
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chemically induced proximity system engineered from the plant auxin signaling pathway†
Weiye
Zhao
,
Huong
Nguyen
,
Guihua
Zeng
,
Dan
Gao
,
Hao
Yan
and
Fu-Sen
Liang
*
Department of Chemistry and Chemical Biology, University of New Mexico, 300 Terrace Street NE, Albuquerque, New Mexico 87131, USA. E-mail: fsliang@unm.edu
First published on 12th June 2018
Abstract
Methods based on chemically induced proximity (CIP) serve as powerful tools to control cellular processes in a temporally specific manner. To expand the repertoire of CIP systems available for studies of cellular processes, we engineered the plant auxin signaling pathway to create a new indole-3-acetic acid (IAA) based CIP method. Auxin-induced protein degradation that occurs in the natural pathway was eliminated in the system. The new IAA based method is both readily inducible and reversible, and used to control the production of therapeutic proteins that induced the apoptosis of cancer cells. The approach is also orthogonal to existing CIP systems and used to construct a biological Boolean logic gate controlling gene expression system. We believe that the new CIP method will be applicable to the artificial control and dissection of complex cellular functions.
Introduction
Controlling the proximity between biological molecules is a key regulatory strategy used by nature. Chemically induced proximity (CIP), or chemically induced dimerization (CID), methods have been proven to be powerful strategies in the design of novel approaches to gain artificial control of protein–protein interactions in a temporally specific manner. As such, these methods have been widely applied in biological research and therapeutic applications. In the CIP method, a small molecule inducer promotes heterodimerization between two inducer-binding proteins that are individually fused to proteins of interest (POIs). The binding process in turn triggers specific downstream biological events.1–6Several CIP methods have been developed, which utilize either natural or synthetic ligands as inducers.1–18 Each system has unique characteristics in dosage dependency and dimerization/disassociation kinetics, which has to be considered to design optimal CIP-controlled studies and applications. Furthermore, the combinatorial use of orthogonal CIP systems enables independent and simultaneous interrogations of multiple biological pathways in complex cellular environments. However, the available number of CIP systems is still limited, which impedes the broader application of CIPs.
To expand the CIP toolkit, we developed a new indole-3-acetic acid (IAA) based CIP system by engineering the plant auxin signaling pathway in the study described below. We demonstrated that the system is readily inducible, reversible and dosage dependent, and that it is orthogonal to existing CIPs, thus enabling its use with other CIPs in constructing a biological “AND” Boolean logic gate to control gene expression. We also used this new CIP system to control the production of an anticancer therapeutic protein, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which induced the apoptosis of cancer cells. This proof-of-principle study showed the potential utility of this method in therapeutic applications.
Phytohormone auxins (e.g. indole-3-acetic acid (IAA)) are a class of critical hormones that control the growth and development of plants.19 In this signaling pathway, auxins induce heterodimerization between F-box protein transport inhibitor response 1 (TIR1) and transcriptional corepressor auxin/indole-3-acetic acid (AUX/IAA) proteins (termed AIDs).19 Meanwhile, TIR1 recruits the Skp–Cullin–F-box containing protein complex (SCF complex), which leads to the ubiquitination of AIDs and their subsequent degradation by the proteasome.20,21 Novel auxin-inducible protein degradation systems have been developed, which rely on the use of auxin to control the level of AID-fused POIs temporally in both yeast and mammalian cells.22–24 However, for the utilization of this pathway in a new CIP method, the degradation of POIs following their heterodimerization is not desirable. We reasoned that an auxin (i.e. IAA) based CIP system that triggers biological effects and not protein degradation could be created by disrupting the interaction between TIR1 and the SCF complex while retaining the auxin-induced dimerization capability between TIR1 and AID (Fig. 1).
 | ||
| Fig. 1 Engineering the auxin pathway for the development of a chemically induced proximity method. By engineering TIR1 to disrupt the association of TIR1 with a SCF protein complex, the auxin-induced protein degradation capability is abolished while the auxin-induced dimerization of TIR1 and AID remains unaffected. | ||
Results and discussion
Recent studies have shown that the N-terminal H1 helix of Arabidopsis TIR1 is responsible for binding to the Cullin1 (CUL1) subunit of the SCF complex.25 It has also been reported that the E12 and E15 residues of Arabidopsis TIR1 are critical for such interaction, as reflected in the fact that the E12K/E15K double mutation disrupts TIR1-SCF binding.25 It is known that Arabidopsis TIR1 does not possess thermal stability under the physiological conditions in mammalian systems. As a result, Oryza sativa TIR1 (osTIR1) is utilized in place of auxin-induced protein degradation in mammalian cells.22Based on previous studies with Arabidopsis TIR1, we hypothesized that the interaction between osTIR1 and the SCF complex could be abolished by truncating or mutating the N-terminal regions of osTIR1. To gain information about this issue, a sequence alignment was carried out between Arabidopsis TIR1 and osTIR1 (Fig. S1†). The results suggest that, like E12 and E15 in Arabidopsis TIR1, E7 and E10 of osTIR1 should be critical for binding to the SCF complex and that the E7K/E10K double mutant of osTIR1 should not bind to the SCF complex. In addition to point mutations, we anticipated that the removal of the N-terminal region of osTIR1 would be another possible approach to block osTIR1/SCF binding. Although the structure of Arabidopsis TIR1 is known,26 that of osTIR1 is not. To identify possible critical regions on osTIR1 that are responsible for binding to CUL1 of the SCF complex, computational analysis using the Phyre2 protein fold recognition program27 was carried out to generate the three dimensional structure of osTIR1 based on that reported for Arabidopsis TIR1 (ref. 26) (Fig. S2†). The inspection of the modeled structure of osTIR1 enabled the identification of the first three α-helixes and the subsequent β-sheet of osTIR1 from its N-terminus as potential regions for truncation to interrupt binding to the SCF complex.
