
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming conservation in TALE–DNA interactions: a minimal repeat scaffold enables selective recognition of an oxidized 5-methylcytosine†
Sara
Maurer
,
Benjamin
Buchmuller
 ,
Christiane
Ehrt
,
Julia
Jasper
,
Oliver
Koch
and
Daniel
Summerer
*
,
Christiane
Ehrt
,
Julia
Jasper
,
Oliver
Koch
and
Daniel
Summerer
*
Faculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Str. 4a, 44227 Dortmund, Germany. E-mail: Daniel.summerer@tu-dortmund.de
First published on 2nd August 2018
Abstract
Transcription-activator-like effectors (TALEs) are repeat-based proteins featuring programmable DNA binding. The repulsion of TALE repeats by 5-methylcytosine (5mC) and its oxidized forms makes TALEs potential probes for their programmable analysis. However, this potential has been limited by the inability to engineer repeats capable of actual, fully selective binding of an (oxidized) 5mC: the extremely conserved and simple nucleobase recognition mode of TALE repeats and their extensive involvement in inter-repeat interactions that stabilize the TALE fold represent major engineering hurdles. We evaluated libraries of alternative, strongly truncated repeat scaffolds and discovered a repeat that selectively recognizes 5-carboxylcytosine (5caC), enabling construction of the first programmable receptors for an oxidized 5mC. In computational studies, this unusual scaffold executes a dual function via a critical arginine that provides inter-repeat stabilization and selectively interacts with the 5caC carboxyl group via a salt-bridge. These findings argue for an unexpected adaptability of TALE repeats and provide a new impulse for the design of programmable probes for nucleobases beyond A, G, T and C.
5-Methylcytosine (5mC, Fig. 1a) is a dynamic epigenetic nucleobase with key roles in mammalian development and disease.1 Ten-eleven translocation (TET) dioxygenases can oxidize 5mC to the nucleobases 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC, Fig. 1a), which are intermediates of active DNA demethylation.2–7 Oxidized 5mCs further exhibit unique genomic levels and distributions,1 uniquely interact with important nuclear proteins,8–12 may influence nucleosome flexibility,13 and 5fC can form lysine-mediated Schiff-base crosslinks with proteins in vitro.14,15
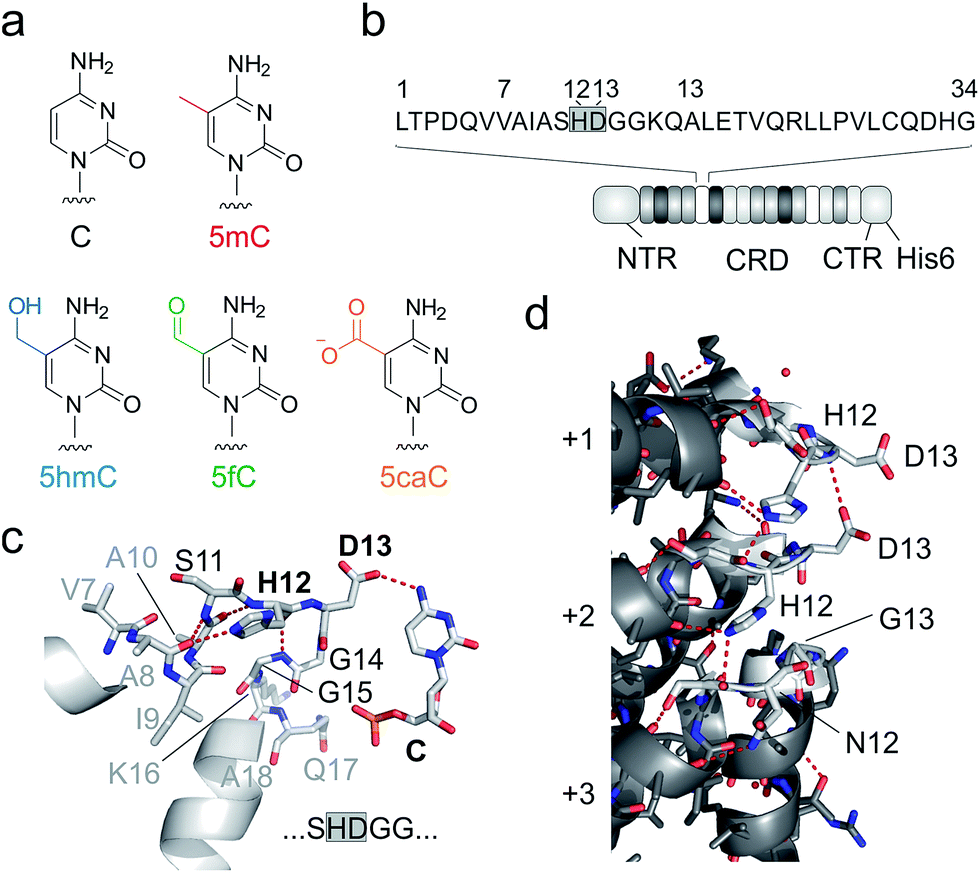 | ||
| Fig. 1 DNA recognition by TALEs. (a) Chemical structures of human cytosine nucleobases. (b) Features of used TALE constructs. Sequence of one TALE repeat on top with RVD in grey box. NTR: N-terminal region (including an N-terminal GFP domain), CRD: central repeat domain, CTR: C-terminal region. (c) Crystal structure of TALE repeat with RVD HD bound to C.28,29 Hydrogen bonds are shown as dotted red lines. (d) Inter-repeat interactions in a crystal structure of DNA-unbound TALE (pdb entry 3V6P).28 for three exemplary CRD repeats (+1–+3). Residues 11–15 of each repeat targeted in this study for deletion or randomization are shown as white sticks, others in grey. Hydrogen bonds are shown as dotted red lines. | ||
