
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining traditional 2D and modern physical organic-derived descriptors to predict enhanced enantioselectivity for the key aza-Michael conjugate addition in the synthesis of Prevymis™ (letermovir)†
Toni T.
Metsänen‡
 a,
Katrina W.
Lexa‡
*b,
Celine B.
Santiago
a,
Cheol K.
Chung
c,
Yingju
Xu
c,
Zhijian
Liu
c,
Guy R.
Humphrey
c,
Rebecca T.
Ruck
c,
Edward C.
Sherer
b and
Matthew S.
Sigman
*a
a,
Katrina W.
Lexa‡
*b,
Celine B.
Santiago
a,
Cheol K.
Chung
c,
Yingju
Xu
c,
Zhijian
Liu
c,
Guy R.
Humphrey
c,
Rebecca T.
Ruck
c,
Edward C.
Sherer
b and
Matthew S.
Sigman
*a
aDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA. E-mail: sigman@chem.utah.edu
bModeling and Informatics, MRL, Merck Sharp & Dohme, Rahway, New Jersey 07065, USA. E-mail: katrina.lexa@gmail.com
cProcess Research and Development, MRL, Merck Sharp & Dohme, Rahway, New Jersey 07065, USA
First published on 18th July 2018
Abstract
Quantitative structure–activity relationships have an extensive history for optimizing drug candidates, yet they have only recently been applied in reaction development. In this report, the predictive power of multivariate parameterization has been explored toward the optimization of a catalyst promoting an aza-Michael conjugate addition for the asymmetric synthesis of letermovir. A hybrid approach combining 2D QSAR and modern 3D physical organic parameters performed better than either approach in isolation. Using these predictive models, a series of new catalysts were identified, which catalyzed the reaction to provide the desired product in improved enantioselectivity relative to the parent catalyst.
Introduction
Catalyst design and optimization for enantioselective syntheses remains largely guided by meticulous and costly empirically-driven experimentation. As such, the development of robust methods for accurately predicting the relationship between catalyst structure and reactivity would have a tremendous impact on reaction development. Since catalyst activity and selectivity are fundamentally tied to structure, one possible approach is to apply quantitative structure–activity relationship (QSAR) modeling, which has traditionally been applied to drug development campaigns.1 Once identified, QSAR models can rapidly provide activity and selectivity estimates for new structures, thus enabling high-throughput virtual assessment of key structural components for the target of interest. Furthermore, highly weighted descriptors in the QSAR model may indicate structural features that influence the desired activity.2,3QSAR in the context of catalysis remains in its infancy.4–11 In the reported studies, two distinct approaches have been investigated: (1) the use of traditional QSAR-type descriptors evolved in the medicinal chemistry field, which are often 2D;4 and (2) the more recent use of parameters derived from quantum mechanics (QM) to accurately model key structural features involved in the hypothesized reaction mechanism.11 Examples of these descriptor types in the context of catalysis are depicted in Fig. 1. Clearly, these descriptors are innately unique and, if combined, may synergize to provide better statistical fitting of a dataset. Thus, we hypothesized that integrating these two descriptor sets had the potential to improve a recently developed enantioselective aza-Michael conjugate addition, representing a key step in the industrial synthesis of the approved pharmaceutical, Prevymis™ (letermovir; MK-8228).12,13
 | ||
| Fig. 1 Descriptors used to establish structure–activity relationships. | ||
In the initial disclosure, the product enantioselectivity from this key step was 88.3% ee using catalyst 2c (Scheme 1) for the commercial process. Importantly, as part of the initial optimization effort, a library of 29 bistriflamide catalysts was generated and evaluated under the same reaction conditions with the resultant enantioselectivity (ee) and conversions measured in triplicate. This library provides a wide range of ee's, offering an excellent foundation to examine the potential benefits of QSAR (Scheme 1). Despite the range in ee, this library failed to demonstrate any intuitive SAR trends. For example, during library design, it was hypothesized that amine pKa would track with ee given the reaction mechanism; however, no relationship was observed. Furthermore, while both electron-withdrawing and -donating substituents (e.g., 2h and 2l, respectively) as well as steric bulk (2f and 2c) were tolerated, only four catalysts (2a–2d) provided improvement to the parent unsubstituted catalyst 2e.