To test our hypothesis, we prepared the E7K/E10K osTIR1 mutant (TIR1*) and different truncated versions of osTIR1 in which the first α-helix (TIR1-1a), the first two (TIR1-2a) and first three α-helixes (TIR1-3a), and the first three α-helixes plus the following β-sheet secondary structure (TIR1-4b) are removed from the N-terminus (Fig. 2a). We then assessed whether the mutated or truncated osTIR1 maintains IAA-induced dimerization capability towards AID but loses the ability for SCF recruiting and AID degradation. For this purpose, the VP16 activation domain (VP16AD) was fused to wild type Arabidopsis AID (i.e. the IAA/AUX17 protein), and the GAL4 DNA binding domain (GAL4DBD) was fused to the wild type or different mutated or truncated osTIR1s to construct an IAA inducible split transcriptional activator that can activate an inducible EGFP reporter10 (Fig. 2b, S3†). The expression of EGFP upon IAA induction would indicate the existence of a CIP compatible version of engineered osTIR1 that responds to IAA and heterodimerizes with AID to activate gene expression while not degrading the AID-fused protein. To determine which of the engineered osTIR1 fusion proteins has this desired property, HEK293T cells were transfected with the constructed DNA plasmids (including VP16AD-AID, different GAL4DBD-TIR1s, and the EGFP reporter) for 24 h followed by treatment with 500 μM IAA22,23 for another 24 h. EGFP expression was monitored utilizing fluorescence microscopy. The results show that only the E7K/E10K mutant, TIR1*, but not the wild type or any truncated osTIR1 induces EGFP expression (Fig. 2c and S4†). Consequently, TIR1* should be an ideal IAA-inducible heterodimerization domain for CIP.
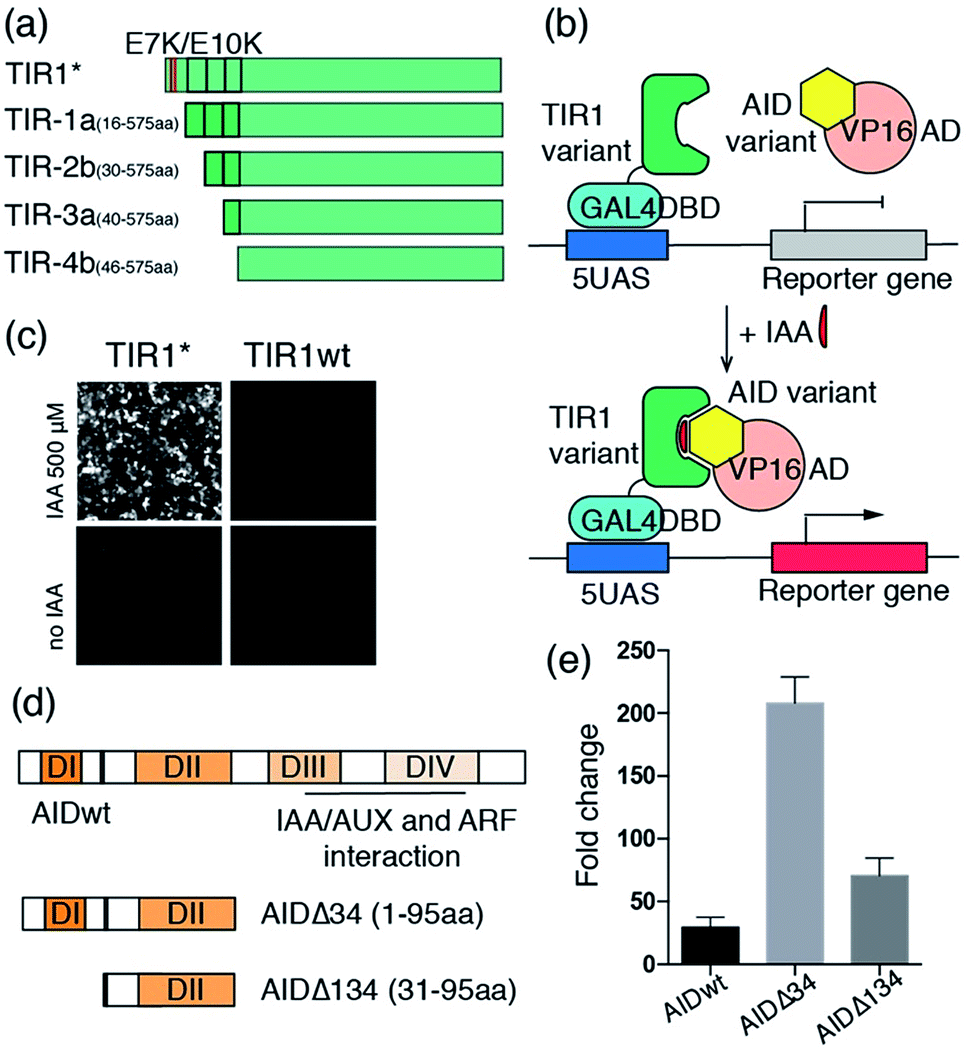 | ||
| Fig. 2 Engineering osTIR1 and Arabidopsis AID for CIP applications. (a) Point mutations and truncations of osTIR1. (b) The IAA inducible reporter assay. (c) IAA-induced EGFP expression using wild type or mutant osTIR1. (d) Wild type and truncations of AID. (e) IAA-induced luciferase expression. Fold change of induced luciferase expression calculated based on DMSO treated samples. Error bars represent ±s.e.m. from independent cell assays (n = 3). | ||
Our efforts next focused on optimizing the AID protein for CIP applications. Wild type Arabidopsis AID used in the initial studies described above is comprised of four functional domains (Fig. 2d).19 Domain II contains a conserved motif responsible for auxin-mediated TIR1 binding in Arabidopsis.19 Because domains III and IV are not involved in TIR1 binding but instead interact with the auxin response factor (ARF) proteins for transcriptional regulation,19 we reasoned that their removal would not affect dimerization capability. Moreover, the reduced size of the resulting truncated AID should minimize perturbations on fused POIs and also eliminate potential undesired side-interactions with other endogenous proteins. To this end, we prepared two truncated versions of AID in which either domains III and IV (AIDΔ34) or domains I, III and IV (AIDΔ134) are deleted (Fig. 2d), and constructed plasmids encoding VP16AD-AIDΔ34 and