Unlike canonical nucleobases that can be targeted and analysed by programmable hybridization probes in a wide range of applications, 5mC and its oxidized derivatives are “transparent” to such probes, since they do not selectively influence Watson–Crick hybridization. Instead, these nucleobases are analysed by utilizing their unique chemical reactivities, e.g. in modified bisulfite sequencing protocols and in affinity enrichments via the incorporation of reactive handles.16–21 In addition, receptors and enzymes with selectivity for oxidized 5mC nucleobases are available for analysis,22,23 and single molecule sequencing approaches are under development24,25 (for reviews, see ref. 1 and 26). Transcription-activator-like effector (TALE) proteins from Xanthomonas species27 are candidate scaffolds for the engineering of probes with epigenetic nucleobase selectivity. These proteins feature a fully programmable DNA binding mode occurring via the major groove28,29 that displays chemically unique information for each canonical and epigenetic nucleobase.30 TALEs consist of concatenated repeats (Fig. 1b), each recognizing one nucleobase via a single amino acid of the so-called repeat variable di-residue (RVD, Fig. 1c).28,29 The RVD amino acids NI, NN, NG and HD thereby predictably bind to A, G/A, T, and C, respectively.31
Natural and engineered TALE repeats can be differentially repulsed by 5mC and its oxidized derivatives, offering potential for their detection at single, user-defined genomic positions.32–35 However, besides being “negative”, these selectivities have typically been ambiguous (for more than one nucleobase or not studied in the range of all five cytosines). In fact, the ultimate requirement for using TALEs as probes for selective, programmable targeting and analysis – positive, fully selective recognition of an (oxidized) 5mC nucleobase – has not yet been reported.
Indeed, the engineering potential of Xanthomonas TALE repeats appears poor: they are highly conserved in both sequence36 and structure,28,29 including amino acid side chain conformations (ESI Fig. 1†). RVD loops are involved in inter-repeat interactions that stabilize the overall TALE fold (Fig. 1d). Moreover, the RVD loop is preorganized by a conserved intra-loop hydrogen bond (via N/H12, Fig. 1c shows one example), and its main chain is in close proximity to the pyrimidine 5-position, offering little room for the recognition of 5-substituents.28,29 Only a single amino acid is involved in canonical nucleobase recognition, and only two different interaction modes are known: hydrogen bonding (by the isosteric D and N in RVDs HD and NN) or mere steric accommodation (RVDs NG and NI). Truncated loop designs could add room to enable new side chain-mediated interactions to oxidized 5mC. However, attempts to delete more than one loop amino acid have led to impaired DNA binding, suggesting interference with essential functions such as inter-repeat interactions.33,37
To identify potential starting points for alternative truncated designs, we performed sequence analyses of naturally evolved TALE repeats. Strikingly, of all amino acid sequences with annotated (or predicted) TALE repeat domains deposited in NCBI and UniProt (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 983 alignments in total), we identified 1396 naturally occurring unique TALE repeat sequences of which however only five had deletions in their loop region (see ESI†). Not a single Xanthomonas repeat had more than one deletion in its loop region, corroborating previous studies, including analyses of TALE repeat subsets based on available long read DNA sequencing data.27,36,38 Given this considerable evolutionary conservation of loop length, we wanted to explore the DNA recognition potential of alternative, even further truncated repeat scaffolds experimentally.