 | ||
| Scheme 1 Asymmetric bistriflamide route for Prevymis™ (letermovir). | ||
On this basis, we set out to identify improved catalysts through the evaluation of QSAR models and hypothesized that the resultant models could provide a more detailed understanding of the salient features required for enantioselective catalysis. Herein, we present a stepwise analysis of both 2D and modern physical organic descriptor-based models as drivers of this optimization campaign. Ultimately, we determined that combining these descriptor sets enabled identification of enhanced catalysts with non-intuitive structural features through a highly predictive virtual screening campaign.
Results and discussion
Training set
In order to accurately model catalyst performance, bistriflamides producing less than 25% conversion were excluded from the training set. Additionally, 2-OH substituted catalyst 2w was excluded because this catalyst with additional hydrogen bond donating sites may occupy unique transition states. This reduced training set totaled 21 bistriflamides, catalyzing reactions with product ee's ranging from 22.9% to 89.7%. To establish a baseline for the performance of 2D QSAR applied to catalysis, several linear models were built based on method/descriptor combinations with demonstrated successes in the literature and earlier MSD (Merck Sharpe & Dohme) QSAR models related to discovery lead optimization. These include random forest (RF)14 or support vector machines15 as the machine learning method, coupled with atom pair substructural descriptors or fingerprint descriptors. In our initial model of the bistriflamide library, we found that RF with Carhart atom pairs (APC)16 gave the best cross-validated (50/50 split) model of ee, with a cv-R2 = 0.34 (model A, Fig. 2).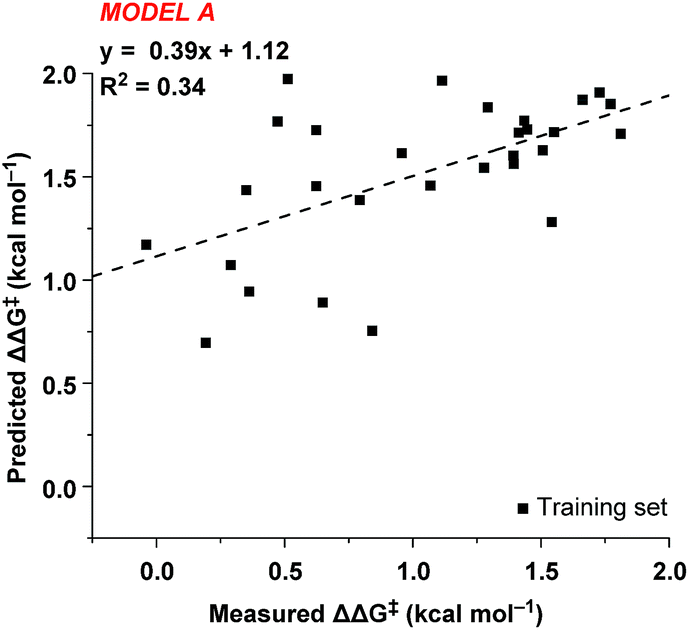 | ||
| Fig. 2 Cross-validated random forest QSAR model of the training set. | ||
As all of the catalysts share a common C2-symmetric bistriflamide core, we envisaged that the parent arene could be used as an efficient surrogate structure to rapidly explore quantum mechanical-derived physical organic descriptor space. Importantly, a simple arene proxy significantly limited the number of low energy conformations to be considered. Multi-variate linear regression modeling17,18 produced a relatively simple model using two parameters (NBOC2 and P19) expressed in three terms (NBOC2, P, and NBOC2 × P) to achieve a good statistical fit between the predicted and the measured enantioselectivity for the initial training set of bistriflamides (R2 = 0.80, LOO-Q2 = 0.77, model B) (Fig. 3). Given the physical organic descriptors identified by our simple arene model, we could interrogate several new structural hypotheses. The unsubstituted C2 NBO charge (NBOC2) represents a general electronic parameter describing the overall electron density of the arene.20–22 Additionally, the NBOC2 term could describe a hydrogen bond interaction between the ortho C–H and the substrate. The partition-coefficient (P) describes the hydrophobicity of the arene. This parameter is often used in drug design to predict water solubility of molecules and due to its broad numeric range, is typically reported as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. We postulated that, in the current case, the partition-coefficient could describe complex attractive interactions between the catalyst and the substrate.
