of the as-deposited NHCs display a preferential orientation perpendicular to the surface.
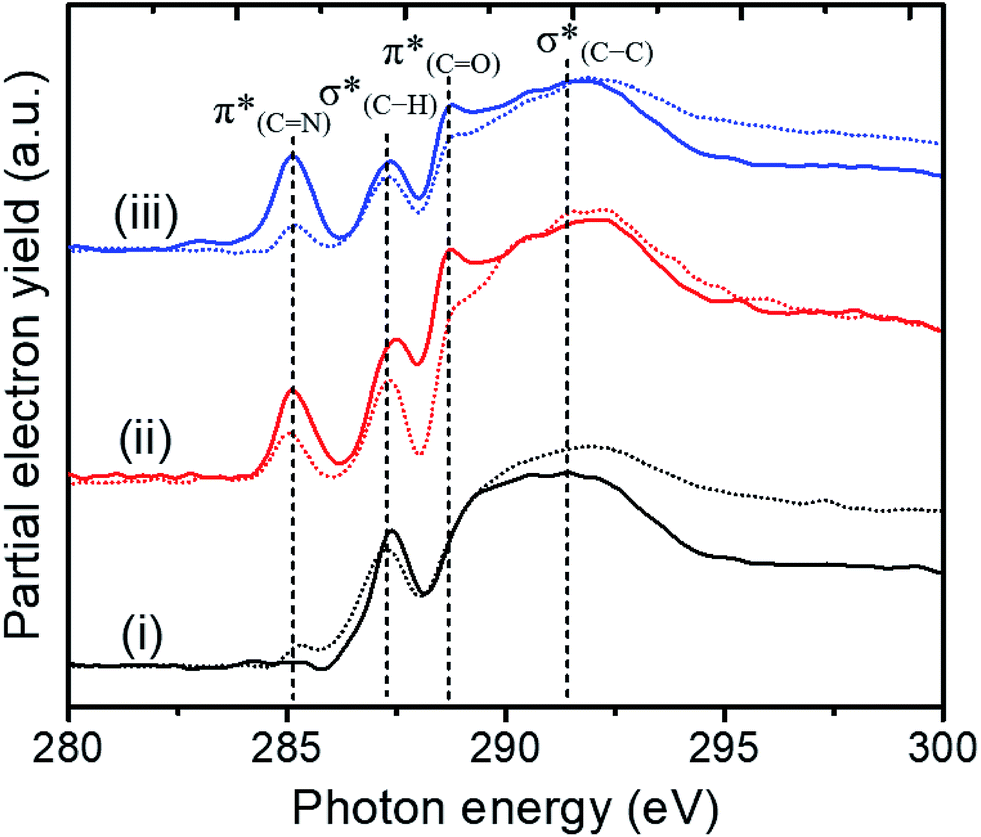 | ||
| Fig. 5 Carbon K-edge NEXAFS spectra of OH-functionalized NHCs that were anchored to the surface of Si-supported Pt particles. p- and s-polarized NEXAFS spectra (marked by solid and dotted lines, respectively) were acquired by changing the orientation of the probed surface with respect to the X-ray beam. NEXAFS measurements were performed on the as-deposited NHCs (i) and after exposure of the sample to gas phase (1 atm O2, 80 °C, 10 h) (ii) and liquid phase (0.1% w/w H2O2, rt, 10 h) (iii) oxidizing conditions. | ||
Exposure of the sample to gas-phase oxidizing conditions (1 atm O2, 80 °C, 10 h) induced the formation of a new IR peak at 1600–1730 cm−1 (Fig. 4a, spectrum (ii)). The detected IRRAS spectrum is complementary to the measured SINS spectrum (Fig. 3, spectrum (ii)) and indicates that exposure of the sample to gas phase oxidizing conditions induced a dominant IR absorption peak in the CO region.
The oxidation of the NHCs' hydroxyl groups was also identified in the C1s XP-spectrum (Fig. 4c, spectrum (ii)). The C1s XP spectrum was fit to three Gaussians, centered at 285.5 and 286.5 and 288.4 eV, corresponding to C–C, C–O/N and COOH species with spectral weight of 32%, 60% and 8%, respectively (Fig. S6†). The detection of a higher binding energy species in the C1s XP-spectra indicated that oxidation of the NHCs' OH-functional groups has occurred following exposure to gas phase oxidizing conditions.