983 alignments in total), we identified 1396 naturally occurring unique TALE repeat sequences of which however only five had deletions in their loop region (see ESI†). Not a single Xanthomonas repeat had more than one deletion in its loop region, corroborating previous studies, including analyses of TALE repeat subsets based on available long read DNA sequencing data.27,36,38 Given this considerable evolutionary conservation of loop length, we wanted to explore the DNA recognition potential of alternative, even further truncated repeat scaffolds experimentally.
We constructed five repeat libraries covering 55 different truncated loops by introducing two – four deletions within amino acid positions 11–15. Fig. 2a shows positions in a model of a repeat bearing the small RVD G* opposite 5caC as starting point for truncation.33 In each library, we allowed amino acids with side chains capable of undergoing polar interactions with the 5-substituents of 5hmC, 5fC and 5caC at one random position. We chose position 11 to be S or N and in case of three deletions, we targeted positions 12–14 or 13–15 (Fig. 2b). We assembled39 TALE genes bearing single mutant repeats using vector pGFP-ENTRY,35 allowing for individual expression and purification of each TALE mutant with N-terminal GFP domain, shortened, AvrBs3-type N-terminal region (NTR) and a C-terminal His6 tag in E. coli (Fig. 1b and ESI Fig. 2–4†). We designed TALEs targeting an 18 nt sequence in the zebrafish HEY2 gene with the mutant repeat opposite the C of a single CpG dyad (Fig. 2c). To screen for new nucleobase selectivities, we employed a DNase I competition assay based on a Cy3/Cy5 double-labeled dsDNA oligonucleotide bearing a single C, 5mC, 5hmC, 5fC or 5caC opposite the mutant TALE repeat (Fig. 2d). TALE-binding inhibits DNase I that otherwise catalyzes DNA cleavage and spatial separation of the fragments, leading to decreased Förster resonance energy transfer from Cy3 to Cy5 as read-out.40 Using wild-type (wt) TALE_SHDGG, the assay afforded differential cleavage kinetics for each of the five nucleobases, with C-containing DNA showing the slowest kinetics and high signal/noise (Fig. 2e). This confirmed the selectivity profile of RVD HD and indicated suitability of the assay for the identification of repeats with new nucleobase selectivities.31,40
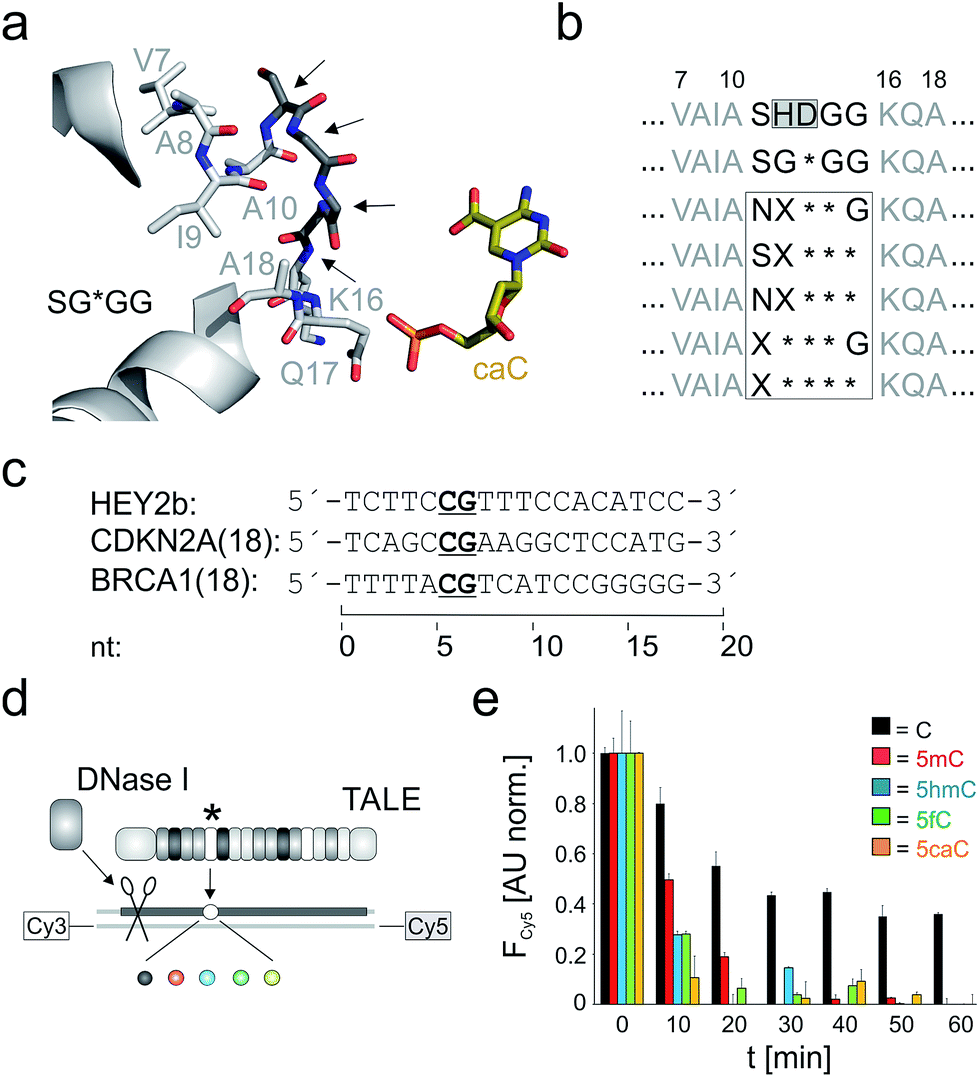 | ||