P. We postulated that, in the current case, the partition-coefficient could describe complex attractive interactions between the catalyst and the substrate.
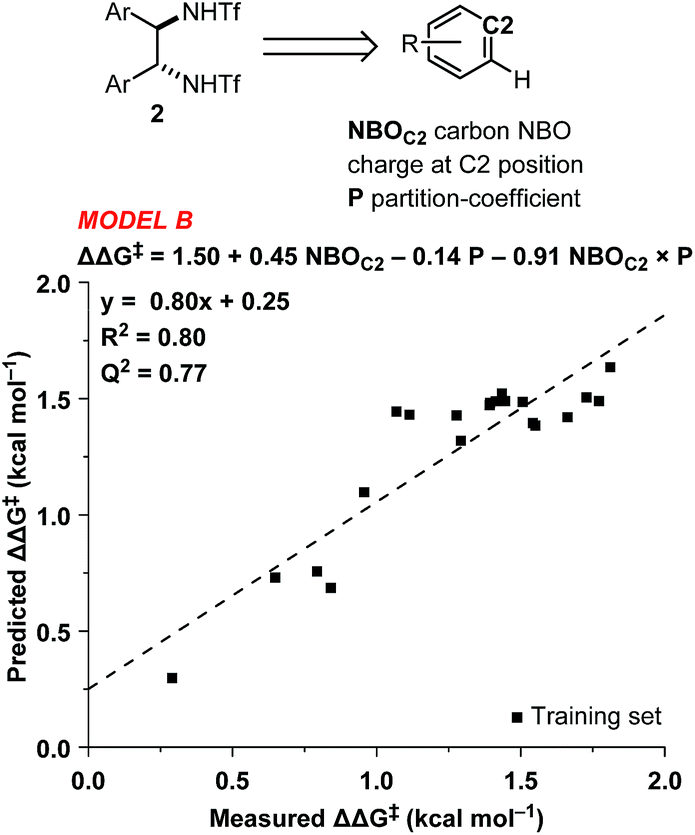 | ||
| Fig. 3 Modern physical organic descriptors and a predictive model for the initial training set. | ||
Model validation screen: round 1
To validate our initial 2D and 3D models, a set of 30 new catalysts was proposed, 10 of which were synthesized based on predicted product ee (VS1, Table 1, see ESI† for details). Since ortho-fluoro substitution seemed a preferred feature, we chose to synthesize several derivatives that incorporated this group, while introducing various para-substituents to probe further subtle effects. Additionally, alternative halide- and CF3-substitution patterns were explored.| 2 | Ar | Predicted% ee (ΔΔG‡) | Measured% ee (ΔΔG‡) | |
|---|---|---|---|---|
| Model A | Model B | |||
| a ΔΔG‡ given in parentheses in kcal mol−1. | ||||
| ab | 2-F-4-Br-C6H3 | 86.7(1.64) | 85.9(1.60) | 90.2(1.85) |
| ac | 2-F-4-Cl-C6H3 | 87.0(1.66) | 85.8(1.60) | 89.3(1.79) |
| ad | 2-F-4-CF3-C6H3 | 87.7(1.70) | 83.6(1.50) | 88.3(1.73) |
| ae | 2,4-Cl2-C6H3 | 73.3(1.16) | 85.3(1.58) | 86.2(1.62) |
| af | 4-CF3-C6H4 | 84.9(1.56) | 81.3(1.43) | 85.3(1.58) |
| ag | 4-F-C6H4 | 82.0(1.44) | 84.0(1.52) | 84.1(1.52) |
| ah | 3-F-C6H4 | 82.8(1.47) | 76.6(1.26) | 79.9(1.36) |
| ai | 3,4-Cl2-C6H3 | 69.8(1.07) | 81.9(1.44) | 78.9(1.33) |
| aj | 2-F-4-CN-C6H3 | 87.1(1.66) | 84.6(1.54) | 78.5(1.32) |
| ak | 3,4,5-F3-C6H2 | 62.3(0.91) | 66.3(0.99) | 75.9(1.24) |
Although both models were able to predict enantioselectivity reasonably well, some catalysts were captured better by one of the two QSAR approaches: 2,4- and 3,4-dichlorinated catalysts (2ae and 2ai, respectively) were more accurately predicted by model B (3D parameters), while model A (2D parameters) gave better predictions for the best three catalysts 2ab–2ad. It is worth noting that all of the best performing catalysts (2ab–2ad) were under-predicted by both models. Overall, the models displayed uneven predictive capability for the poorer performing catalysts (2ah–2ak).