VP16AD-AIDΔ134 (Fig. S3†). After transfecting CHO cells with DNA plasmids encoding the GAL4DBD-TIR1*, VP16AD-AIDs (full-length or truncated) and an inducible luciferase reporter10 for 24 h, the cells were treated with 500 μM IAA for another 24 h, and then harvested and subjected to luciferase assays. The results demonstrate that cells with both truncated versions of AID produce higher induction fold changes when compared to those with full-length AID (Fig. 2e), especially when AIDΔ34 is used.
After identifying TIR1* and AIDΔ34 as optimal IAA-responsive dimerization domains, we characterized the properties of the new CIP method. To test the minimal working concentration of IAA and the dosage dependence, the IAA inducible luciferase expression in CHO cells using the split activator containing TIR1* and AID34 was utilized. The study of the effect of IAA concentration in the range of 0.1 μM to 1 mM showed that the treatment of transfected cells (for 24 h) with a minimal of 10 μM IAA is required to induce luciferase expression and maximum induction is reached at 250 μM (Fig. 3a). An IAA-dependent dosage response was also observed (Fig. 3a). To determine whether the high concentrations of I AA used in these assays are toxic to the cells, we conducted cell viability assays using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).28 No cytotoxicity was observed when treating cells with IAA ranging from 50 to 250 μM (Fig. S5†), which is consistent with previous reports.22,23
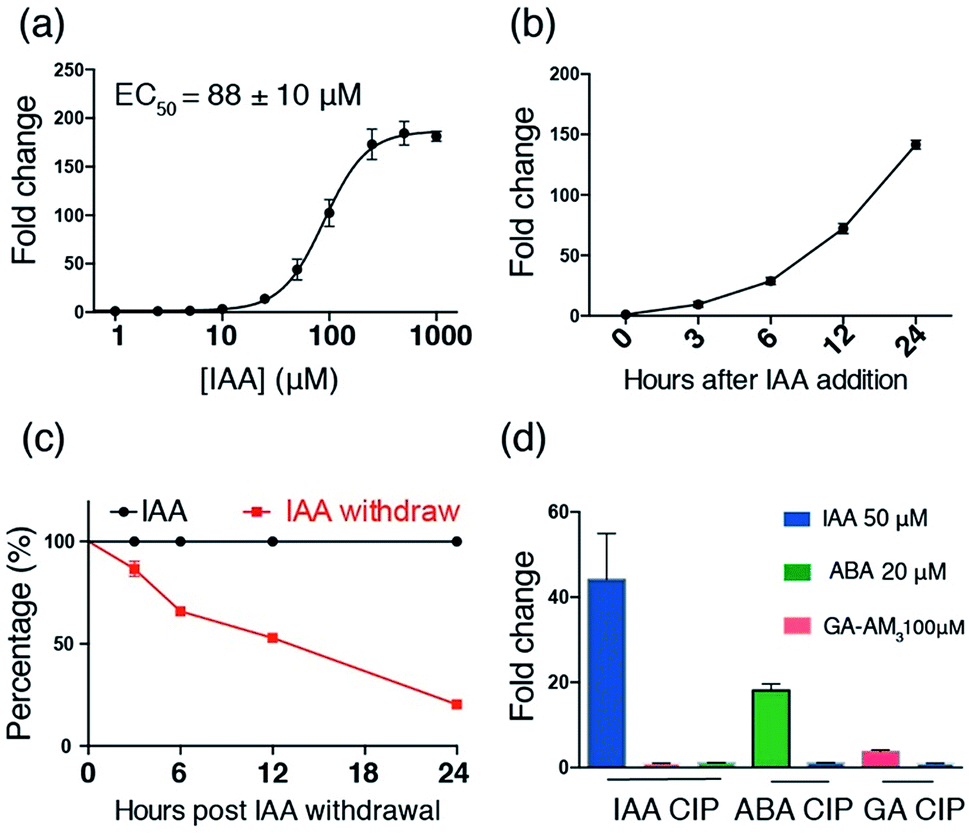 | ||
| Fig. 3 Characterization of the IAA CIP system using inducible luciferase expression. (a) Dosage response of IAA-induced luciferase expression (at 24 h). (b) Time course of IAA (250 μM) induced luciferase expression. (c) Time course of luciferase activity upon IAA withdrawal after induction for 24 h. (d) Orthogonality of IAA and GA-based CIP systems. Error bars represent ±s.e.m. from independent cell assays (n = 3). | ||
It was reported earlier that the D170E/M473L double mutation of Arabidopsis TIR1 facilitates TIR1-AID binding.29 To determine whether these mutations enhance IAA-induced heterodimerization between TIR1* and AIDΔ34, a sequence alignment was first performed, which led to the identification of D165 and M468 as the correspondingly located amino acid residues in osTIR1 (Fig. S6†). Another osTIR1, TIR1**, containing D165E/M468L mutations in addition to those in TIR1* was prepared. We also cloned a new GAL4DBD-TIR1** DNA construct for use with the VP16AD-AIDΔ34 construct (Fig. S3†). Tests of dosage responses of the TIR1** containing construct showed that surprisingly the D165E/M468L mutations significantly reduce the efficiency of IAA induced dimerization (minimal responsive dose of 250 μM and lower induction fold changes) (Fig. S7†).