| Fig. 2 Library design and screening assay for truncated TALE repeat libraries. (a) Positions targeted for deletion or randomization (arrows) in a model of repeat SG*GG opposite 5caC.33 (b) Library designs. Target positions in white box. X: random position, * = deletion. (c) Target sequences of TALEs used in this study. (d) DNase I competition assay using Cy3/Cy5-double labeled DNA oligonucleotides with variable nucleobase (sphere, color-coded as in Fig. 1a) opposite mutant repeat (*). (e) Time course of Cy5 fluorescence from DNase I assay conducted in duplicate with 0.5 μM TALE_SHDGG, 0.1 μM DNA and 1 unit DNase I. Cy5 fluorescence was background-corrected by subtracting a control w/o TALE and normalized first to a control w/o DNase I and then to the reaction with nucleobase C at t = 0. | ||
We conducted screenings in 384-well plate format, covering a total of 275 repeat-nucleobase interactions (Fig. 3a–f). Library NX**G contained the repeats with the highest affinities, with 5mC being a preference of repeats with aromatic amino acids W, H and Y, but also with R, K or S at the random position (Fig. 3b). Other nucleobases were bound less strongly, with W, R and K exhibiting the lowest selectivity, whereas H, S and particularly Y were more 5mC-selective. Strikingly, none of the repeats bound 5caC, that has also exhibited the lowest affinity in the context of previous repeat designs, with even universal repeat SG*GG having the least affinity for this charged and sterically most demanding nucleobase.33 Libraries with three deletions afforded differential profiles with comparably weak and mostly ambiguous nucleobase binding. The presence of an S at position 11 thereby resulted in lower overall binding as compared to N (Fig. 3c and d). Repeat SS*** showed a higher signal for C, whereas other repeats were comparably weak (Fig. 3c). In library NX***, 5mC again was bound most strongly by the majority of repeats, with C and 5fC being the second preference of most repeats. Here, W, T, R and S showed the highest affinities, with W being most 5mC-selective. Repeat NH*** bound C with highest affinity (Fig. 3d). Compared to these two related libraries, shifting the random amino acid to position 11 (library X***G) resulted in low binding, with the exception of W and K that exhibited non-selective binding of four nucleobases as well as R and D exhibiting weaker, non-selective binding (Fig. 3e). An even more pronounced reduction of binding was observed for library X****, covering the smallest repeats of the study. Here, almost no DNA-binding was observed, and importantly – as seen for almost all other mutants in the five libraries – binding to 5caC was completely absent (Fig. 3f).