Focused virtual screen: round 2
The ten new compounds used as external validations in round 1 were incorporated into our existing dataset to inform and improve our QSAR models. Since the dataset is weighted towards high ee catalysts, we split the data into test and training sets by selecting every other catalyst, starting from the best performing catalyst, until we had a third of the data in a new validation set. The updated 3D QSAR model B′ was then used to design and predict a new virtual screening set of 35 bistriflamides (VS2, see ESI† for details). The 2D model was updated likewise. Unfortunately, neither the 2D nor the 3D QSAR model predicted any of the new catalysts would give the product in greater than 90% ee. These modest predictions for VS2 could be the result of the inherent difficulty of extrapolating beyond the best identified catalyst (2ab, 90.2% ee). In fact, RF is recognized for having a ceiling to predicted activity that is set by the highest activities in the training data.14 However, given the orthogonal accuracies observed for the 2D and 3D models within VS1, we hypothesized that combining substructural and physical organic descriptors into a hybrid 2D/3D model might be a beneficial strategy. These two model types have been compared, yet very few studies have investigated their potential for synergy,23–25 and, to the best of our knowledge, none exist in the field of catalyst optimization.We selected the 20 highest-weighted descriptors from model A and added these to the physical organic descriptor set. Both 2D and 3D QSAR parameters were thus available during the process of linear regression modeling.17 Interestingly, one 2D-QSAR parameter, FX1sp3CX2sp207, stood out as highly synergistic with the 3D physical organic descriptors. The FX1sp3CX2sp207 parameter is a substructural atom pair describing fluorine atoms seven bonds away from an sp2-hybridized carbon with two non-hydrogen substituents (Fig. 4). Within the bistriflamide catalyst scaffold, this represents a readout for the unsubstituted ortho and meta positions and, of particular note, ortho- and meta-fluorine substitutions contribute as well. This 2D-descriptor was decisively the best single descriptor (R2 = 0.62) and was found to be a significant predictor in nearly all hybrid models. The fundamental drawback of this parameter is the difficulty to extrapolate beyond current structures within the bisaryltriflamide scaffold.
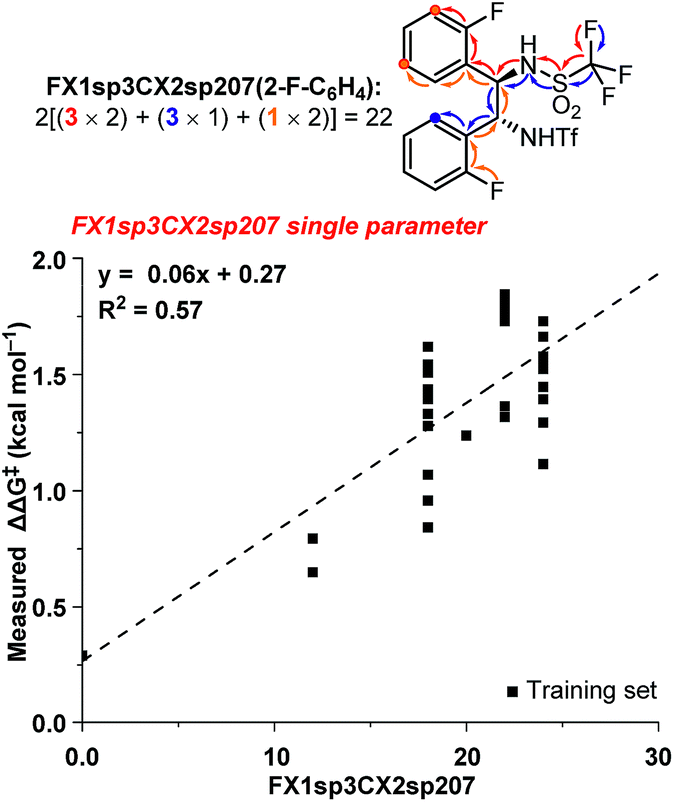 | ||
| Fig. 4 FX1sp3CX2sp207 single parameter model. | ||
From our model building process, we obtained four new hybrid models (C–F) in addition to our standalone models A′ and B′ (Scheme 2C, see ESI† for details). All four new models provided a statistically satisfactory fit to the training set (R2 = 0.78–0.90 and LOO-Q2 = 0.70–0.77) and were well-validated against the test set.
