
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbenes as chiral Brønsted base catalysts: a highly diastereo- and enantioselective 1,6-addition reaction†
Surojit
Santra
,
Arka
Porey
,
Barun
Jana
and
Joyram
Guin
 *
*
Department of Organic Chemistry, Indian Association for the Cultivation of Science, 2A & 2B Raja S. C. Mullick Road, Jadavpur, Kolkata-700032, India. E-mail: ocjg@iacs.res.in
First published on 2nd July 2018
Abstract
Highly diastereo- and enantioselective 1,6-addition of 1,3-ketoamides to p-quinone methides (p-QMs) using chiral NHCs as Brønsted base catalysts is developed. The reaction is based on the utilization of a 1,3-ketoamide having acidic N–H that forms a chiral ion-pair consisting of the enolate and the azolium ion. Different β-ketoamides and functionalized p-QMs are applicable to the reaction. Synthetic application of the method is demonstrated via the preparation of highly enantioenriched β and γ-lactam derivatives.
Introduction
N-Heterocyclic carbenes (NHCs) are the most versatile organocatalysts. Consequently, asymmetric catalysis with chiral NHCs involves covalent interaction with the activated substrate, Lewis acid–base interaction with the reagent (Scheme 1a and 1b)1 and hydrogen bonding interaction with the substrate using a proton shuttle (Scheme 1c).2 Although asymmetric NHC catalysis via ion-pair interaction with substrates by utilizing the intrinsic Brønsted base characteristic of NHCs3 has drawn significant interest, it remains highly challenging (Scheme 1d).4 Herein, we present a catalytic and highly stereoselective 1,6-addition reaction of p-QMs using NHCs as Brønsted base catalysts (Scheme 1e).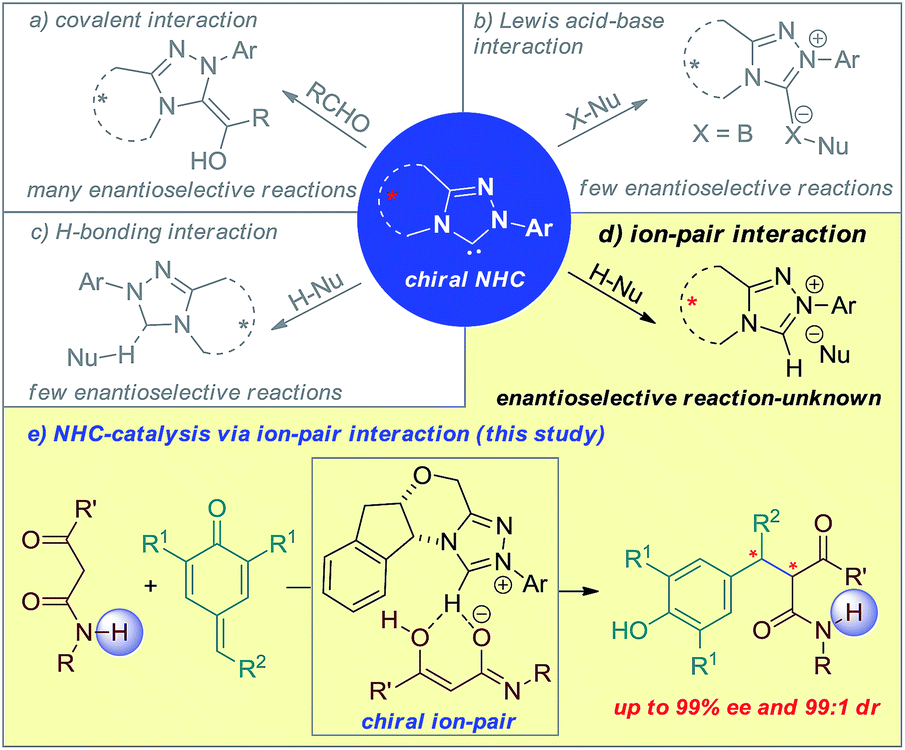 | ||
| Scheme 1 Different modes of substrate activation for asymmetric NHC-catalysis. | ||
p-QM scaffolds are found in many natural products5 and serve as key reactive intermediates in several chemical, medicinal and biological processes.6 Consequently, efforts have been devoted to developing organocatalytic asymmetric nucleophilic addition to p-QM.7 However, attempts at developing enantioselective 1,6-addition of enolizable nucleophiles to p-QMs using chiral NHCs as Brønsted bases remains largely unsuccessful.8 To employ NHCs as Brønsted base catalysts for asymmetric 1,6-addition of p-QMs, we anticipated that the substrate should have a lower pKa value than the NHC. We thus envisioned that easily enolizable 1,3-ketoamides containing an acidic N–H group would be appropriate nucleophiles. It is expected that an in situ generated NHC having a pKa value in the range of 17–19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3a may deprotonate the ketoamide (pKa ≈ 10–12),9 furnishing a chiral ion-pair10 comprising the enolate and the azolium ion. The in situ generated chiral enolate is expected to react with p-QM, thus providing enantioselectivity to the final product.
3a may deprotonate the ketoamide (pKa ≈ 10–12),9 furnishing a chiral ion-pair10 comprising the enolate and the azolium ion. The in situ generated chiral enolate is expected to react with p-QM, thus providing enantioselectivity to the final product.
Results and discussion
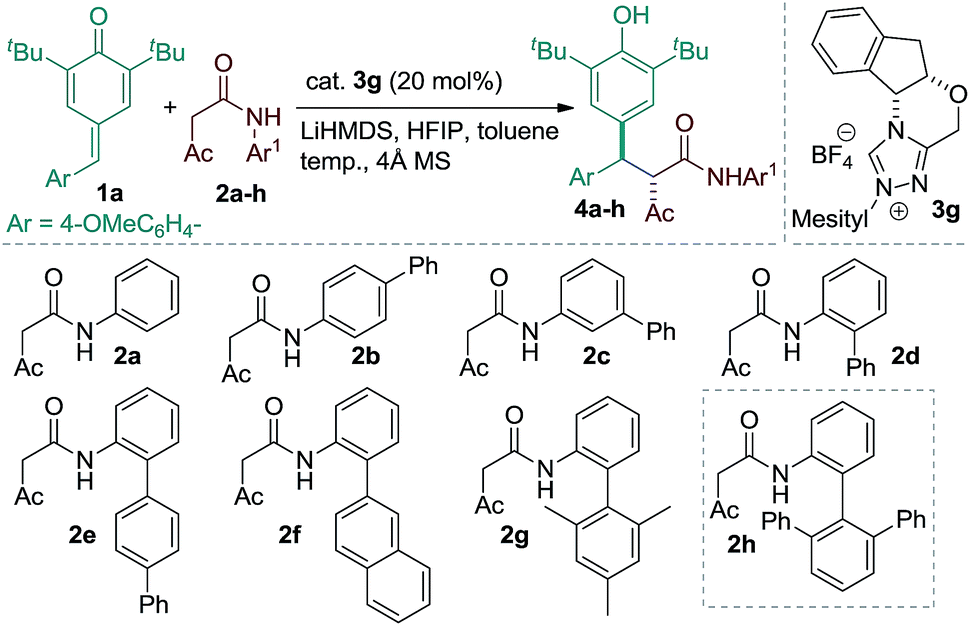
Our studies were commenced using p-QM 1a and the β-ketoamide 2a as model substrates under various reaction conditions with a series of chiral NHCs (see the ESI†). Following initial optimization, a variety of amides possessing different aromatic amines were assayed using 20 mol% of 3g, 16 mol% of LiHMDS, 20 mol% of HFIP and 4 Å MS in toluene (Table 1). The β-ketoamide derived from aniline afforded product 4a with poor stereoselectivity (entry 1). Enantioselectivity of the reaction could not be improved using β-ketoamide 4b or 4c having a phenyl substituent at the para or meta position of the aniline ring (entries 2 and 3). However, introducing a phenyl group at the ortho position of the aniline moiety of β-ketoamide was found to be beneficial, delivering product 4d in 68% ee with the amide 2d (entry 4). Based on this result, different β-ketoamides 2e–h containing bulky substituents at the ortho position of the aniline unit were prepared and examined. No considerable improvement in the stereoselectivity of the products, 4e–g, was realized with the ketoamides 2e–g (entries 5–7). The sterically demanding ketoamide 2h, however, delivered product 4h with a slight improvement in stereoselectivity (entry 8). Interestingly, the reaction efficacy was improved further with the amide 2h, when the reaction was performed in the absence of HFIP (entry 9). This result may indicate that HFIP interferes in the formation of a tight ion-pair comprising the enolate and the azolium ion.11 By carrying out the reaction at reduced temperature, product 4h was obtained in excellent yield and stereoselectivity (entries 10 and 11). Importantly, the loading of precatalyst 3g could also be reduced to 15 mol% without diminishing the overall reaction efficiency (entry 12).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amide | Temp. (°C) | h | dr | ee (%) | Conv. (%) |
| a Reaction conditions: 1a (0.05 mmol), 2a–h (0.05 mmol), LiHMDS (16 mol%), HFIP (20 mol%), 4 Å MS (35 mg) in toluene (1.0 mL); dr determined by 1H NMR and HPLC analysis; ee determined by HPLC analysis on a chiral stationary phase. b ee of the major diastereoisomer is given. c Reaction performed without using HFIP. d Isolated yield. e Reaction performed on a 0.1 mmol scale using 15 mol% of 3g. ND = not determined. | ||||||
| 1 | 2a | 0 | 12 | 64:36 |
44, 48 | 95 |
| 2 | 2b | 0 | 12 | 73:27 |
27, ND | 50 |
| 3 | 2c | 0 | 12 | 65:35 |
30, 12 | 70 |
| 4 | 2d | 0 | 12 | 80:20 |
68, 14 | 70 |
| 5 | 2e | 0 | 12 | 70:30 |
70, 10 | 50 |
| 6 | 2f | 0 | 12 | 67:33 |
66, 20 | 60 |
| 7 | 2g | 0 | 12 | 83:17 |
70, 15 | 70 |
| 8 | 2h | 0 | 12 | 95:5 |
73b | 50 |
| 9c | 2h | 0 | 12 | 97:3 |
85b | 75 |
| 10c | 2h | −20 | 14 | 98:2 |
95b | 70 |
| 11c | 2h | −40 | 20 | 98:2 |
98b | 97d |
| 12c,e | 2h | −40 | 24 | 98:2 |
98b | 93d |
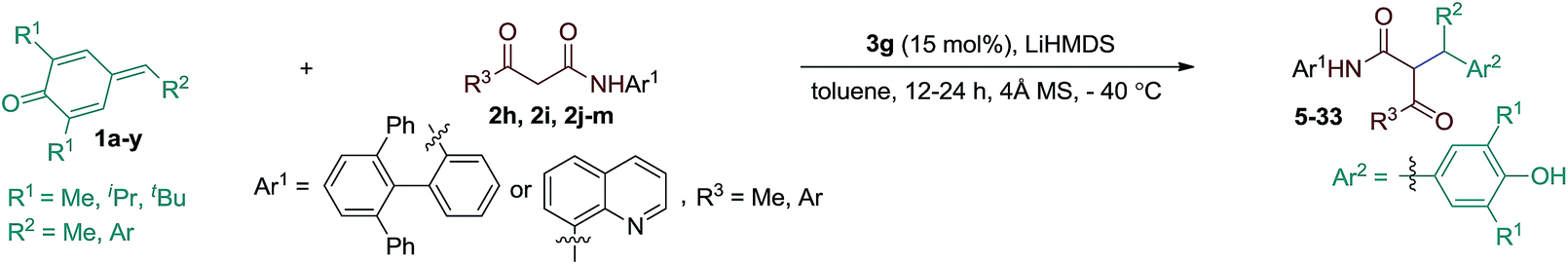
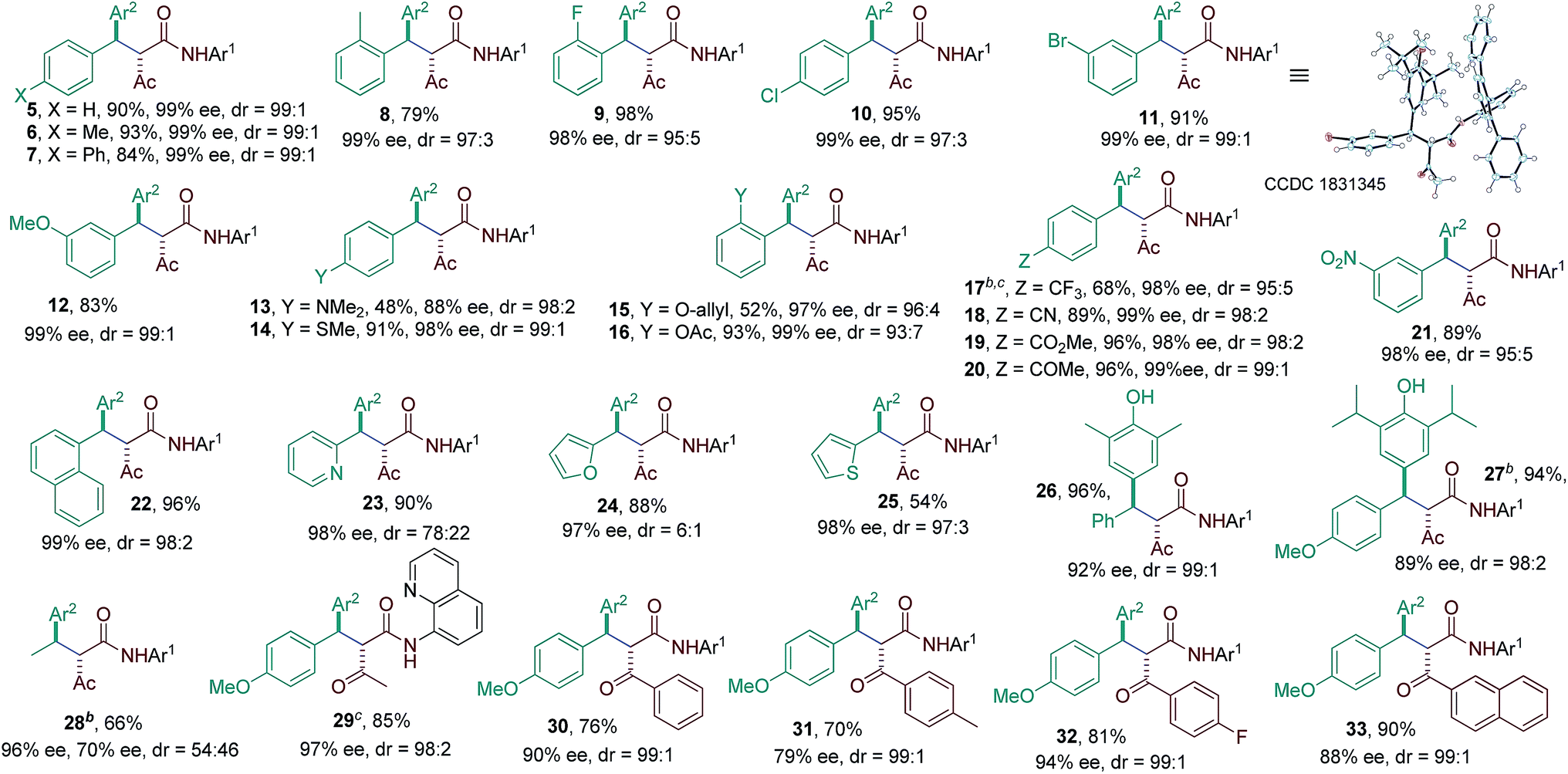
The substrate scope of the reaction was then evaluated varying both p-QMs and amides (Table 2). A series of p-QMs with different substituents at the aromatic ring and quinone moiety were first investigated with the amide 2h. Accordingly, the reaction furnished the desired products 5–7 in excellent yields and stereoselectivity with p-QMs having benzene, p-tolyl and p-biphenyl substituents. While a moderate yield of product 8 was obtained with o-tolyl substituted p-QM, the stereoselectivity remained excellent. p-QMs bearing F, Cl and Br substituents at any position of the aromatic ring were easily transformed into the corresponding products 9–11 in excellent yields and stereoselectivity. Substrates having electron rich substituents like OMe (12), NMe2 (13), SMe (14), O-allyl (15) and OAc (16) or electron deficient functional groups such as CF3 (17), cyano (18), ester (19), keto (20) and nitro (21), irrespective of their position, were applicable to the reaction. Naphthyl substituted p-QM was converted to product 22. Heteroaromatic rings, for instance, pyridine (23), furan (24) and thiophene (25), were tolerated in this catalytic reaction. A slight decrease in ee values was realized by replacing the tert-butyl group of the quinone ring with methyl or isopropyl substituents (products 26 and 27). Interestingly, the catalytic asymmetric process could be extended to the challenging alkylated p-QM, providing the expected product 28 in good yield, albeit with moderate stereoselectivity. Substrate scope studies were then carried out using different amides with 1a. The amide 2i (R3 = Me and Ar1 = quinolin-8-yl) afforded product 29 in good yield and stereoselectivity. Likewise, different α-aroylacetamides 2j–m were found to be suitable for the reaction, furnishing the desired products 30–33 in good yields and enantioselectivity. Absolute configurations of 2S, 3R were determined for compound 11 using single-crystal X-ray analysis.12
| a Reaction conditions: 1a–y (0.1 mmol), amides (0.1 mmol), 3g (15 mol%), LiHMDS (12 mol%), 4 Å MS (70 mg) in toluene (2.0 mL); isolated yields. b Reaction performed at −78 °C. c Reaction performed with amide 2i. Diastereoisomeric ratio (dr) determined by 1H NMR and HPLC analysis. Enantiomeric excess (ee) determined by HPLC analysis on a chiral stationary phase. |
|---|
|
|
To illustrate the synthetic utility of the reaction, a preparative scale experiment using 0.97 g of p-QM 1a and 0.68 g of amide 2i was performed, which resulted in 1.25 g of product 29 in good yield and stereoselectivity (dr = 98:2, ee = 97%, Scheme 2a). Furthermore, a variety of valuable enantioenriched compounds were prepared from product 29 through simple chemical transformations (Scheme 2b, see the ESI†). Accordingly, NaBH4 reduction of ketone 29 afforded the alcohol 34a having three contiguous stereogenic centres in good yield and excellent ee along with a minor diastereoisomer 34b (see the ESI†). Compound 34b was separated by column chromatography. The major diastereoisomer 34a, whose structure was confirmed by single crystal X-ray analysis, was transformed into the spirocyclic γ-lactam 35 in one step via PIDA oxidation. The removal of the tert-butyl group from the alcohol 34a with AlCl3 afforded the phenol 36 in good yield without diminishing its original stereoselectivity. The mesyl protection of the alcohol 34a provided compound 37 in good yield. The absolute configuration of compound 37 was unambiguously determined by single-crystal X-ray analysis.12 Compound 37 was then converted to the enantioenriched β-lactam 38 in good yield with excellent stereocontrol. Furthermore, the synthesis of biologically relevant enantioenriched triarylmethane 39 was achieved with excellent yield and ee value upon the treatment of 29 with hydrazine hydrate.
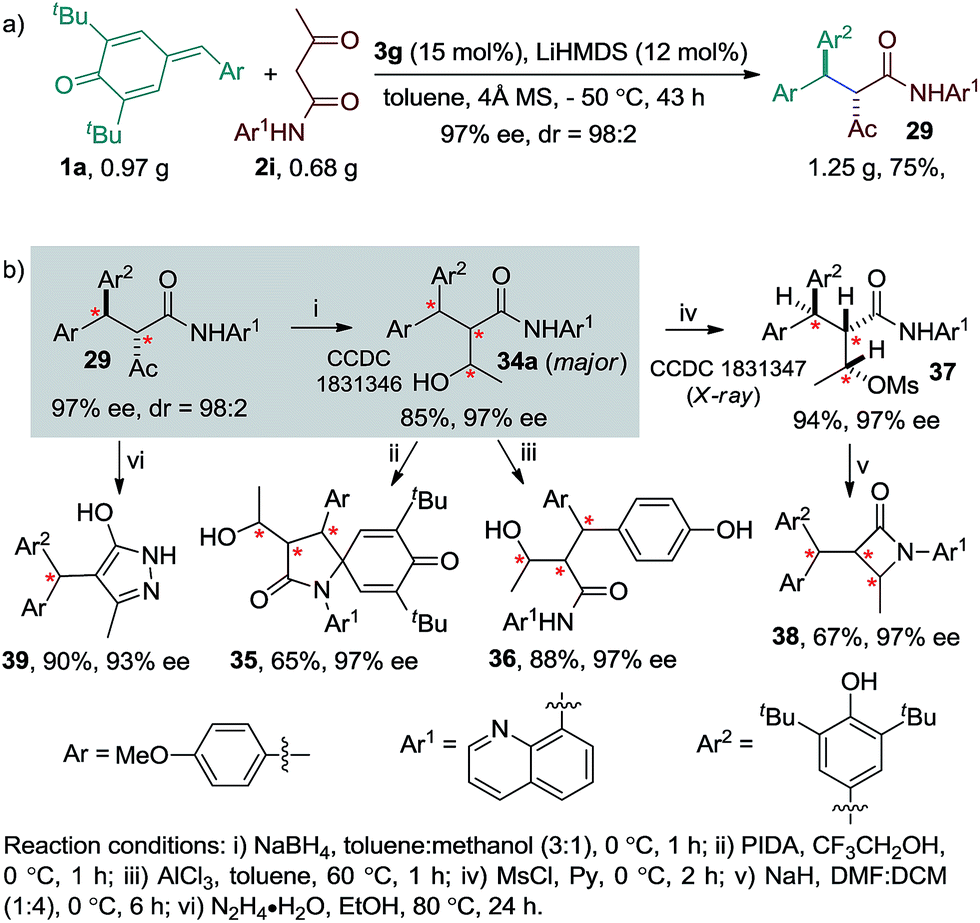 | ||
| Scheme 2 Synthetic applications. | ||
For a better understanding of the reaction mechanism, several other experiments were carried out (see the ESI†). It was observed that the reaction afforded the desired product with a similar reactivity and ee value using NaHMDS/KHMDS in lieu of LiHMDS as the base. These results indicate that the metal ion of the base may not have any considerable effect on the reaction outcome. Using a preformed NHC as the catalyst, the reaction afforded the desired product with an identical stereoselectivity albeit with a very low reactivity. The importance of the acidic N–H group of β-ketoamide was established by carrying out the reaction using N-methylated amide, which resulted in complete inhibition of the catalytic process (see the ESI†). Additionally, the in situ formation of the NHC under our reaction conditions was confirmed by performing the well-established oxidative annulation reaction between cinnamaldehyde and acetylacetone using precatalyst 3g (see the ESI†). This was further established by 1H NMR spectroscopy studies. It was found that the 1H NMR resonance at 10.76 ppm corresponding to the iminium C2–H of 3g disappeared when 3g was treated with LiHMDS. However, the 1H NMR signal reappeared upon adding an equimolar amount of the β-ketoamide 2h into the solution (see the ESI†). These observations may indicate that the initially formed NHC deprotonates the acidic N–H of 2h, thus forming a chiral ion-pair involving the azolium ion and enolate.
For mechanistic consideration, two possible reaction pathways are proposed for the reaction (Scheme 3). The reaction may involve hydrogen bonding interaction between the NHC and the β-ketoamide (Scheme 3, A). Alternatively, it may proceed through a chiral ion-pair intermediate consisting of the enolate and the azolium ion (Scheme 3, B). By reconciling the reported pKa values of the similar NHC and β-ketoamide with the results obtained in our preliminary mechanistic studies, we tend towards the ion-pair interaction between the NHC and the substrate. Finally, the activated nucleophile undergoes addition reaction with the p-QM to deliver the desired product with excellent stereocontrol. Further studies are surely required to establish the actual mode of NHC-catalysis for this reaction.
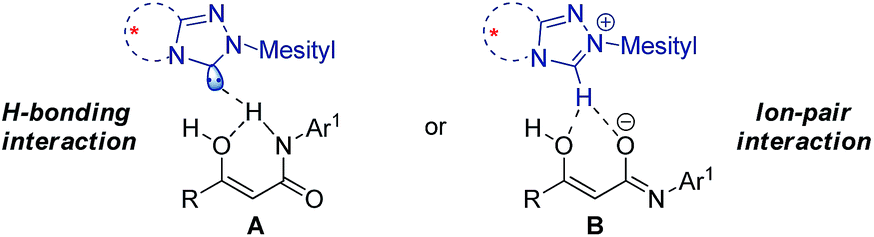 | ||
| Scheme 3 Proposed activation pathway. | ||
Based on the crystal structure analysis of product 11, the observed high stereoselectivity of the addition product is explained through the proposed transition state (TS) in which the Re-face of p-QM 1h approaches the chiral enolate due to hydrogen bonding activation and π–π interaction as shown in Fig. 1.
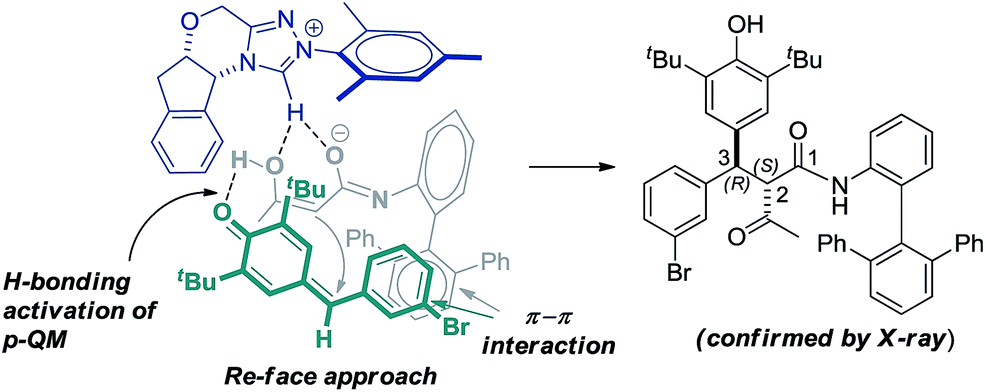 | ||
| Fig. 1 Probable transition state of the reaction. | ||
Conclusions
In conclusion, we have introduced a new mode of asymmetric NHC-catalysis through ion-pair interaction between a catalyst and a nucleophile. The novel catalytic method enables highly diastereo- and enantioselective 1,6-conjugate addition of 1,3-ketoamides to p-QMs using NHCs as Brønsted bases. The reaction furnishes the desired products with excellent stereoselectivity (ee and dr) and yields. Synthetic application of the method is demonstrated by the preparation of several valuable materials with excellent stereoselectivity.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the generous financial support from the Science and Engineering Research Board, India (EMR/2016/006344) and CSIR for fellowship to SS and AP.Notes and references
- For some selected reviews on asymmetric NHC-catalysis with covalent interaction, see: (a) M. Zhao, Y.-T. Zhang, J. Chen and L. Zhou, Asian J. Org. Chem., 2018, 7, 54 CrossRef; (b) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912 CrossRef PubMed; (c) S. R. Yetra, A. Patra and A. T. Biju, Synthesis, 2015, 47, 1357 CrossRef; (d) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC; (e) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef PubMed; (f) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef PubMed; (g) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef PubMed; (h) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC; (i) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664 CrossRef PubMed; (j) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef PubMed; (k) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef PubMed.
- (a) J. Chen and Y. Huang, Sci. China: Chem., 2016, 59, 251 CrossRef; (b) L. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 15414 CrossRef PubMed; (c) J. Chen, S. Meng, L. Wang, H. Tang and Y. Huang, Chem. Sci., 2015, 6, 4184 RSC; (d) J. Chen and Y. Huang, Nat. Commun., 2014, 5, 3437 CrossRef PubMed.
- (a) R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421 CrossRef PubMed; (b) T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366 CrossRef PubMed; (c) Y.-J. Kim and A. Streitwieser, J. Am. Chem. Soc., 2002, 124, 5757 CrossRef PubMed.
- (a) F. Perez, Y. Ren, T. Boddaert, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2015, 80, 1092 CrossRef PubMed; (b) Q. Kang and Y. Zhang, Org. Biomol. Chem., 2011, 9, 6715 RSC; (c) T. Boddaert, Y. Coquerel and J. Rodriguez, Chem.–Eur. J., 2011, 17, 2266 CrossRef PubMed; (d) E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179 CrossRef PubMed; (e) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190 CrossRef PubMed; (f) M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453 CrossRef PubMed; (g) J. A. Cowan, J. A. C. Clyburne, M. G. Davidson, R. L. W. Harris, J. A. K. Howard, P. Küpper, M. A. Leech and S. P. Richards, Angew. Chem., Int. Ed., 2002, 41, 1432 CrossRef.
- (a) R. Jansen, K. Gerth, H. Steinmetz, S. Reinecke, W. Kessler, A. Kirschning and R. Müller, Chem.–Eur. J., 2011, 17, 7739 CrossRef PubMed; (b) H. J. Martin, T. Magauer and J. Mulzer, Angew. Chem., Int. Ed., 2010, 49, 5614 CrossRef PubMed.
- (a) C. Sridar, J. D'Agostino and P. F. Hollenberg, Drug Metab. Dispos., 2012, 40, 2280 CrossRef PubMed; (b) R. Dehn, Y. Katsuyama, A. Weber, K. Gerth, R. Jansen, H. Steinmetz, G. Höfle, R. Müller and A. Kirschning, Angew. Chem., Int. Ed., 2011, 50, 3882 CrossRef PubMed.
- For some recent examples on asymmetric organocatalytic 1,6-addition of p-QM, see: (a) W. Li, X. Xu, Y. Liu, H. Gao, Y. Cheng and P. Li, Org. Lett., 2018, 20, 1142 CrossRef PubMed; (b) T.-C. Kang, L.-P. Wu, Q.-W. Yu and X.-Y. Wu, Chem.–Eur. J., 2017, 23, 6509 CrossRef PubMed; (c) M. Chen and J. Sun, Angew. Chem., Int. Ed., 2017, 56, 4583 CrossRef PubMed; (d) K. Zhao, Y. Zhi, A. Wang and D. Enders, ACS Catal., 2016, 6, 657 CrossRef; (e) Y. F. Wong, Z. Wang and J. Sun, Org. Biomol. Chem., 2016, 14, 5751 RSC; (f) X. Li, X. Xu, W. Wei, A. Lin and H. Yao, Org. Lett., 2016, 18, 428 CrossRef PubMed; (g) L. Ge, X. Lu, C. Cheng, J. Chen, W. Cao, X. Wu and G. Zhao, J. Org. Chem., 2016, 81, 9315 CrossRef PubMed; (h) N. Dong, Z.-P. Zhang, X.-S. Xue, X. Li and J.-P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1460 CrossRef PubMed; (i) L. Caruana, F. Kniep, T. K. Johansen, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2014, 136, 15929 CrossRef PubMed; (j) W.-D. Chu, L.-F. Zhang, X. Bao, X.-H. Zhao, C. Zeng, J.-Y. Du, G.-B. Zhang, F.-X. Wang, X.-Y. Ma and C.-A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9229 CrossRef PubMed.
- (a) S. Santra, A. Porey and J. Guin, Asian J. Org. Chem., 2018, 7, 477 CrossRef; (b) P. Arde and R. V. Anand, RSC Adv., 2016, 6, 77111 RSC.
- (a) M. d. M. Sanchez Duque, O. Baslé, N. Isambert, A. Gaudel-Siri, Y. Génisson, J.-C. Plaquevent, J. Rodriguez and T. Constantieux, Org. Lett., 2011, 13, 3296 CrossRef PubMed; (b) J. W. Bunting, J. P. Kanter, R. Nelander and Z. Wu, Can. J. Chem., 1995, 73, 1305 CrossRef.
- For some selected reviews on asymmetric ion-pair organocatalysis, see: (a) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2017, 117, 10608 CrossRef PubMed; (b) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef PubMed; (c) S. Shirakawa, S. Liu, S. Kaneko, Y. Kumatabara, A. Fukuda, Y. Omagari and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 15767 CrossRef PubMed; (d) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388 CrossRef PubMed; (e) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef PubMed; (f) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef PubMed; (g) M. Terada, Synthesis, 2010, 1929 CrossRef; (h) D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120 CrossRef PubMed; (i) C. Palomo, M. Oiarbide and R. Lopez, Chem. Soc. Rev., 2009, 38, 632 RSC; (j) J. Lacour and D. Moraleda, Chem. Commun., 2009, 7073 RSC; (k) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef PubMed.
- The addition of a catalytic amount of a proton shuttle like HFIP was essential for the reported hydrogen bonding asymmetric NHC-catalysis (see ref. 2). In contrast, the addition of HFIP was found to be detrimental in this study. This may indicate that the reaction involves an alternative mode of asymmetric NHC-catalysis via ion-pair interaction.
- CCDC 1831345, 1831346 and 1831347 contain the supplementary crystallographic data for 11, 34a and 37, respectively (see also the ESI†).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831345–1831347. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02138d |
| This journal is © The Royal Society of Chemistry 2018 |