 | ||
| Fig. 3 Screening of truncated TALE repeat libraries for selective recognition of oxidized 5mC nucleobases. (a) Data of control reactions. Conditions as in Fig. 2e. (b–f) Screening data for libraries as indicated above each heat map. Amino acid at position X indicated for each lane on the right, target nucleobase for each column below. Reactions were conducted in duplicate with 5 μM TALE, 0.1 μM DNA and 1 unit DNase I. Cy5 fluorescence data were recorded at t = 25 min and normalized as in Fig. 2e. | ||
However, a surprisingly different profile was observed for the single repeat mutant R**** that exhibited the strongest binding of 5caC among all 55 tested repeats, but did not bind to any other nucleobase (Fig. 3f). This intriguing “inverted” selectivity was confirmed in DNase I time-course experiments over 30 min (Fig. 4a). To get more quantitative insights, we performed titrations with this TALE and five DNA duplexes containing one of the cytosine nucleobases opposite the mutant repeat in DNA polymerase accessibility assays. In this reference assay, TALE binding inhibits primer extension by the Klenow fragment of E. coli DNA polymerase I (3′-5′-exo−, KF(exo−)) enabling quantification of binding via measuring the extension product after PAGE separation (Fig. 4b and ESI†).41 We obtained a Ki of 420.5 nM for binding of 5caC-containing DNA, whereas other nucleobases were only weakly bound even at the highest applicable TALE concentrations (Fig. 4c, close to the solubility limit). To assess potential context dependencies of this 5caC selectivity, we designed TALEs targeting 18 nt sequences in the human CDKN2A and BRCA1 genes with a R**** repeat opposite the C-position of the single CpG dyade (Fig. 2c). DNase I competition assays confirmed the 5caC selectivity of the repeat in both sequence contexts (Fig. 4d and e), suggesting programmability.
 | ||
| Fig. 4 5caC-selective DNA binding of TALE repeat R****. (a) Time course of DNase I competition assay as in Fig. 2e with 5 μM TALE_R****. (b) Principle of DNA polymerase accessibility assay using primer–template oligonucleotide duplexes with variable nucleobase (sphere, color-coded as in Fig. 1a) opposite mutant repeat (*). (c) Ki determination for TALE_R**** by DNA polymerase accessibility assay. Reactions contained 8.3 nM primer/template complex, 25 mU KF(exo−), 100 μM dNTP and were run for 15 min. Line: dose response fit. (d) Nucleobase selectivity of TALE_R**** targeting sequence BRCA1(18) (Fig. 2c) evaluated by DNase I assay as in (a) with 7.5 μM TALE and monitoring of Cy5 fluorescence at t = 30 min after addition of DNase I. (e) as Fig. 3d, but with target sequence CDKN2A (Fig. 2c) and use of 3.5 μM TALE. (f–h) Visualization of conformational changes from MD studies for TALEs bearing a single repeat SHDGG (wt), S**** or R**** (red arrows). White cartoons show the initial structures for the MD production run after equilibration, grey cartoons show representative structures of the cluster that represents the final simulation models at the end of the production run. Small arrows indicate the first three principal components (green, PC1; red, PC2; blue, PC3) resulting from a principal component analysis of the MD trajectory based on the backbone atoms' movements. (i) RMSF (root mean square fluctuations) for the protein residues sampled over the 250 ns production run. (j) Crystal structure of TALE–DNA complex bearing wt repeat SHDGG (white sticks) opposite C (not shown) superimposed to homology model of TALE–DNA complex bearing repeat R**** opposite 5caC (yellow sticks). Hydrogen bonds are shown as dotted red lines. | ||
We were next interested, how this minimal repeat scaffold with four deletions can retain DNA-binding without loss of essential inter-repeat interactions. TALE repeats of the CRD extensively interact with each other via van-der Waals and polar interactions involving both helices and RVD loops (Fig. 1d shows part of these interactions).28,29 This preorganizes the overall TALE fold, while still offering a likely essential conformational plasticity that is illustrated by compression along the superhelical axis upon DNA binding.28 We performed molecular dynamics (MD) simulations with 250 ns trajectories for DNA-unbound TALEs42–45 (designed for sequence CDKN2A(18), Fig. 2c) bearing at the fifth repeat position the wt repeat SHDGG, its truncated form S****, or the truncated substitution mutant R****. The wt TALE showed only subtle movements over the whole trajectory with an overall rigid superhelical topology (Fig. 4f and i, see ESI† for movies), whereas the presence of a single S**** repeat resulted in extensive kinking at the deletion site (Fig. 4g and i and ESI†). Since the conformations of the individual repeats were conserved in simulations with all TALEs, this loss of preorganization was likely due to