NEXAFS measurements revealed that exposure of the sample to gas phase oxidizing conditions led to the development of a new peak in the p-polarized spectrum at 288.8 eV (Fig. 5, spectra (ii), solid line), which was associated to a 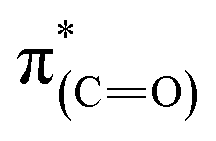 transition.48 The appearance of the
transition.48 The appearance of the 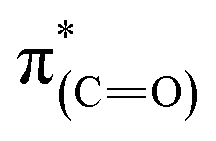 resonance and the decrease of the
resonance and the decrease of the 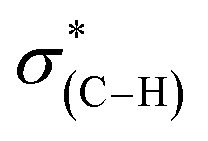 one in the NEXAFS spectra indicates that the NHCs' hydroxyl functional groups were oxidized following exposure of the sample to gas-phase oxidizing conditions. Thus, the analysis of the X-ray spectroscopy shows that exposure of the sample to gas phase oxidizing conditions induced oxidation of the NHCs' hydroxyl functional groups into a carboxylic acid. These results nicely correlate to SINS measurements, which identified enhanced hydroxyl oxidation rate on the periphery of the particles (Fig. 3, spectrum (ii)).
one in the NEXAFS spectra indicates that the NHCs' hydroxyl functional groups were oxidized following exposure of the sample to gas-phase oxidizing conditions. Thus, the analysis of the X-ray spectroscopy shows that exposure of the sample to gas phase oxidizing conditions induced oxidation of the NHCs' hydroxyl functional groups into a carboxylic acid. These results nicely correlate to SINS measurements, which identified enhanced hydroxyl oxidation rate on the periphery of the particles (Fig. 3, spectrum (ii)).
The N1s XP-spectrum (Fig. 4b, spectrum (ii)) included a tail toward lower binding energy, which was not observed in the as-deposited NHCs (Fig. 4b, spectrum (i)). The latter feature was fitted by an additional Gaussian component centered at 398.5 eV (weight of 5%) and was associated to CN species. In addition, the center of the high binding energy component in the N1s XP-spectrum shifted to lower energy (402.7 eV), in comparison to the location of this component in the as-deposited sample (Fig. S6†).
Exposure of the sample to oxidizing conditions also led to a noticeable increase in the intensity of the 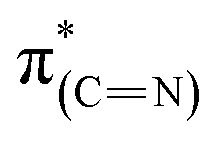 resonance at 285.2 eV. Transitions in this region are usually attributed to aromatic rings.49 We therefore conclude that exposure of the sample to oxidizing conditions facilitated oxidative dehydrogenation reaction of the imidazoline ring, resulting in increased unsaturation and aromaticity of the molecule.50 The increased aromaticity of the sample can potentially induce the observed shift in the position of the high binding energy fraction of the N1s spectra to lower energy values.
resonance at 285.2 eV. Transitions in this region are usually attributed to aromatic rings.49 We therefore conclude that exposure of the sample to oxidizing conditions facilitated oxidative dehydrogenation reaction of the imidazoline ring, resulting in increased unsaturation and aromaticity of the molecule.50 The increased aromaticity of the sample can potentially induce the observed shift in the position of the high binding energy fraction of the N1s spectra to lower energy values.
Comparison of the p- and s-polarized NEXAFS spectra (Fig. 5, spectra (ii), solid and dotted lines, respectively) reveals that both 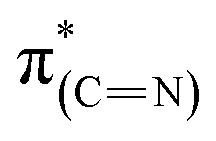 and
and 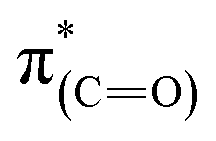 resonances (285.2 and 288.8 eV, respectively) are more intense in the p-polarized NEXAFS spectrum. This dichroism implies that after exposure of the sample to gas-phase oxidizing conditions, NHCs display a preferential orientation parallel to the Pt surface.51
resonances (285.2 and 288.8 eV, respectively) are more intense in the p-polarized NEXAFS spectrum. This dichroism implies that after exposure of the sample to gas-phase oxidizing conditions, NHCs display a preferential orientation parallel to the Pt surface.51
It should be noted that spatially averaged IRRAS, XPS and NEXAFS measurements were performed after exposure of the sample to gas-phase oxidizing conditions (1 atm O2, 80 °C, 10 h) which were harsher than those adopted prior to SINS measurement (1 atm O2, 50 °C, 10 h). Exposure of the sample to harsher oxidizing conditions ensured that most of the surface anchored NHCs were oxidized into acid.23 Thus, the spatially averaged spectroscopic data can be directly compared to SINS measurements performed at the nanoparticle's periphery under milder oxidizing conditions.