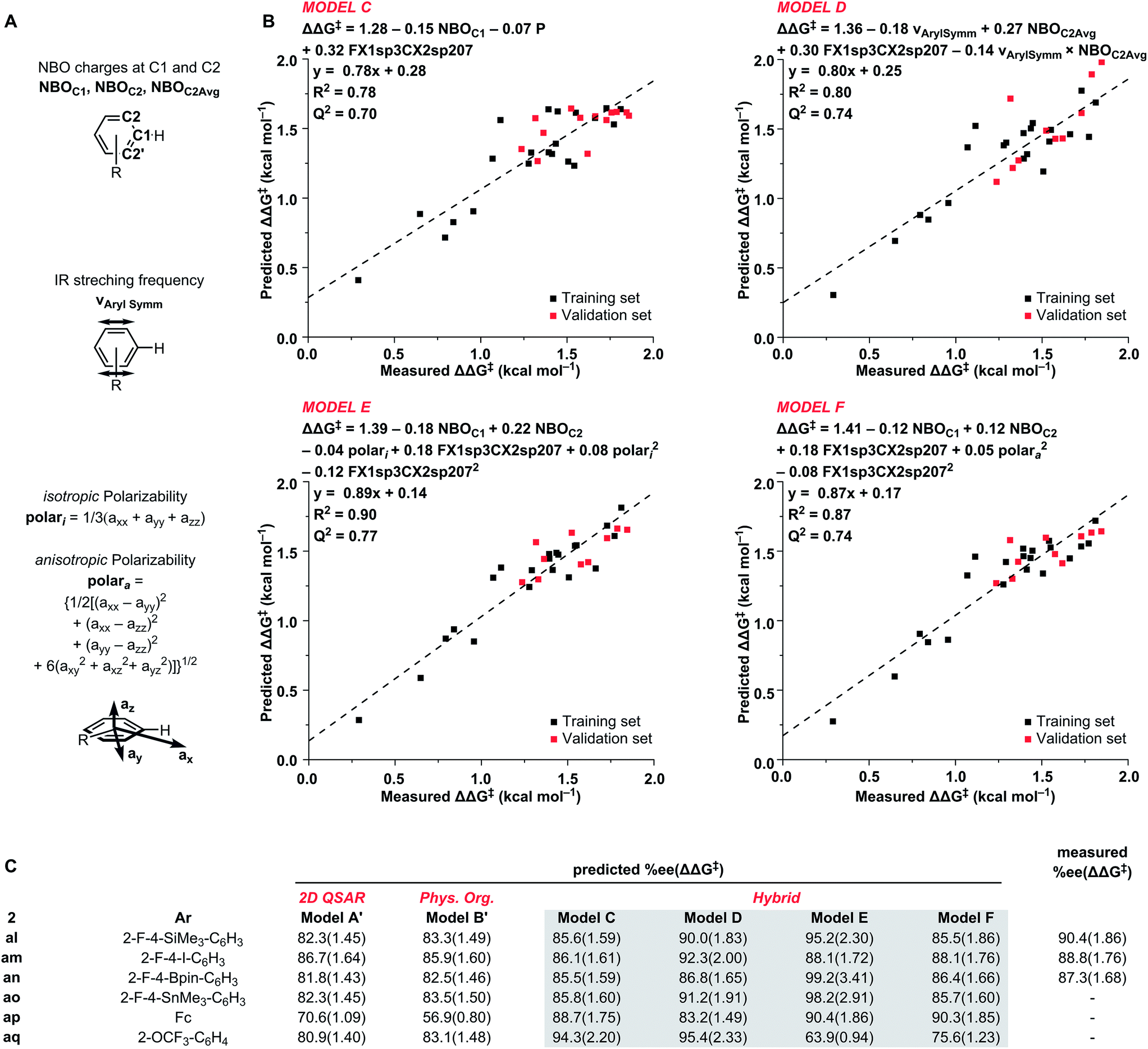 | ||
| Scheme 2 Physical organic descriptors (A), hybrid models (B), and virtual screen 2 (C); ΔΔG‡ given in parentheses in kcal mol−1. | ||
Hybrid model C incorporates the C1 NBO charge as an electronic parameter and the previously used partition-coefficient together with FX1sp3CX2sp207 parameter. Out of the virtual screening set, model C predicted the OCF3-substituted catalyst 2aq to be the most selective (94.3% ee).