In the studies described above, the POIs, GAL4DBD and VP16AD, are fused to the N-termini of TIR1* and AIDΔ34. To assess the effects of fusing POIs to different ends of TIR1* and AIDΔ34 on heterodimerization efficiencies, additional plasmids encoding proteins with POIs fused to the C-termini of TIR1* and AIDΔ34 (i.e. TIR1*-GAL4DBD and AIDΔ34-VP16AD, Fig. S3†) were prepared and subjected to IAA inducible luciferase assays. The results show that while induced luciferase expression is not dependent on which terminus of TIR1* GAL4DBD is fused to, expression is significantly reduced when VP16AD is fused to the C-terminus of AIDΔ34 (Fig. S8†). This finding suggests that TIR1*-binding of AIDΔ34 is sensitive to how POI is tagged and that the N-terminus of AID34 is less susceptible to POI tagging. The observations indicate that optimization might be required when constructing POI-AIDΔ34.
The induction rate and the reversibility of the new CIP system were determined next by carrying out luciferase assays on transfected CHO cells treated with 250 μM of IAA for varied time periods within 24 h. The results show that IAA rapidly induces luciferase expression within 3 h (Fig. 3b). To compare the induction rate of IAA to that of existing CIPs including abscisic acid (ABA) and rapamycin (Rap) systems, we transfected CHO cells with plasmids encoding IAA, ABA or a Rap-responsive split transcriptional activator10 and an inducible luciferase reporter and treated the cells with each corresponding CIP inducer for short time periods within 3 h. We observed that the induction rate of IAA is faster than that of ABA and Rap (Fig. S9†), indicating a rapid “on” rate of dimerization. In addition, the results of experiments, in which the transfected cells were treated with 250 μM of IAA for 24 h followed by washing with fresh media without IAA, show that IAA-induced luciferase expression is readily reversible at a rate comparable to that of the ABA system, which is reported to have fast on and off rates in dimerization (Fig. 3c, S10†).10,30
To assess the independence and orthogonality of the IAA- and other established induced proximity systems, we tested IAA CIP versus other plant hormone based CIPs (i.e. ABA and gibberellic acid (GA) systems).11 CHO cells were transfected with the GAL4DBD-TIR1*/VP16AD-AIDΔ34 (IAA responsive) constructs along with the inducible luciferase reporter for 24 h. Cells were then treated with either IAA (50 μM), ABA (20 μM) or GA-AM3 (ref. 11) (100 μM) for another 24 h before the cells were harvested and subjected to luciferase assays. The results show that luciferase expression is only induced in IAA treated cells (Fig. 3d). In separate experiments, cells were transfected with either the GAL4DBD-ABI/VP16AD-PYL10 (ABA responsive) or newly cloned GAL4DBD-GAI/VP16AD-GID1 (GA responsive) constructs (Fig. S3†) along with the inducible luciferase reporter for 24 h. The transfected cells were then treated with either IAA or ABA, or IAA or GA-AM3 for 24 h before being harvested for assays. The results show that IAA does not induce luciferase expression in either the ABA or GA-based systems (Fig. 3d). In addition, when similar experiments were carried out using plasmids encoding mismatched dimerizable protein pairs (i.e. VP16AD-PYL with GAL4DBD-TIR1* or VP16AD-GAI with GAL4DBD-TIR1*), no luciferase expression was induced when IAA, GA or ABA was added (Fig. S11†). These results further confirm the orthogonality of IAA to other CIP systems.
To demonstrate the viability of the combinatorial use of IAA and other orthogonal CIP systems in the cells, we constructed an IAA and ABA-dependent “AND” Boolean logic gate system, which activates luciferase expression only when both IAA and ABA are present (Fig. 4a). For this purpose, plasmids encoding fusion proteins of VP16AD-PYL, ABI-AIDΔ34 and GAL4DBD-TIR1* (Fig. S3†) were prepared and transfected (along with the luciferase reporter) into HEK293T cells for 24 h. The transfected cells, treated with no inducers, only IAA (250 μM) or both IAA (250 μM) and ABA (50 μM) for 24 h, were harvested and subjected to luciferase assays. Luciferase expression was found to occur only in cells to which both IAA and ABA were added, but not in those treated with only IAA or ABA (Fig. 4b).
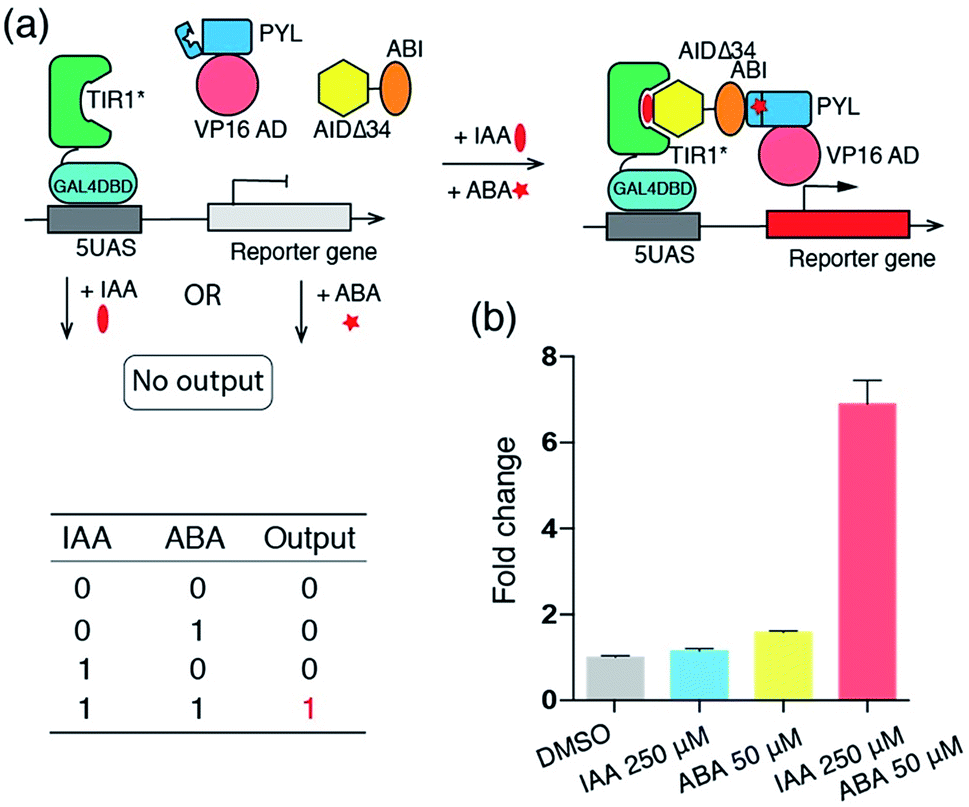 | ||
| Fig. 4 (a) Biological “AND” logic gate based on the IAA and ABA CIP systems. (b) Induced luciferase expression output of the “AND” logic gate. Fold changes of induced luciferase expression were calculated based on DMSO treated samples. Error bars represent ±s.e.m. from replicates (n = 4). | ||
To explore the possibility of applying the IAA inducible system in a therapeutic setting, we tested the effectiveness of using IAA to induce the production of a therapeutic protein TRAIL, which has been used to induce apoptosis in a wide range of cancer cells bearing death receptors while not affecting non-cancer cells.31–33 We made a DNA construct that can be induced to produce secretable TRAIL (sTRAIL) (Fig. S3†). We co-transfected HEK293T cells with this plasmid and the ones encoding the IAA-responsive split transcriptional activator. After 24 h treatment with IAA (or DMSO as a control), the secreted sTRAIL in the cell culture supernatant was quantified by the enzyme-linked immunosorbent assay (ELISA). Only when transfected cells were treated with IAA, we observed a high yield of sTRAIL production, which is comparable to that of previously reported systems (Fig. 5a).34,35 To confirm that the produced sTRAIL is biologically active, we treated MDA-MD-231 breast cancer cells with the sTRAIL-containing supernatant or under other control conditions for 20 h. The viability and apoptosis of MDA-MD-231 cells were then analyzed using flow cytometry. We observed that IAA-induced sTRAIL effectively led to the apoptosis of cancer cells, while treating MDA-MD-231 cells with IAA alone, or with the supernatant from transfected HEK293T cells treated with DMSO, did not show significant effects (Fig. 5b). These results demonstrate the potential of using the IAA system to control the production of anticancer agents for therapeutic applications.
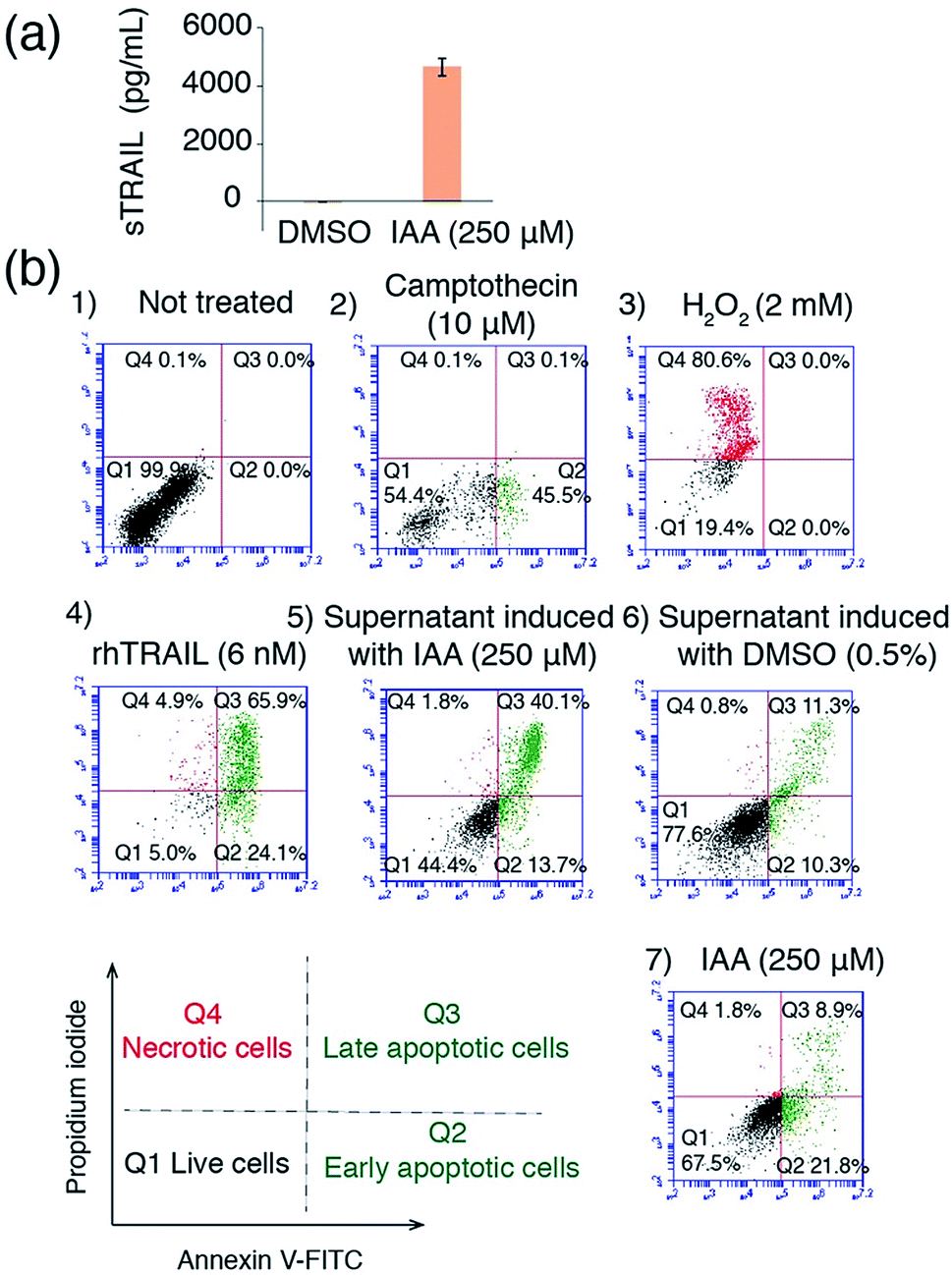 | ||
| Fig. 5 Applying the IAA CIP system to control sTRAIL production and induce apoptosis. (a) The quantification of sTRAIL produced from HEK293T cells using ELISA. Error bars represent ±s.e.m. from replicates (n = 4). (b) Apoptosis and death of MDA-MB-231 cells analyzed by flow cytometry. MDA-MB-231 cells were (1) not treated or treated with (2) camptothecin (inducing apoptosis), (3) H2O2 (inducing necrosis), (4) purified recombinant human TRAIL (rhTRAIL), (5) supernatant from transfected HEK293T cells treated with IAA (containing sTRAIL), (6) supernatant from transfected cells treated with DMSO (no sTRAIL) or (7) IAA only, and then labelled with annexin V-FITC (labeling apoptotic cells, in green) and propidium iodide (labeling necrotic cells, in red). | ||
Conclusions
In the effort described above, we successfully engineered a new IAA CIP system with optimized osTIR1 (i.e. TIR1*) and Arabidopsis AID (i.e. AIDΔ34). The new CIP system rapidly responds to IAA in a dosage-dependent and reversible manner, and it is orthogonal to other plant hormone-based CIPs. It can be used to control the production of the anticancer agent TRAIL in high yield and induce the apoptosis of cancer cells. Several studies have shown that caged CIP inducers can be developed to become activatable by different artificial or cellular signals (e.g. light, H2O2, and Fe2+).17,30,36–43 We expect that similar caging strategies can be extended to IAA as IAA caged by photo-labile groups has been used to introduce light control in the auxin regulated plant phenotype.44,45 This new method will expand the powerful tool kit of CIP technology and should find applications in synthetic biology and the development of novel gene and cell therapies.46–50Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health R21 HG008776.Notes and references
- J. E. Gestwicki and P. S. Marinec, Comb. Chem. High Throughput Screening, 2007, 10, 667–675 CrossRef PubMed.
- A. Fegan, B. White, J. C. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315–3336 CrossRef PubMed.
- S. S. Shekhawat and I. Ghosh, Curr. Opin. Chem. Biol., 2011, 15, 789–797 CrossRef PubMed.
- R. DeRose, T. Miyamoto and T. Inoue, Pflügers Archiv: European Journal of Physiology, 2013, 465, 409–417 CrossRef PubMed.
- S. Voß, L. Klewer and Y.-W. Wu, Curr. Opin. Chem. Biol., 2015, 28, 194–201 CrossRef PubMed.
- B. Z. Stanton, E. J. Chory and G. R. Crabtree, Science, 2018, 3, 359 Search PubMed.
- D. M. Spencer, T. J. Wandless, S. L. Schreiber and G. R. Crabtree, Science, 1993, 262, 1019–1024 CrossRef PubMed.
- H. Lin, W. M. Abida, R. T. Sauer and V. W. Cornish, J. Am. Chem. Soc., 2000, 122, 4247–4248 CrossRef.
- J. H. Bayle, J. S. Grimley, K. Stankunas, J. E. Gestwicki, T. J. Wandless and G. R. Crabtree, Chem. Biol., 2006, 13, 99–107 CrossRef PubMed.
- F.-S. Liang, W. Q. Ho and G. R. Crabtree, Sci. Signaling, 2011, 4, rs2 CrossRef PubMed.
- T. Miyamoto, R. DeRose, A. Suarez, T. Ueno, M. Chen, T. P. Sun, M. J. Wolfgang, C. Mukherjee, D. J. Meyers and T. Inoue, Nat. Chem. Biol., 2012, 8, 465–470 CrossRef PubMed.
- P. Mateusz and S. Carsten, FEBS Lett., 2012, 586, 2097–2105 CrossRef PubMed.
- M. Skwarczynska, M. Molzan and C. Ottmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E377–E386 CrossRef PubMed.
- D. Erhart, M. Zimmermann, O. Jacques, M. B. Wittwer, B. Ernst, E. Constable, M. Zvelebil, F. Beaufils and M. P. Wymann, Chem. Biol., 2013, 20, 549–557 CrossRef PubMed.
- S. Feng, V. Laketa, F. Stein, A. Rutkowska, A. MacNamara, S. Depner, U. Klingmüller, J. Saez-Rodriguez and C. Schultz, Angew. Chem., Int. Ed., 2014, 53, 6720–6723 CrossRef PubMed.
- P. Liu, A. Calderon, G. Konstantinidis, J. Hou, S. Voss, X. Chen, F. Li, S. Banerjee, J. E. Hoffmann, C. Theiss, L. Dehmelt and Y. W. Wu, Angew. Chem., Int. Ed., 2014, 53, 10049–10055 CrossRef PubMed.
- E. R. Ballister, C. Aonbangkhen, A. M. Mayo, M. A. Lampson and D. M. Chenoweth, Nat. Commun., 2014, 5, 5475 CrossRef PubMed.
- Z. B. Hill, A. J. Martinko, D. P. Nguyen and J. A. Wells, Nat. Chem. Biol., 2017, 14, 112 CrossRef PubMed.
- D. Weijers and D. Wagner, Annu. Rev. Plant Biol., 2016, 67, 539–574 CrossRef PubMed.
- S. Kepinski and O. Leyser, Nature, 2005, 435, 446–451 CrossRef PubMed.
- N. Dharmasiri, S. Dharmasiri and M. Estelle, Nature, 2005, 435, 441–445 CrossRef PubMed.
- K. Nishimura, T. Fukagawa, H. Takisawa, T. Kakimoto and M. Kanemaki, Nat. Methods, 2009, 6, 917–922 CrossRef PubMed.
- A. J. Holland, D. Fachinetti, J. S. Han and D. W. Cleveland, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3350–E3357 CrossRef PubMed.
- A. Khakhar, N. J. Bolten, J. Nemhauser and E. Klavins, ACS Synth. Biol., 2016, 5, 279–286 CrossRef PubMed.
- H. Yu, Y. Zhang, B. L. Moss, B. O. Bargmann, R. Wang, M. Prigge, J. L. Nemhauser and M. Estelle, Nat. Plants, 2015, 1, 14030 CrossRef PubMed.
- X. Tan, L. I. Calderon-Villalobos, M. Sharon, C. Zheng, C. V. Robinson, M. Estelle and N. Zheng, Nature, 2007, 446, 640–645 CrossRef PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef PubMed.
- H. Yu, B. L. Moss, S. S. Jang, M. Prigge, E. Klavins, J. L. Nemhauser and M. Estelle, Plant Physiol., 2013, 162, 295 CrossRef PubMed.
- C. W. Wright, Z. F. Guo and F. S. Liang, ChemBioChem, 2015, 16, 254–261 CrossRef PubMed.
- D. W. Stuckey and K. Shah, Trends Mol. Med., 2013, 19, 685–694 CrossRef PubMed.
- J. Lemke, S. von Karstedt, J. Zinngrebe and H. Walczak, Cell Death Differ., 2014, 21, 1350 CrossRef PubMed.
- A. Refaat, A. Abd-Rabou and A. Reda, Oncol. Lett., 2014, 7, 1327–1332 CrossRef PubMed.
- K. T. Roybal, J. Z. Williams, L. Morsut, L. J. Rupp, I. Kolinko, J. H. Choe, W. J. Walker, K. A. McNally and W. A. Lim, Cell, 2016, 167, 419–432 CrossRef PubMed.
- K. T. Roybal, L. J. Rupp, L. Morsut, W. J. Walker, K. A. McNally, J. S. Park and W. A. Lim, Cell, 2016, 164, 770–779 CrossRef PubMed.
- N. Umeda, T. Ueno, C. Pohlmeyer, T. Nagano and T. Inoue, J. Am. Chem. Soc., 2011, 133, 12–14 CrossRef PubMed.
- A. V. Karginov, Y. Zou, D. Shirvanyants, P. Kota, N. V. Dokholyan, D. D. Young, K. M. Hahn and A. Deiters, J. Am. Chem. Soc., 2011, 133, 420–423 CrossRef PubMed.
- K. M. Schelkle, T. Griesbaum, D. Ollech, S. Becht, T. Buckup, M. Hamburger and R. Wombacher, Angew. Chem., Int. Ed., 2015, 54, 2825–2829 CrossRef PubMed.
- K. A. Brown, Y. Zou, D. Shirvanyants, J. Zhang, S. Samanta, P. K. Mantravadi, N. V. Dokholyan and A. Deiters, Chem. Commun., 2015, 51, 5702–5705 RSC.
- G. Zeng, R. Zhang, W. Xuan, W. Wang and F. S. Liang, ACS Chem. Biol., 2015, 10, 1404–1410 CrossRef PubMed.
- G. Zeng, H. Li, Y. Wei, W. Xuan, R. Zhang, L. E. Breden, W. Wang and F. S. Liang, ACS Synth. Biol., 2017, 6, 921–927 CrossRef PubMed.
- X. Chen, M. Venkatachalapathy, D. Kamps, S. Weigel, R. Kumar, M. Orlich, R. Garrecht, M. Hirtz, M. Niemeyer Christof, Y. W. Wu and L. Dehmelt, Angew. Chem., Int. Ed., 2017, 56, 5916–5920 CrossRef PubMed.
- H. Zhang, C. Aonbangkhen, E. V. Tarasovetc, E. R. Ballister, D. M. Chenoweth and M. A. Lampson, Nat. Chem. Biol., 2017, 13, 1096 CrossRef PubMed.
- K. Hayashi, N. Kusaka, S. Yamasaki, Y. Zhao and H. Nozaki, Bioorg. Med. Chem. Lett., 2015, 25, 4464–4471 CrossRef PubMed.
- Q. Delacour, C. Li, M.-A. Plamont, E. Billon-Denis, I. Aujard, T. Le Saux, L. Jullien and A. Gautier, ACS Chem. Biol., 2015, 10, 1643–1647 CrossRef PubMed.
- C. J. Bashor, A. A. Horwitz, S. G. Peisajovich and W. A. Lim, Annu. Rev. Biophys., 2010, 39, 515–537 CrossRef PubMed.
- D. Aubel and M. Fussenegger, BioEssays, 2010, 32, 332–345 CrossRef PubMed.
- W. C. Ruder, T. Lu and J. J. Collins, Science, 2011, 333, 1248–1252 CrossRef PubMed.
- F. Lienert, J. J. Lohmueller, A. Garg and P. A. Silver, Nat. Rev. Mol. Cell Biol., 2014, 15, 95 CrossRef PubMed.
- C. Y. Wu, K. T. Roybal, E. M. Puchner, J. Onuffer and W. A. Lim, Science, 2015, 350, aab4077 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02353k |
| This journal is © The Royal Society of Chemistry 2018 |