Exposure of the sample to liquid phase oxidizing conditions induced a broadening of the IRRAS peak at high frequency (1520–1700 cm−1) and a shift of the N1s XPS peak to higher binding energy (spectrum (iii) in Fig. 4a and b, respectively). The N1s XPS peak was fit to three Gaussians, centered at 398.5, 401.2 and 402.7 eV, which were associated to NC, N–C and N–Ox/amide species with a weight of 10%, 30% and 60%, respectively. The corresponding C1s XPS peak was fit to three Gaussians, centered at 285.5, 286.6 and 288.4 eV, which were associated to C–C, C–O/N and COOH species with a weight of 33%, 60% and 7%, respectively (Fig. 4c, curve (iii)). The C1s XP-spectrum from the sample exposed to liquid phase oxidizing conditions provides results qualitatively similar to those obtained after exposure of the sample to gas phase oxidizing conditions, however the total area of the former peak decreased by ∼15% in comparison to the one measured after gas phase reaction conditions.
The shift to higher energy observed in the N1s XPS peak and that to lower energy of the high frequency IR peak of the liquid phase oxidized sample is consistent with decarboxylation followed by oxidation of the NHCs. The decarboxylation results in the observed decrease in the C1s XPS peak area (spectrum (iii) in Fig. 4c) and the overall decrease of the  (287.5 eV) and
(287.5 eV) and 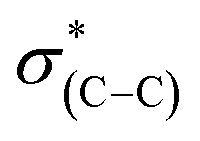 (at 289–293 eV) resonances in the NEXAFS spectrum (Fig. 5, spectrum (iii)).
(at 289–293 eV) resonances in the NEXAFS spectrum (Fig. 5, spectrum (iii)).
Analysis of the N1s to Pt4f peaks area ratio (Fig. S7†) revealed that exposure of the sample to either gas or liquid phase oxidizing conditions induced relatively small changes (<10%) in the N1s/Pt4f peaks ratio. This analysis verifies that exposure of the sample to different oxidizing conditions did not lead to a noticeable detachment of the NHCs' imidazoline ring from the Pt surface, in agreement with a previous report.32
Exposure of the sample to liquid phase oxidizing conditions led to an overall increase of the 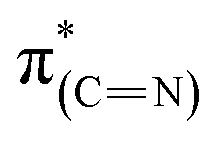 resonance. This result can be attributed to an increase in the aromatization of the imidazole ring resulting from oxidative dehydrogenation of NHC imidazoline. This hypothesis is supported by the detection of a higher fraction of CN species in the N1s XPS peak (Table 1).52–54 The dichroism of the NEXAFS spectra, in particular of the
resonance. This result can be attributed to an increase in the aromatization of the imidazole ring resulting from oxidative dehydrogenation of NHC imidazoline. This hypothesis is supported by the detection of a higher fraction of CN species in the N1s XPS peak (Table 1).52–54 The dichroism of the NEXAFS spectra, in particular of the 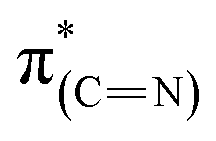 and
and 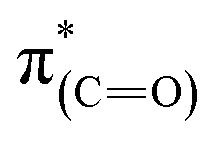 resonances, was increased upon exposure of the sample to liquid phase oxidizing conditions (Fig. 5, spectrum (iii), solid and dotted lines, respectively). This large dichroism indicates that exposure of the sample to liquid phase oxidizing conditions favored a tilting of the oxidized NHCs' imidazole rings to an orientation preferentially parallel to the Pt surface.
resonances, was increased upon exposure of the sample to liquid phase oxidizing conditions (Fig. 5, spectrum (iii), solid and dotted lines, respectively). This large dichroism indicates that exposure of the sample to liquid phase oxidizing conditions favored a tilting of the oxidized NHCs' imidazole rings to an orientation preferentially parallel to the Pt surface.
From the analysis of the spectroscopic data, we conclude that exposure of the sample to liquid phase oxidizing conditions induced two main chemical changes: (1) decarboxylation and oxidation of the NHCs' functional group, which preferentially occurs on the nanoparticles' periphery. (2) Enhanced oxidative dehydrogenation reaction that increased the aromaticity of the molecule. These two effects were coupled to a change of the adsorption geometry corresponding to a change of the preferential orientation of the oxidized NHCs' imidazole ring and of the carbonyl group from perpendicular to parallel to the Pt surface.
Conclusions
In this work surface anchored NHCs as chemical indicators were employed to interrogate the site-dependent reactivity of oxidation reactions on single Pt particles. Thus, the integration of spatially localized and averaged measurements provided a detailed analysis of the influence of reaction conditions on the site-dependent reactivity and on the geometry of NHCs on Pt particles. More specifically, integration of high spatial resolution SINS measurements and spatially averaged XPS, NEXAFS and IRRAS measurements provide evidence that, under gas phase oxidizing conditions, the hydroxyl groups of surface-anchored NHCs at the periphery of Pt particles showed enhanced reactivity relative to those located at the center, thus leading to the formation of carboxylic acid substituted NHCs. Importantly, this enhancement in reactivity was detected at NHCs located at the periphery of the particles; however, this differential reactivity was highly dependent on the reaction conditions. On the other hand, no differences were observed in the chemical properties of NHCs that were anchored on the center of the Pt particles and were exposed to either gas or liquid phase oxidizing conditions. Exposure of the sample to harsher, liquid phase oxidizing conditions, using H2O2 as oxidant, resulted in modification of the NHCs located at the periphery of the nanoparticles. Moreover, exposure of the sample to either gas or liquid phase oxidizing conditions induced oxidative dehydrogenation reaction of the imidazoline ring that led to a favorable preferential orientation of the now oxidized NHC imidazole rings parallel to the surface. The dehydrogenation reaction was not observed in the hydroxyl functionalized NHCs prior to their exposure to oxidizing conditions. While NHC-substituted surfaces are gaining prominence in catalysis, this observation highlights that in some cases, these ligands may be reactive under reaction condition. Finally, this work highlights that the observation of differential reactivity at sites in heterogeneous catalysts should be examined and expressed in, and maybe limited to, the context of given reaction conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Grant No. 2016344 from the United States-Israel Binational Science Foundation (BSF). S. D. acknowledges the Israeli Ministry of Energy and the Rudin fellowship for their financial support. F. D. T. thanks the Director, Office of Science, Office of Basic Energy Sciences and the Division of Chemical Sciences, Geosciences, and Biosciences of the US Department of Energy at LBNL (DE-AC02-05CH11231) for partial support of this work. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract number DE-AC02-05CH11231. We acknowledge the assistance of Dr Hans Bechtel (Advanced Light Source, Lawrence Berkeley National Lab) in conducting the SINS experiments.References
- G. A. Somorjai and Y. Li, Introduction to surface chemistry and catalysis, John Wiley & Sons, 2010 Search PubMed.
- H. J. Freund, Chemistry, 2010, 16, 9384–9397 CrossRef PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef PubMed.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of modern catalysis and kinetics, Wiley-VCH, Weinheim, 2nd edn, 2007 Search PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Glaser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef PubMed.
- L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212–9228 CrossRef PubMed.
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef PubMed.
- W. Wang and N. J. Tao, Anal. Chem., 2014, 86, 2–14 CrossRef PubMed.
- W. L. Xu, J. S. Kong, Y. T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef PubMed.
- M. A. Bañares, Catal. Today, 2005, 100, 71–77 CrossRef.
- E. Stavitski and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4615–4625 RSC.
- E. Gross, X. Z. Shu, S. Alayoglu, H. A. Bechtel, M. C. Martin, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 3624–3629 CrossRef PubMed.
- Y. Levartovsky and E. Gross, Top. Catal., 2016, 59, 1700–1711 CrossRef.
- E. Gross, Surf. Sci., 2016, 648, 136–140 CrossRef.
- R. W. Johns, H. A. Bechtel, E. L. Runnerstrom, A. Agrawal, S. D. Lounis and D. J. Milliron, Nat. Commun., 2016, 7, 11583 CrossRef PubMed.
- F. Huth, A. Govyadinov, S. Amarie, W. Nuansing, F. Keilmann and R. Hillenbrand, Nano Lett., 2012, 12, 3973–3978 CrossRef PubMed.
- N. Kumar, B. Stephanidis, R. Zenobi, A. J. Wain and D. Roy, Nanoscale, 2015, 7, 7133–7137 RSC.
- H. A. Bechtel, E. A. Muller, R. L. Olmon, M. C. Martin and M. B. Raschke, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7191–7196 CrossRef PubMed.
- A. B. Zrimsek, N. H. Chiang, M. Mattei, S. Zaleski, M. O. McAnally, C. T. Chapman, A. I. Henry, G. C. Schatz and R. P. Van Duyne, Chem. Rev., 2017, 117, 7583–7613 CrossRef PubMed.
- E. M. van Schrojenstein Lantman, T. Deckert-Gaudig, A. J. G. Mank, V. Deckert and B. M. Weckhuysen, Nat. Nanotechnol., 2012, 7, 583–586 CrossRef PubMed.
- J. H. Zhong, X. Jin, L. Y. Meng, X. Wang, H. S. Su, Z. L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136 CrossRef PubMed.
- C. Y. Wu, W. J. Wolf, Y. Levartovsky, H. A. Bechtel, M. C. Martin, F. D. Toste and E. Gross, Nature, 2017, 541, 511–515 CrossRef PubMed.
- D. L. Fu, K. Park, G. Delen, O. Attila, F. Meirer, D. Nowak, S. Park, J. E. Schmidt and B. M. Weckhuysen, Chem. Commun., 2017, 53, 13012–13014 RSC.
- Y. Levratovsky and E. Gross, Faraday Discuss., 2016, 188, 345–353 RSC.
- W. Wang, Chem. Soc. Rev., 2018, 47, 2485–2508 RSC.
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef PubMed.
- H. Wang, A. Sapi, C. M. Thompson, F. Liu, D. Zherebetskyy, J. M. Krier, L. M. Carl, X. Cai, L.-W. Wang and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 10515–10520 CrossRef PubMed.
- Z. Ristanovic, A. V. Kubarev, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2016, 138, 13586–13596 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef PubMed.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H. B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef PubMed.
- A. V. Zhukhovitskiy, M. G. Mavros, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418–7421 CrossRef PubMed.
- A. Ruhling, K. Schaepe, L. Rakers, B. Vonhoren, P. Tegeder, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5856–5860 CrossRef PubMed.
- J. B. Ernst, C. Schwermann, G. I. Yokota, M. Tada, S. Muratsugu, N. L. Doltsinis and F. Glorius, J. Am. Chem. Soc., 2017, 139, 9144–9147 CrossRef PubMed.
- C. R. Larrea, C. J. Baddeley, M. R. Narouz, N. J. Mosey, J. H. Horton and C. M. Crudden, ChemPhysChem, 2017, 18, 3536–3539 CrossRef PubMed.
- G. Q. Wang, A. Ruhling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H. J. Gao, A. Timmer, H. Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2017, 9, 152–156 CrossRef PubMed.
- L. Jiang, B. D. Zhang, G. Medard, A. P. Seitsonen, F. Haag, F. Allegretti, J. Reichert, B. Kuster, J. V. Barth and A. C. Papageorgiou, Chem. Sci., 2017, 8, 8301–8308 RSC.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Norskov, J. Catal., 2004, 223, 232–235 CrossRef.
- G. A. Somorjai and D. W. Blakely, Nature, 1975, 258, 580–583 CrossRef.
- J. B. Ernst, S. Muratsugu, F. Wang, M. Tada and F. Glorius, J. Am. Chem. Soc., 2016, 138, 10718–10721 CrossRef PubMed.
- Y. H. La, Y. J. Jung, T. H. Kang, K. Ihm, K. J. Kim, B. Kim and J. W. Park, Langmuir, 2003, 19, 9984–9987 CrossRef.
- D. A. Outka, J. Stohr, J. P. Rabe and J. D. Swalen, J. Chem. Phys., 1988, 88, 4076–4087 CrossRef.
- M. Beccari, A. Kanjilal, M. G. Betti, C. Mariani, L. Floreano, A. Cossaro and V. Di Castro, J. Electron Spectrosc. Relat. Phenom., 2009, 172, 64–68 CrossRef.
- A. L. Johnson, E. L. Muetterties, J. Stohr and F. Sette, J. Phys. Chem., 1985, 89, 4071–4075 CrossRef.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef.
- A. Cossaro, S. Terreni, O. Cavalleri, M. Prato, D. Cvetko, A. Morgante, L. Floreano and M. Canepa, Langmuir, 2006, 22, 11193–11198 CrossRef PubMed.
- K. G. Latham, M. I. Simone, W. M. Dose, J. A. Allen and S. W. Donne, Carbon, 2017, 114, 566–578 CrossRef.
- D. W. Blakely and G. A. Somorjai, J. Catal., 1976, 42, 181–196 CrossRef.
- U. Siemeling, H. Memczak, C. Bruhn, F. Vogel, F. Trager, J. E. Baio and T. Weidner, Dalton Trans., 2012, 41, 2986–2994 RSC.
- R. J. J. Jansen and H. Vanbekkum, Carbon, 1995, 33, 1021–1027 CrossRef.
- R. V. Duevel and R. M. Corn, Anal. Chem., 1992, 64, 337–342 CrossRef.
- B. L. Frey and R. M. Corn, Anal. Chem., 1996, 68, 3187–3193 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01956h |
| This journal is © The Royal Society of Chemistry 2018 |