The second hybrid model D combines the symmetrical aryl IR vibrational frequency26 and the average of C2 NBO charges as physical organic descriptors for the arene ring, and the substructural descriptor FX1sp3CX2sp207. Model D accurately predicted the best performing catalyst in the training and validation sets. Prediction of ee in VS2 with model D identified four catalysts with potential outputs above 90% ee. In agreement with model C, 2aq was predicted at 95.4% ee. Additionally, catalysts 2al, 2an, and 2ao were predicted to give 90.0%, 92.3%, and 91.2% ee, respectively.
The models E and F both include C1 and C2 NBO charge, FX1sp3CX2sp207, and a polarizability parameter. Additionally, both models show an excellent fit to the training and validation sets. The only difference between the two models was the means by which the polarizability was expressed. For model E, isotropic polarizability (polari) was used, which is the calculated average of the ax, ay, az polarizability vectors. In the arene models used in this study, the az term was typically small and the isotropic polarizability was effectively the average polarizability along the xy-plane. In the model F, anisotropic polarizability (polara) is used instead. Whereas isotropic polarizability can be readily used to compare the polarizability of equally spherical (or planar) molecules, anisotropic polarizability can be used to better describe asymmetrical structures.27 As expected, where most of the training set consists of flat structures with negligible polarizability along the z-axis, the two terms are collinear (Fig. 5). However, the situation changes dramatically when bulkier substituents that increase polarizability along the z-axis are incorporated: the 2-OTf substituted catalyst 2c is the greatest outlier from the training set. Due to this difference in the polarizability term, the predictions between the two models are dramatically different. While isotropic model E predicts the silicon (2al), tin (2ao), and boron (2an) substituted catalysts at 95.2–99.2% ee, the anisotropic model F predicts them to give only 85.5–86.4% ee. Interestingly, both models E and F predict the ferrocene catalyst 2ap at 90% ee.
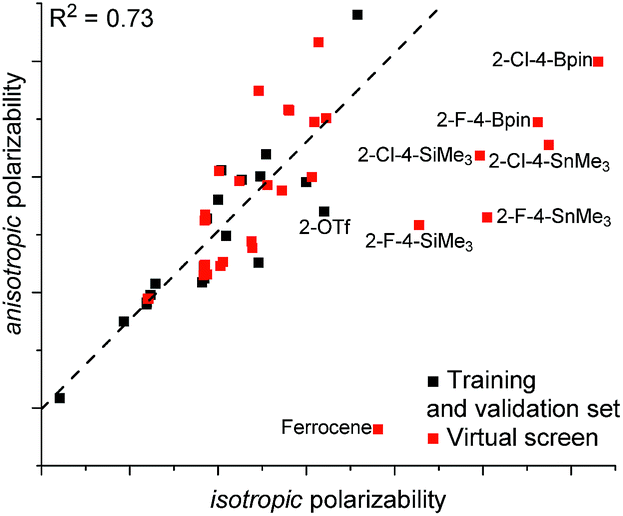 | ||
| Fig. 5 Correlation between different polarizability terms. | ||
Based on the predictions from the six models (Scheme 2c), as well as synthetic accessibility, we synthesized three catalysts that were expected to yield high product enantioselectivities: 2-F-4-SiMe3 (2al), 2-F-4-I (2am), and 2-F-4-Bpin (2an). Gratifyingly, the three catalysts all gave full conversion and good ee, with 2-F-4-SiMe3 giving the highest enantioselectivity observed for this reaction to date, 90.4% ee.