the removed inter-loop interactions, and may account for the low affinity of this and other truncated repeats (Fig. 3f). Surprisingly, exchange of repeat S**** with R**** resulted in a final equilibrated structure similar to the one of the wt TALE (Fig. 4h and i and ESI†). Analysis of side chain dynamics revealed that a new stabilizing inter-loop interaction was established via a salt bridge of the R11 side chain and D13 of the preceding HD repeat (Fig. 2c and ESI Fig. 11 and 13†). Since RVD HD binds to nucleobase C, this suggested sequence-dependence of the stabilization. However, we observed 5caC-selectivity also for sequence BRCA1(18) with a preceding A bound by an NI RVD that is incapable of undergoing polar interactions with its I13 side chain (Fig. 2c and 4d). MD studies with this TALE revealed alternative stabilizations as explanation for the sequence-independence of the effect. These included hydrogen bonds between R11 of repeat R**** and the (not nucleobase-specific) N12 side chain of the preceding NI RVD, as well as between K12 of repeat R**** and the I13 backbone of the NI repeat (ESI Fig. 12 and 14†). This prompted us to evaluate the selectivity of repeat R**** also for a sequence with a preceding G or T bound by RVD NN or NG. Both RVDs bear the same identified hydrogen bond donors and acceptors as RVD NI. We constructed and tested both TALEs in the sequence context CDKN2A(18) and indeed observed comparable 5caC selectivity in both cases (ESI Fig. 5†). Taken together, repeat R**** exhibited 5caC selectivity independently of the preceding repeat, indicating its applicability for programmable 5caC-targeting.
To gain insights into how repeat R**** structurally differs from the evolutionary conserved repeat architecture and how it achieves selective binding of 5caC, we performed modeling studies. Alignment of a CDKN2A(18) targeting TALE–DNA complex bearing repeat R**** opposite 5caC with the crystal structure of a TALE bearing the natural HD repeat29 revealed a completely different organization of repeat R****. The conserved loop (residues 11 to 14) is cut off in this designer repeat, resulting in a large cavity. Compared to the close proximity of natural repeats to bound nucleobases (e.g. Cα atom of residue 13 is in ∼3.4 Å to the 5-methyl group of T for RVD NG28,29), the Cα atom of the closest residue in repeat R**** (R11) is positioned far away from the 5caC carboxyl C-atom (8.6 Å, Fig. 4j). Despite this drastic difference in loop architecture, the conformations of adjacent helix residues are almost fully retained: the R11 Cα atom exactly occupies the Cα-position of G15, and the Cα-position of A10 is unchanged. This leads to a repeat geometry that allows for regular helix orientations and interactions (Fig. 4j). Notably, the conformations of Q16 and K17 side chains that are critical for DNA phosphate interactions are also retained.28,29 5caC is recognized by R11 via electrostatic interactions, enabled by an extended side chain that is oriented similarly to the backbone of G14 and G15 in natural repeats. Both the 5-carboxyl group and the backbone phosphate of 5caC are thereby bound via the guanidinium group, providing a plausible explanation for the 5caC selectivity of repeat R****.
Conclusions
In conclusion, we report the first programmable receptor capable of directly and selectively recognizing an oxidized 5mC nucleobase in DNA. By strongly deviating from evolutionary conserved repeat designs, we accessed an unexplored structure space and discovered a truncated scaffold exhibiting full selectivity for 5caC. Independently of the RVD of the preceding repeat, this scaffold seems to execute a dual function via a critical arginine that both stabilized the TALE fold via loop-mediated repeat–repeat interactions and recognizing 5caC. This reveals an unexpected adaptability of the TALE repeat and sheds new light on its recognition potential for nucleobases that strongly deviate from A, G, T and C. Though the observed nucleobase selectivity of repeat R**** is lower than the one of the well-studied RVD HD, it is still in the range of natural RVDs (such as NG and NN) that have been characterized by comparable in vitro assays32,34,46 and are widely used for genome targeting.27 Evolution experiments with multiple random sites allowing for combinatorial effects will further expand the applicability of TALEs as versatile probes for the programmable targeting and analysis of epigenetic nucleobases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (Grant EPICODE). J. Jasper is supported by the Chemical Industry Fund (FCI). We thank A. J. Bogdanove and D. F. Voytas for TALE plasmids obtained via Addgene. Structures have been prepared with PyMOL, Version 1.7.4 Schrödinger, LLC.References
- X. Wu and Y. Zhang, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef PubMed.
- T. Pfaffeneder, B. Hackner, M. Truss, M. Munzel, M. Muller, C. A. Deiml, C. Hagemeier and T. Carell, Angew. Chem., Int. Ed. Engl., 2011, 50, 7008–7012 CrossRef PubMed.
- K. Iwan, R. Rahimoff, A. Kirchner, F. Spada, A. S. Schroder, O. Kosmatchev, S. Ferizaj, J. Steinbacher, E. Parsa, M. Muller and T. Carell, Nat. Chem. Biol., 2018, 14, 72–78 CrossRef PubMed.
- C. G. Spruijt, F. Gnerlich, A. H. Smits, T. Pfaffeneder, P. W. Jansen, C. Bauer, M. Munzel, M. Wagner, M. Muller, F. Khan, H. C. Eberl, A. Mensinga, A. B. Brinkman, K. Lephikov, U. Muller, J. Walter, R. Boelens, H. van Ingen, H. Leonhardt, T. Carell and M. Vermeulen, Cell, 2013, 152, 1146–1159 CrossRef PubMed.
- M. Iurlaro, G. Ficz, D. Oxley, E. A. Raiber, M. Bachman, M. J. Booth, S. Andrews, S. Balasubramanian and W. Reik, Genome Biol., 2013, 14, R119 CrossRef PubMed.
- Q. Du, P. L. Luu, C. Stirzaker and S. J. Clark, Epigenomics, 2015, 7, 1051–1073 CrossRef PubMed.
- H. Hashimoto, Y. O. Olanrewaju, Y. Zheng, G. G. Wilson, X. Zhang and X. D. Cheng, Gene Dev., 2014, 28, 2304–2313 CrossRef PubMed.
- M. W. Kellinger, C. X. Song, J. Chong, X. Y. Lu, C. He and D. Wang, Nat. Struct. Mol. Biol., 2012, 19, 831–833 CrossRef PubMed.
- T. T. Ngo, J. Yoo, Q. Dai, Q. Zhang, C. He, A. Aksimentiev and T. Ha, Nat. Commun., 2016, 7, 10813 CrossRef PubMed.
- F. Li, Y. Zhang, J. Bai, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 10617–10620 CrossRef PubMed.
- S. Ji, H. Shao, Q. Han, C. L. Seiler and N. Tretyakova, Angew. Chem., Int. Ed. Engl., 2017, 56, 14130–14134 CrossRef PubMed.
- W. A. Pastor, U. J. Pape, Y. Huang, H. R. Henderson, R. Lister, M. Ko, E. M. McLoughlin, Y. Brudno, S. Mahapatra, P. Kapranov, M. Tahiliani, G. Q. Daley, X. S. Liu, J. R. Ecker, P. M. Milos, S. Agarwal and A. Rao, Nature, 2011, 473, 394–397 CrossRef PubMed.
- C. X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C. H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Nat. Biotechnol., 2011, 29, 68–72 CrossRef PubMed.
- M. Su, A. Kirchner, S. Stazzoni, M. Muller, M. Wagner, A. Schroder and T. Carell, Angew. Chem., Int. Ed. Engl., 2016, 55, 11797–11800 CrossRef PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C. X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J. H. Min, P. Jin, B. Ren and C. He, Cell, 2012, 149, 1368–1380 CrossRef PubMed.
- B. Samanta, J. Seikowski and C. Hobartner, Angew. Chem., Int. Ed. Engl., 2016, 55, 1912–1916 CrossRef PubMed.
- G. Ficz, M. R. Branco, S. Seisenberger, F. Santos, F. Krueger, T. A. Hore, C. J. Marques, S. Andrews and W. Reik, Nature, 2011, 473, 398–402 CrossRef PubMed.
- J. Josse and A. Kornberg, J. Biol. Chem., 1962, 237, 1968–1976 Search PubMed.
- Z. L. Wescoe, J. Schreiber and M. Akeson, J. Am. Chem. Soc., 2014, 136, 16582–16587 CrossRef PubMed.
- B. A. Flusberg, D. R. Webster, J. H. Lee, K. J. Travers, E. C. Olivares, T. A. Clark, J. Korlach and S. W. Turner, Nat. Methods, 2010, 7, 461–465 CrossRef PubMed.
- M. J. Booth, E. A. Raiber and S. Balasubramanian, Chem. Rev., 2015, 115, 2240–2254 CrossRef PubMed.
- J. Boch and U. Bonas, Annu. Rev. Phytopathol., 2010, 48, 419–436 CrossRef PubMed.
- D. Deng, C. Yan, X. Pan, M. Mahfouz, J. Wang, J. K. Zhu, Y. Shi and N. Yan, Science, 2012, 335, 720–723 CrossRef PubMed.
- A. N. S. Mak, P. Bradley, R. A. Cernadas, A. J. Bogdanove and B. L. Stoddard, Science, 2012, 335, 716–719 CrossRef PubMed.
- M. W. Szulik, P. S. Pallan, B. Nocek, M. Voehler, S. Banerjee, S. Brooks, A. Joachimiak, M. Egli, B. F. Eichman and M. P. Stone, Biochemistry, 2015, 54, 1294–1305 CrossRef PubMed.
- J. Boch, H. Scholze, S. Schornack, A. Landgraf, S. Hahn, S. Kay, T. Lahaye, A. Nickstadt and U. Bonas, Science, 2009, 326, 1509–1512 CrossRef PubMed.
- G. Kubik, S. Batke and D. Summerer, J. Am. Chem. Soc., 2015, 137, 2–5 CrossRef PubMed.
- S. Maurer, M. Giess, O. Koch and D. Summerer, ACS Chem. Biol., 2016, 11, 3294–3299 CrossRef PubMed.
- Y. Zhang, L. Liu, S. Guo, J. Song, C. Zhu, Z. Yue, W. Wei and C. Yi, Nat. Commun., 2017, 8, 901 CrossRef PubMed.
- P. Rathi, S. Maurer, G. Kubik and D. Summerer, J. Am. Chem. Soc., 2016, 138, 9910–9918 CrossRef PubMed.
- J. Grau, M. Reschke, A. Erkes, J. Streubel, R. D. Morgan, G. G. Wilson, R. Koebnik and J. Boch, Sci. Rep., 2016, 6, 21077 CrossRef PubMed.
- M. Giess, A. Witte, J. Jasper, O. Koch and D. Summerer, J. Am. Chem. Soc., 2018, 140(18), 5904–5908 CrossRef PubMed.
- A. Erkes, M. Reschke, J. Boch and J. Grau, Genome Biol. Evol., 2017, 9, 1599–1615 CrossRef PubMed.
- T. Cermak, E. L. Doyle, M. Christian, L. Wang, Y. Zhang, C. Schmidt, J. A. Baller, N. V. Somia, A. J. Bogdanove and D. F. Voytas, Nucleic Acids Res., 2011, 39, e82 CrossRef PubMed.
- P. Rathi, S. Maurer and D. Summerer, Philos. Trans. R. Soc. Lond. B Biol. Sci., 2018, 373, 1748 CrossRef PubMed.
- G. Kubik, M. J. Schmidt, J. E. Penner and D. Summerer, Angew. Chem., Int. Ed. Engl., 2014, 53, 6002–6006 CrossRef PubMed.
- H. Wan, J. P. Hu, K. S. Li, X. H. Tian and S. Chang, PLoS One, 2013, 8, e76045 CrossRef PubMed.
- N. Tochio, K. Umehara, J. I. Uewaki, H. Flechsig, M. Kondo, T. Dewa, T. Sakuma, T. Yamamoto, T. Saitoh, Y. Togashi and S. I. Tate, Sci. Rep., 2016, 6, 37887 CrossRef PubMed.
- H. Flechsig, PLoS One, 2014, 9, e109919 CrossRef PubMed.
- B. I. Wicky, M. Stenta and M. Dal Peraro, PLoS One, 2013, 8, e80261 CrossRef PubMed.
- A. Juillerat, G. Dubois, J. Valton, S. Thomas, S. Stella, A. Marechal, S. Langevin, N. Benomari, C. Bertonati, G. H. Silva, F. Daboussi, J. C. Epinat, G. Montoya, A. Duclert and P. Duchateau, Nucleic Acids Res., 2014, 42, 5390–5402 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01958d |
| This journal is © The Royal Society of Chemistry 2018 |