Of the six models used for the virtual screening, both the pure 2D and pure 3D models failed to accurately predict the behavior of the best catalysts. Although models E and F containing the polarizability terms accurately predicted the iodine-substituted catalyst 2am, they over- or underestimated the silicon (2al) and boron (2an) catalysts, respectively. The best effective predictions for VS2 were obtained with the hybrid model D.
Focused solvent screening
In our earlier investigation,13 we had observed that the choice of solvent could have a significant effect on the reaction. Therefore, we performed a focused solvent screen on the best performing catalysts located in this study. While most catalysts performed equally or worse in ethereal solvents compared to the standard toluene conditions (Table 2), the selectivity with best performing catalyst 2al was further increased giving the highest ee (93.7%) in methyl tert-butyl ether (MTBE).| 2 | Ar | Measured% ee | |||||
|---|---|---|---|---|---|---|---|
| Anhydr. toluene | Wet toluene | MTBE | CPME | EtOAc | 2-MeTHF | ||
| a ΔΔG‡ given in parentheses in kcal mol−1; MTBE = methyl tert-butyl ether; CPME = cyclopentyl methyl ether. | |||||||
| ab | 2-F-4-Br-C6H3 | 90.2 | 90.2 | 86.8 | 88.6 | 77.8 | 79.0 |
| al | 2-F-4-SiMe3-C6H3 | 90.4 | 91.8 | 93.7 | 92.3 | 88.9 | 89.2 |
| am | 2-F-4-l-C6H3 | 88.8 | 88.6 | 87.1 | 87.0 | 78.1 | 80.5 |
| an | 2-F-4-Bpin-C6H3 | 87.3 | 87.8 | 89.4 | 88.7 | 83.2 | 84.9 |
Structural interpretation of the hybrid models
While the combinations of parameters used in the hybrid models do not lend themselves to immediate 3D interpretation, our results made it clear that the strongest-performing models of this reaction require: (1) an electronic parameter, NBO charge or IR frequency; and (2) the substructural parameter FX1sp3CX2sp207. While we anticipated the observed importance of an electronic parameter, its combination with the strong predictive power of the substructural descriptor confirmed our hypothesis that 2D and 3D information could provide orthogonally beneficial information for guiding structure-based design. The atom-pair substructural parameters condense a steric estimate into one numerical value and, in this case, simultaneously amplified the score for fluorine-substituted catalysts. The positive effect of an ortho-fluorine substituent is undeniable, but the precise explanation for this effect remains uncertain. We propose that certain ortho-substituents, especially fluorine, lock the catalyst in a favorable conformation for the transition state.Our previous investigation of this reaction mechanism revealed that hydrogen bonding between the catalyst and substrate is a key driver for enantioselectivity.13 Furthermore, structural modifications of the backbone demonstrated that both sulfonamide N–H's are critical for conversion. DFT (density functional theory) calculations with 2c showed that ring closure most probably occurs via aza-Michael conjugate addition; however, we believe that, following substrate tautomerization, a 6-π electrocyclization mechanism can be energetically accessible with some catalysts as well. This structural information from the mechanistic work is consistent with our 2D, 3D, and hybrid models, indicating the utility of performing both machine learning as well as density functional calculations to fully capture the dynamics of a catalyst system.
Conclusions
QSAR offers advantages for catalyst design because well-validated models can facilitate rapid selection and optimization of catalyst properties while lowering labour and resource cost. Here, the best catalyst was found using a hybrid 2D/3D modelling approach. Although only modest gains in enantioselectivity were achieved, it is unlikely that the unusual 2-F-4-SiMe3 catalyst 2al would have been identified in a traditional intuition-driven screening. This hybrid 2D/3D QSAR modeling strategy should enable guided design and rapid screening of large catalyst libraries, thus facilitating faster route optimization at substantially reduced cost.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Robert Sheridan (MSD) for helpful discussions regarding QSAR strategy and Qunsheng Guo for assistance in catalyst characterization. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged. T. T. M. and M. S. S. would like to thank MSD for support. M. S. S. thanks the National Science Foundation (CHE-1361296) for partial support of C. B. S.References
- M. L. Drummond and B. G. Sumpter, Inorg. Chem., 2007, 46, 8613 CrossRef PubMed.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803 CrossRef PubMed.
- M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688 CrossRef PubMed.
- A. Cherkasov, E. N. Muratov, D. Fourches, A. Varnek, I. I. Baskin, M. Cronin, J. Dearden, P. Gramatica, Y. C. Martin, R. Todeschini, V. Consonni, V. E. Kuz'min, R. Cramer, R. Benigni, C. Yang, J. Rathman, L. Terfloth, J. Gasteiger, A. Richard and A. Tropsha, J. Med. Chem., 2014, 57, 4977 CrossRef PubMed.
- K. C. Harper and M. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2179 CrossRef PubMed.
- S. E. Denmark, N. D. Gould and L. M. Wolf, J. Org. Chem., 2011, 76, 4337 CrossRef PubMed.
- K. C. Harper and M. S. Sigman, Science, 2011, 333, 1875 CrossRef PubMed.
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366 CrossRef PubMed.
- S. E. Denmark, R. C. Weintraub and N. D. Gould, J. Am. Chem. Soc., 2012, 134, 13415 CrossRef PubMed.
- C. Yang, E.-G. Zhang, X. Li and J.-P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 6506 CrossRef PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292 CrossRef PubMed.
- G. R. Humphrey, S. M. Dalby, T. Andreani, B. Xiang, M. R. Luzung, Z. J. Song, M. Shevlin, M. Christensen, K. M. Belyk and D. M. Tschaen, Org. Process Res. Dev., 2016, 20, 1097 CrossRef.
- C. K. Chung, Z. Liu, K. W. Lexa, T. Andreani, Y. Xu, Y. Ji, D. A. DiRocco, G. R. Humphrey and R. T. Ruck, J. Am. Chem. Soc., 2017, 139, 10637 CrossRef PubMed.
- V. Svetnik, A. Liaw, C. Tong, C. J. C. Culberson, R. P. Sheridan and B. P. Feuston, J. Chem. Inf. Comput. Sci., 2003, 43, 1947 CrossRef PubMed.
- C. Cortes and V. N. Vapnik, Mach. Learn., 1995, 20, 273 Search PubMed.
- R. E. Carhart, D. H. Smith and R. Ventkataraghavan, J. Chem. Inf. Comput. Sci., 1985, 25, 64 CrossRef.
- J.-Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144 CrossRef.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398 RSC.
- Calculated using Moriguchi's method: I. Moriguchi, S. Hirono, Q. Liu, I. Nakagome and Y. Matsushita, Chem. Pharm. Bull., 1992, 40, 127 CrossRef , see ESI† for details.
- F. Weinhold and C. R. Landis, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, Hoboken, NJ, 2012 Search PubMed.
- C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90, 1396 CrossRef.
- C. B. Santiago, A. Milo and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 13424 CrossRef PubMed.
- H. Matter and T. Pötter, J. Chem. Inf. Comput. Sci., 1999, 39, 1211 CrossRef.
- E. Estrada, I. Perdomo-López and J. J. Torres-Labandeira, J. Chem. Inf. Model., 2001, 41, 1561 CrossRef PubMed.
- S. Vilar, E. Estrada, E. Uriarte, L. Santana and Y. Gutierrez, J. Chem. Inf. Model., 2005, 45, 502 CrossRef PubMed.
- A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210 CrossRef PubMed.
- R. J. W. Le Fèvre, Adv. Phys. Org. Chem., 1965, 3, 1 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, computational details and characterization data. See DOI: 10.1039/c8sc02089b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |