
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling reaction networks behind the catalytic oxidation of methane with H2O2 over a mixed-metal MIL-53(Al,Fe) MOF catalyst†
Ágnes
Szécsényi
 ab,
Guanna
Li
ab,
Jorge
Gascon
c and
Evgeny A.
Pidko
*a
ab,
Guanna
Li
ab,
Jorge
Gascon
c and
Evgeny A.
Pidko
*a
aInorganic Systems Engineering Group, Chemical Engineering Department, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: e.a.pidko@tudelft.nl; Tel: +31 1527 81938
bCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, the Netherlands
cKing Abdullah University of Science and Technology, KAUST Catalysis Center, Advanced Catalytic Materials, Thuwal 23955, Saudi Arabia
First published on 20th July 2018
Abstract
Reaction paths underlying the catalytic oxidation of methane with H2O2 over an Fe containing MIL-53(Al) metal–organic framework were studied by periodic DFT calculations. Not only the activation of methane, but the full reaction network was considered, which includes the formation of the active site, the overoxidation of methane to CO2 and the decomposition of H2O2 to H2O and O2. Calculations indicate that the activation barrier for the initial activation of the Fe sites upon reaction with H2O2 is comparable to that of the subsequent C–H activation and also of the reaction steps involved in the undesirable overoxidation processes. The pronounced selectivity of the oxidation reaction over MIL-53(Al,Fe) towards the target mono-oxygenated CH3OH and CH3OOH products is attributed to the limited coordination freedom of the Fe species encapsulated in the extended octahedral [AlO6] structure-forming chains, which effectively prevents the direct overoxidation paths prior to product desorption from the active sites. Importantly, our computational analysis reveals that the active sites for the desired methane oxidation are able to much more efficiently promote the direct catalytic H2O2 decomposition reaction, rendering thus the current combination of the active site and the reactants undesirable for the prospective methane valorization process.
Introduction
The oxidation of methane to methanol is regarded a promising route for the on-site valorization of natural gas.1,2 Despite decades of research and technological attractiveness, the establishment of a catalytic route for the selective low-temperature oxidation of methane is still regarded as one of the grand challenges for catalysis sciences and technologies.3–5 The limited efficiency of the available approaches is directly related to the fundamental characteristics of both the substrate and the target product of the title catalytic reaction. Methane is a symmetric apolar molecule with a large HOMO–LUMO gap making it unsuitable for ordinary redox or acid–base chemistry. Therefore, a substantial energy input is required to activate its quite inert C–H bonds. On the other hand, the target product – methanol – contains much more reactive bonds making it more susceptible to further oxidation reactions. The fundamental challenge here is to find a catalyst that is able to cleave the C–H bonds of methane and promote its oxo-functionalization, but at the same time provide a mechanism for the fast release of the methanol product from the active site to ensure that it does not oxidise further to the undesirable carbon oxides that limit the overall selectivity and efficiency of the catalytic process.Previous studies have revealed that C–H bond activation is the rate-determining step of the catalytic methane oxidation.6–11 Therefore most of the research reported so far aimed at revealing the factors that facilitate this reaction step, and attempted to establish structure–activity relationships that would guide the development and optimization of the promising catalytic systems.12–16 Such factors as the radical character of the oxygen centre17,18 in the reactive metal-oxo active site or its hydrogen affinity12 were proposed as the suitable activity descriptors for the successful C–H activation catalyst.
Despite the impressive progress made in the last decade in understanding the fundamentals of C–H activation and revealing crucial structure–activity relationships for this pivotal reaction step, there is much less insight into the mechanistic factors that influence other steps of the catalytic methane oxo-functionalization cycle as well as the numerous competing reaction channels that do not determine the rate of the target methane activation but all contribute potentially to the decreased selectivity of the target process. In particular, the formation of the actual reaction centre capable of C–H cleavage and, the activation of the oxidant molecule are much less understood than the C–H dissociation step. Given the crucial role of these secondary processes in the overall efficiency of the catalytic methane oxidation, a systematic approach to the mechanistic analysis of the underlying reaction networks is highly desirable.
An inspiration to solving the selectivity problem in methane oxidation can be obtained from Nature. Methanotropic bacteria evolved to promote this reaction with a high efficiency.19 They utilize very effective and complex enzymatic systems, called methane monooxygenases (MMO) to ensure a high selectivity of the methane oxidation process. The natural systems prevent the overoxidation reactions by providing a steric hindrance to the CH3OH product formed at the MMO active site. The catalytic ensemble of MMOs contains Fe or Cu centres that activate molecular O2 to create the highly reactive oxygen species capable of cleaving the C–H bonds in methane. Indeed, O2 is the greenest, cheapest, most abundant and desirable oxidant for any industrial oxidation process. However the controlled utilization of O2 for a selective chemo-catalytic oxidation mediated by transition metal complexes is very challenging. Such an oxidation process is fundamentally hampered by (a) the need to transfer 4 electrons to the O2 molecule and (b) the need to change the spin configuration of the system from triplet to singlet along the reaction.20 That is why even the MMO systems show only a limited efficiency in terms of O2 utilization, from which only one oxygen atom is incorporated into the CH3OH product, while the other one is consumed to form H2O by stoichiometric co-reductants NADH and FADH2. These reactants supply electrons and protons necessary for the overall biological process to proceed under very mild conditions.
In chemo-catalysis, zeolites are often regarded as the synthetic mimics to enzymatic systems. The introduction of various metal species such as Fe,21–23 Cu,24,25 Co,26 Zn,27,28 and Ni29 into these microporous aluminosilicate matrices gives rise to reactive sites capable of selectively oxidizing methane30 with such oxidants as O2,24,29 H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and N2O.31,32 Besides the nature of the metal and the type of oxidant, the topology of the confinement matrix, the nuclearity of the intra-zeolite active sites and the oxidation state of the reactive metal centres were found to be important factors affecting the activity and selectivity of the zeolite-based oxidation catalysts.13,30,33 In the context of the chemo-catalytic conceptual mimic of the enzymatic process, the development of an integrated catalyst system employing different co-reductants including H2 to promote the activation of the O2 oxidant has been discussed in the literature.34 However, the performance of such integrated systems for methane oxofunctionalization fell short for industrial application. To circumvent the problem of simultaneous use of the stoichiometric oxidant and reductant for methane oxidation, it was proposed to split the partial reduction of O2 and the CH4 oxidation steps and utilize H2O2 as the partially reduced oxidant in the latter step.35,36 Such a process has been realized experimentally by Hutchings23et al. The authors developed a Cu–Fe/ZSM-5 zeolite catalyst capable of selectively converting methane to mono-oxygenated products in the presence of H2O2. Extensive characterization studies on this system revealed oxygen-containing extra-framework Fe species to be the active sites for the catalytic process, while the role of Cu in the system was attributed to the suppression of the undesirable free radical reaction paths.
23 and N2O.31,32 Besides the nature of the metal and the type of oxidant, the topology of the confinement matrix, the nuclearity of the intra-zeolite active sites and the oxidation state of the reactive metal centres were found to be important factors affecting the activity and selectivity of the zeolite-based oxidation catalysts.13,30,33 In the context of the chemo-catalytic conceptual mimic of the enzymatic process, the development of an integrated catalyst system employing different co-reductants including H2 to promote the activation of the O2 oxidant has been discussed in the literature.34 However, the performance of such integrated systems for methane oxofunctionalization fell short for industrial application. To circumvent the problem of simultaneous use of the stoichiometric oxidant and reductant for methane oxidation, it was proposed to split the partial reduction of O2 and the CH4 oxidation steps and utilize H2O2 as the partially reduced oxidant in the latter step.35,36 Such a process has been realized experimentally by Hutchings23et al. The authors developed a Cu–Fe/ZSM-5 zeolite catalyst capable of selectively converting methane to mono-oxygenated products in the presence of H2O2. Extensive characterization studies on this system revealed oxygen-containing extra-framework Fe species to be the active sites for the catalytic process, while the role of Cu in the system was attributed to the suppression of the undesirable free radical reaction paths.
The main limitations of transition metal-containing zeolite-based catalysts are the limited number of practical topologies suitable for acting as the stabilizing microporous matrices and the extra-framework nature of the deposited catalytic metal species.37 The latter factor inevitably causes a heterogeneous metal speciation in practical catalysts38–40 and the inherent flexibility of the coordination environment as the metal centres are bound to rather weak donor sites of the aluminosilicate zeolite lattice. Whereas the formation of well-defined metal species in zeolite pores can potentially be achieved through the optimization of the synthetic approaches,18,41 the coordination flexibility of the intra-zeolite complexes is the inherent property. In the context of methane oxidation catalysis, both these factors may contribute to the decreased selectivity of the overall reaction.
A promising alternative to the conventional pure inorganic zeolite-based catalysts is hybrid metal–organic frameworks (MOF) that are crystalline porous materials, which structures are made of metal ions or clusters connected by organic linkers.42 The well-defined nature of the inorganic nodes and their established coordination chemistry together with the great versatility of the organic linkers allow tailoring the structural, electronic and catalytic properties of MOFs towards a specific application.43–50
In particular, recently MOFs have emerged as promising catalyst platforms for selective methane oxidation. Lercher51et al. described a Zr-based MOF, NU-1000 containing copper oxide cluster synthesized via atomic layer deposition that is active for the selective oxidation of methane with O2 under mild reaction conditions. Very recently, we reported that the introduction of isolated Fe species into the well-defined inorganic structure of an Al-terephthalate-based MOF gives rise to a MIL-53(Fe,Al) mixed-metal catalyst showing a high activity and selectivity in the oxidation of methane to methanol with H2O2.52 The parent MIL-53(Al) microporous MOF matrix is made up of [AlO6] octahedral chains connected by 1,4-benzodicarboxylic acid struts to form well-defined 1D channels (Fig. 1). The catalytic function can be introduced into this material by creating well-dispersed Fe sites inside these inorganic structure-forming chains. The extensive characterization of the catalysts revealed the predominant speciation of the reactive Fe as dimeric (Fe2-MIL-53(Al)) and monomeric (Fe1-MIL-53(Al)) complexes isomorphously substituting Al ions in the MIL-53 crystalline lattice. Such Fe sites are placed in a unique octahedral weak ligand field environment that is expected to be favourable for the C–H bond activation in methane.53,54 The isolated nature of the Fe sites in MIL-53(Fe,Al) is crucial for the structural stability of the material towards the oxidizing aqueous environment of the catalytic reaction.
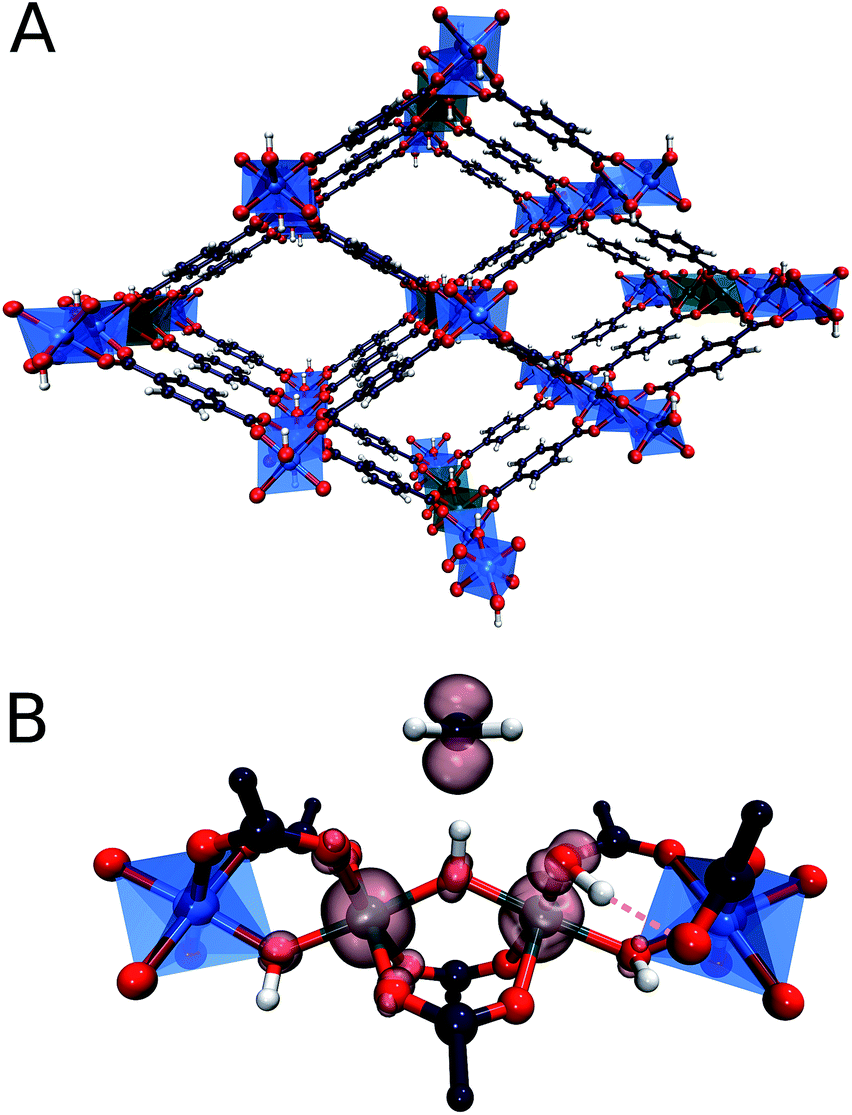 | ||
| Fig. 1 (A) Fe2-MIL-53(Al) metal organic framework, containing dimeric Fe species substituting Al ions in the Al–O chain. (B) Methyl radical formed upon C–H bond cleavage of methane by the bridging oxygen of the active site. Spin density is visualized at the isosurface of 0.1. | ||
The selectivity of the catalytic reaction did not exceed 85% due to overoxidation processes. Furthermore, the overall efficiency of the catalytic system towards methane oxidation was found to be limited by the competing direct H2O2 decomposition reaction to H2O and O2. Therefore, to improve the efficiency and selectivity of the methane oxidation process, a control over these competing secondary reaction channels is needed, which cannot be achieved without a detailed understanding of the underlying mechanistic characteristics.
Herein we present the results of a comprehensive computational analysis of the reaction paths behind methane oxidation with H2O2 over the MIL-53(Fe,Al) catalyst. The computational results presented here indicate promising routes for the optimization of the title selective oxidation process.
Computational details
The spin polarized periodic DFT calculations were carried out using the Vienna Ab Initio Simulation Package (VASP,55–58 version 5.3.5.). The PBE exchange-correlation functional,59,60 plane wave basis set with a cut-off energy of 400 eV and the projector augmented wave (PAW) method61,62 were applied. To account for the van der Waals interactions Grimme's dispersion correction with the Becke-Johnson damping (DFT-D3(BJ))63 method was used. A Gaussian smearing of the population of partial occupancies with a width of 0.05 eV was used during iterative diagonalization of the Kohn–Sham Hamiltonian. Brillouin zone sampling was restricted to the Gamma point.64 We have previously successfully employed this methodology for studying various zeolite-catalysed processes including chemical transformations in spin-polarized systems.14,18,38 Convergence was assumed to be reached when the force on each atom was below 0.04 eV Å−1. Geometry optimization was completed for all considered spin states.During the calculations, the supercell approach was applied, which means that the basic unit cell is multiplied by an integer, in this case 3 times to the x direction. The supercell catalyst model contained 228 atoms. Firstly, the lattice vectors of MIL-53(Al) were optimized, and they were obtained to be orthogonal: a = 20.197 Å, b = 17.808 Å, c = 12.216 Å. To obtain the models of Fe1-MIL-53(Al) and Fe2-MIL-53(Al) one or two neighbouring Al atoms were substituted with Fe respectively. The geometries of the catalyst models as well as all the related intermediates and transition states were fully optimized with the cell parameters fixed to the values determined for the parent Al-form of MIL-53.
To locate the transition state structures, the nudged elastic band method (NEB)65 was used. The maximum energy geometry along the reaction path generated by the NEB method was further optimized using a quasi-Newton algorithm. In this procedure, only the extra-framework atoms, and relevant framework atoms were relaxed. Vibrational frequencies were calculated using the finite difference method (0.02 Å atomic displacements) as implemented in VASP. The transition state showed a single imaginary frequency corresponding to the reaction path.
Dimeric Fe species substituting framework Al atoms were considered as the sites for methane oxidation. In line with the experimental results,52 DFT predicts the antiferromagnetically coupled high spin (AHS) state (S = 0) for the Fe pair to be the most stable one (Table S1 in the ESI†). The ferromagnetic high spin (HS) configuration (S = 5) is however only 10 kJ mol−1 higher in energy. Spin density analysis shows that the absolute value of spin assigned to each atom in the S = 0 state is similar to those in the S = 5 state (for details see Table S2 in the ESI†). The other intermediate spin states have a higher energy than the S = 5 state (Table S1 in the ESI†). In view of the difficulties in convergence of the antiferromagnetically coupled HS systems and given that according to Baerends66et al. a weak exchange coupling does not influence the chemical reactivity significantly, the reaction network analysis was further carried out over the HS potential energy surface. For further confirmation of the validity of this simplification we calculated the AHS state of several intermediates. The relative energies and spin densities are shown in Table S3 in the ESI.† In this work we report relative electronic energies for elementary steps and activation energies computed as the difference of electronic energy of the computed structures.
Images of structures were created using the VMD Software.67,68
Results and discussion
The different conversion paths and competing cycles for methane and H2O2 conversion over Fe-MIL-53(Al) considered in this study are summarized in Fig. 2. The oxidation cycles start with the catalyst activation (Fig. 3A) upon which the initial Fe(III)–μOH–Fe(III) site (1) is oxidized with H2O2 to form an Fe(IV)–μO–Fe(IV)–OH complex (3). This intermediate serves as the active site in all subsequent oxidation reactions (cycle I, Fig. 2) as well as in the competing H2O2 decomposition path (cycle II, Fig. 2). Cycle I depicts a sequential oxidation of methane to CO2 with the intermediate formation of methanol, formaldehyde, formic acid or carbon monoxide. Each of the oxidation steps within the cycle involves the regeneration of the active complex (3) or a similar site (such as 8 or 15) via oxidation with H2O2. We identified two alternative reaction channels for both the methanol and formaldehyde oxidations with distinctly different reaction mechanisms and intermediate products. Below we will first discuss the computational results obtained for each of the individual conversion steps and then use these data to analyse how the complete oxidation network operates and how it can be used to tailor the selectivity of the methane oxo-functionalization process.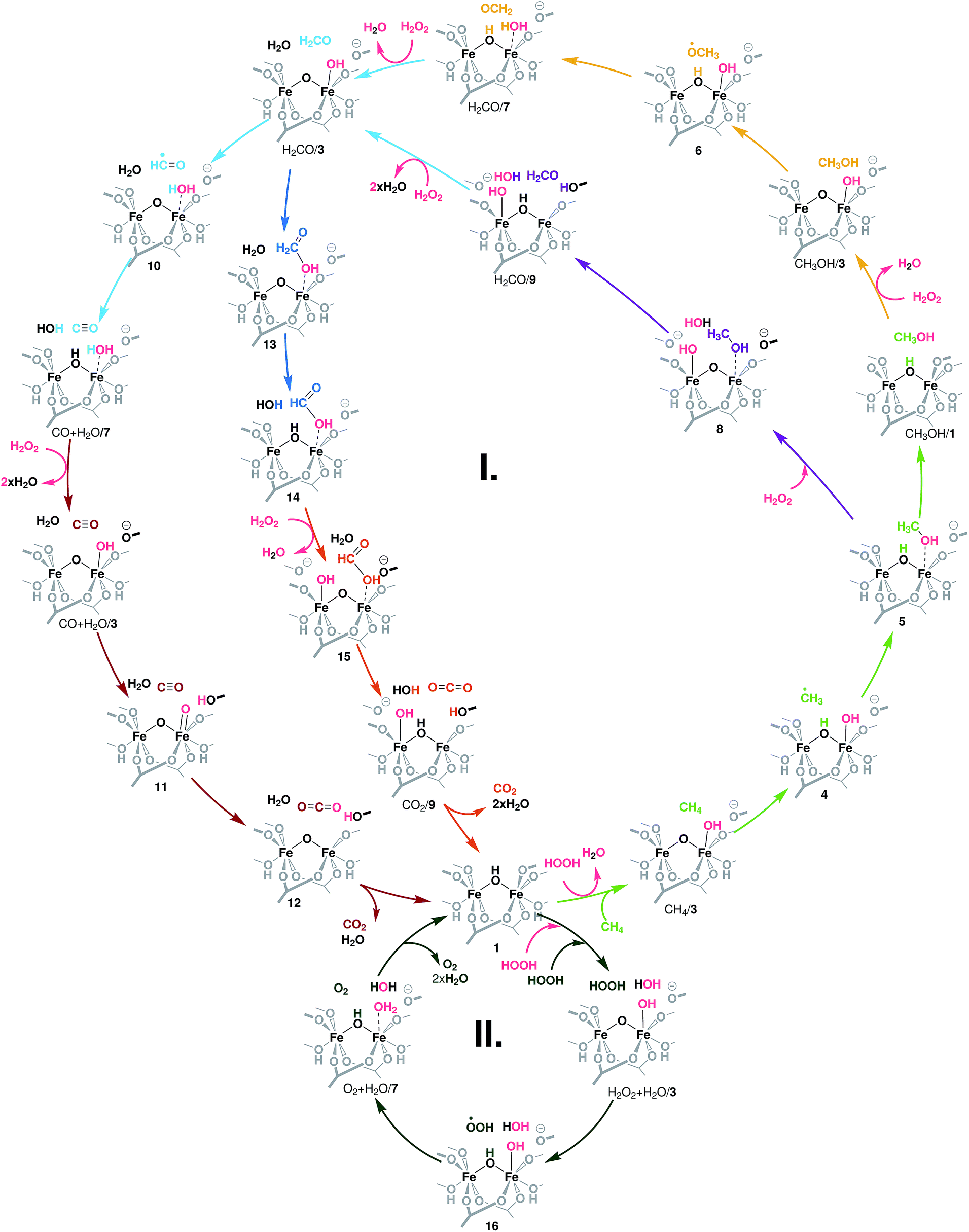 | ||
| Fig. 2 Reaction network underlying the liquid phase methane oxidation with H2O2 by Fe2 sites in MIL-53. The formation of the catalytic species 3 – the reaction of the reduced site 1 with H2O2 – is detailed in Fig. 3A. Cycle I shows the oxidation of methanol to CO2. Each colour indicates an oxidation step: light green: CH4 → CH3OH, yellow and purple: CH3OH → CH2O, light blue: CH2O → CO, dark blue: CH2O → HCOOH, brown: CO → CO2, and orange: HCOOH → CO2. Cycle II depicts the oxidation of a second H2O2 to O2 and the formation of H2O. All oxidation reactions occur over the same active site (3). | ||
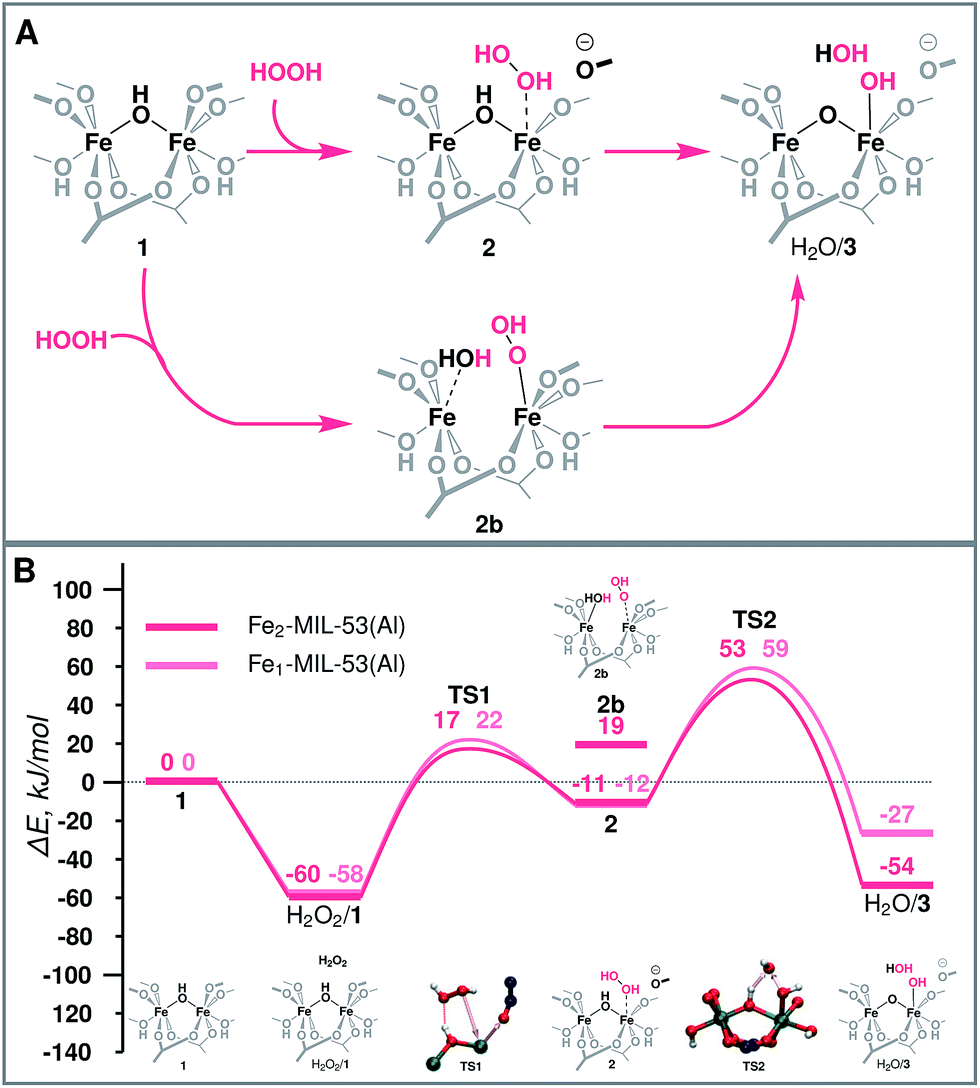 | ||
| Fig. 3 (A) Schematic representation and (B) reaction energy diagram of the formation of the active site over binuclear and mononuclear Fe species. | ||
Formation of the active site
The molecular mechanism of the active species generation and the respective DFT-computed energy diagram are summarized in Fig. 3. The oxidation of the initial Fe(III) dimer with H2O2 starts with the physisorption of H2O2 in the pores of the MOF near the Fe centres (H2O2/1). To proceed further along the reaction path, the coordination environment of the Fe centre has to be opened to allow for H2O2 coordination and activation. The intrusion of H2O2 can take place either via the cleavage of a bond with the bridging O (Fe–μO–Fe) (2b) or with the carboxylate linker O (Fe-OCRO) (2). Both reactions are thermodynamically unfavourable and show reaction energies of 79 and 49 kJ mol−1 (E‡(TS1) = 77 kJ mol−1), respectively. The reaction H2O2/1 → 2 is more favourable than reaction H2O2/1 → 2b rendering it more likely to occur during the catalytic process. The resulting coordinatively unsaturated Fe site promotes the coordination and the dissociation of H2O2 that is converted to an OH ligand bound to an Fe atom and a transient OH radical that readily abstracts an H atom from a bridging hydroxyl group to form an H2O molecule during the same elementary reaction step (ΔE = −42 kJ mol−1, E‡(TS2) = 34 kJ mol−1) (H2O/3). During this redox reaction step the oxygen atoms of H2O2 are reduced, while the Fe(III) sites are oxidized. Partial charge and spin density analysis (Table S4 in the ESI†) indicates the equality of the two Fe(IV) centres. With the oxidation state of Fe the HS state also changes from S = 5 to S = 4, and the spin-crossing transition takes place after the transition state (TS2). This step is exothermic with the calculated ΔE = −42 kJ mol−1, which effectively compensates for the energy losses associated with the initial Fe-OCRO cleavage step making the overall activation sequence thermodynamically neutral (ΔE(H2O2/1 → H2O/3) = 6 kJ mol−1).The increase of the number of Fe atoms in the reactive ensemble may result in a compromised stability of the framework towards the reactive environment. The Fe-rich structure has more sites potentially participating in the reaction with H2O2 that give rise to the formation of defect sites more susceptible to hydrolysis and further decomposition. This has been confirmed experimentally by showing that the pure MIL-53(Fe) sample disintegrates in the presence of H2O2, while when sufficient site-isolation of Fe ions in MIL-53(Fe,Al) materials is ensured, no Fe leaching was observed.52
Methane oxidation and overoxidation (cycle I)
The oxidized species 3 efficiently promotes methane to methanol oxidation.52 The adsorbed methane is activated via a homolytic C–H cleavage over the bridging O centre to yield a bridging hydroxyl group and a CH3 radical weakly coupled with the paramagnetic Fe dimer (4). The CH3 radical formation is endothermic by 62 kJ mol−1 and proceeds with a barrier of only 76 kJ mol−1. Upon the C–H cleavage step one proton and one electron are transferred from the methane to the active site and the cluster is reduced to Fe(IV)–Fe(III). Next, the CH3 radical recombines with the neighbouring OH group to form a methanol molecule. The radical recombination is accompanied by the reduction of the second Fe(IV) atom to Fe(III) obtaining the initial oxidation state of the cluster (5). The release of methanol into the MIL-53 channels regenerates the initial closed Fe dimer site (CH3OH/1).Next CH3OH is oxidized to H2CO . The reaction involves an abstraction of two H atoms that can either occur concurrently, in which case methanol stays adsorbed on the Fe site, while it is being oxidized again by H2O2, or consecutively, which means that the methanol encounters another active site in the MOF (Fig. 4B). In the concurrent case (purple) (E‡(TS6) = 117 kJ mol−1) the transfer of three H atoms takes place simultaneously: one C–H bond of CH3OH dissociates, and the carboxylic O accepts the H atom. At the same time, the OH group of CH3OH is also deprotonated by the bridging O with an H2O molecule acting as a H atom shuttle. The co-adsorbed water molecule accepts a H atom from methanol and simultaneously transfers one of its own to the bridging O (H2CO/9). In the alternative path (yellow path in Fig. 4B), the methanol is converted to CH2O in two steps via a CH3O radical intermediate. This reaction proceeds via an outer-sphere mechanism, in which CH3OH is not coordinated to the Fe centres during the reaction. At first the O–H bond is cleaved by the bridging O (E‡(TS4) = 65 kJ mol−1) and the CH3O radical is formed (6). This is followed by an almost barrierless (E‡(TS5) = 4 kJ mol−1) C–H bond dissociation by a neighbouring Fe–OH moiety to form a coordinated H2O molecule and formaldehyde (H2CO/7). We previously showed that the release of methanol to the MOF pore and the regeneration of the original site (reaction 5 → CH3OH/1) is a thermodynamically favourable reaction (shown in Fig. 5A). This together with a relatively high activation barrier predicted for the one-step mechanism suggests that the conversion of methanol to formaldehyde should take place on an Fe site different from the one where it was originally formed.
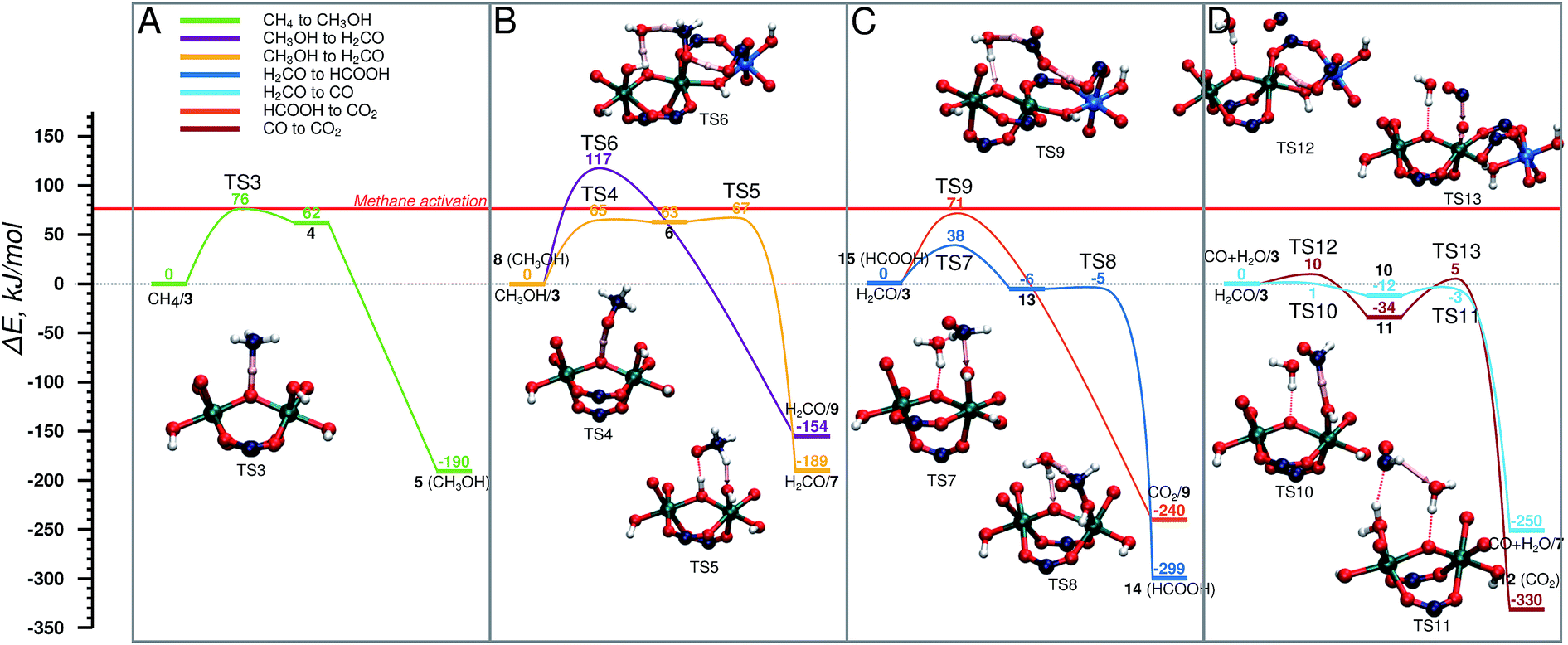 | ||
| Fig. 4 Energy diagram of cycle I, schematic representation is shown in Fig. 2. (A) CH4 → CH3OH, (B) CH3OH → CH2O in two steps (yellow) and in one step (purple), (C) H2CO → HCOOH (dark blue), and its consecutive oxidation to CO2 (orange), (D) CH2O → CO (light blue) and its consecutive oxidation to CO2 (brown). The reference point for all reactions is the active site and the adsorbed molecule as represented in Fig. 2. | ||
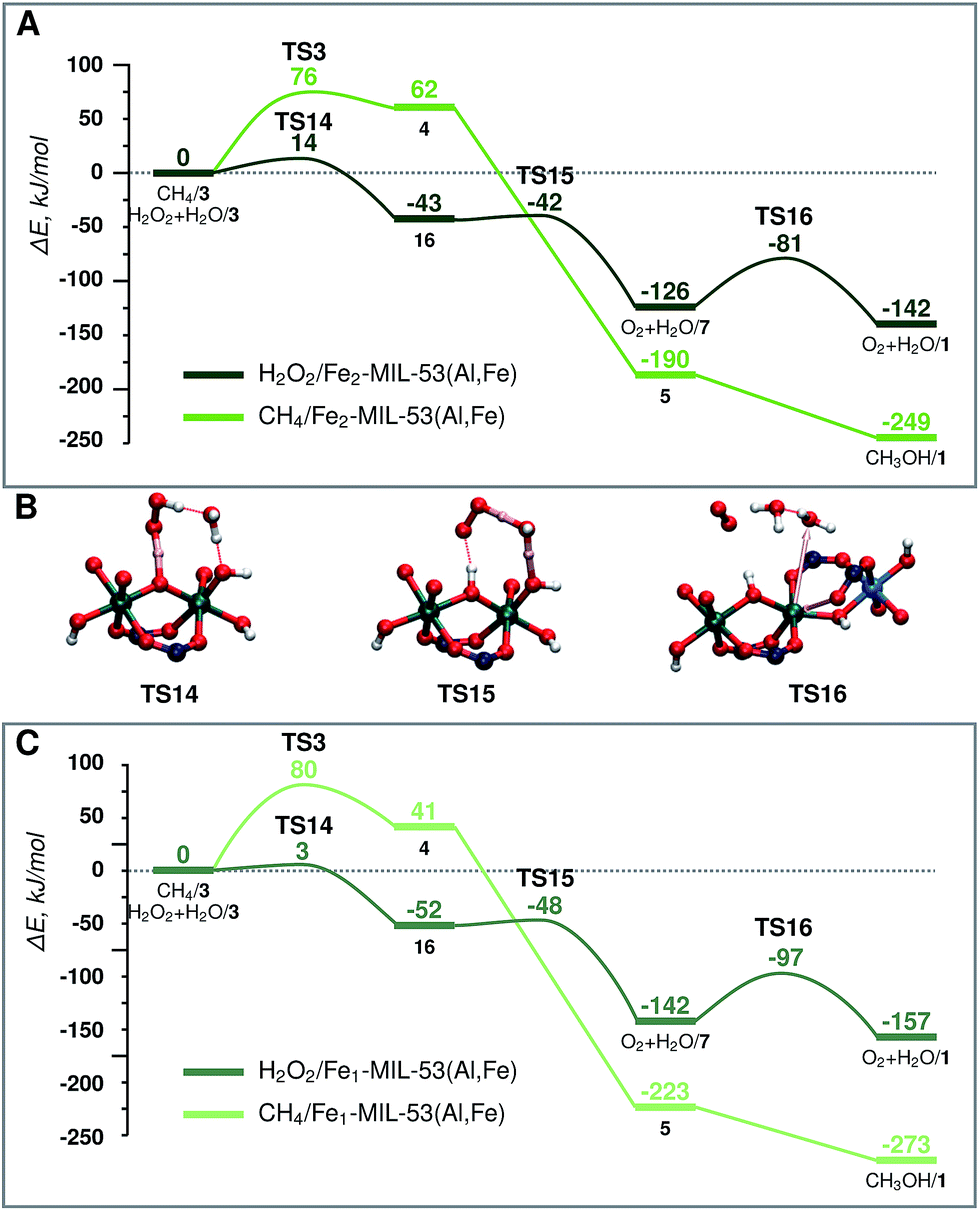 | ||
| Fig. 5 Energy diagram of H2O2 decomposition towards O2 (dark green) and CH4 oxidation towards CH3OH (light green) over (A) Fe2-MIL-53(Al) and (C) Fe1-MIL-53(Al). (B) shows the transition states for part (A) H2O2 decomposition. The reference point is the active site and the adsorbed H2O2 or CH4 molecule as represented in Fig. 2. | ||
In the next step H2CO is oxidized to CO2 . The oxidation can proceed via the HCOOH (Fig. 4C) or CO (Fig. 4D) intermediate. The former H2CO → HCOOH route (H2CO/3 → 14) is a two-step process (Fig. 4C), in which first an H2COOH complex is formed by the addition of H2CO to an OH group of the active site (13) (E‡(TS7) = 38 kJ mol−1) in a slightly exothermic reaction followed by the almost barrierless (E‡(TS8) = 1 kJ mol−1) water assisted C–H bond dissociation yielding HCOOH (14). HCOOH is then oxidized to CO2 (15 → CO2/9) in a one-step water assisted transfer of two H atoms to a carboxylic linker O and the bridging O centre (E‡(TS9) = 71 kJ mol−1).
The other path to convert H2CO to CO2 proceeds via CO as an intermediate and it is shown in Fig. 4D. This path starts with the oxidation of H2CO to CO (H2CO/3 → CO + H2O/7) by a two-step H-abstraction process. The first H atom is accepted by the OH group (E‡(TS10) = 1 kJ mol−1) forming H2O and the CHO radical (10). The second transition state (E‡(TS11) = 12 kJ mol−1) is associated with the rotation of the HCO radical around the C–O axis, which is followed by the cleavage of the C–H bond. The nearby H2O molecule acts as an H shuttle accepting the H atom and donating its own to the bridging O site (CO + H2O/7). CO is further oxidized to CO2via CO + H2O/3 → 12. In the first step, a H atom is transferred from the OH group to the carboxylate ligand to form a terminal oxo-species (11) in a slightly exothermic reaction (E‡(TS12) = 10 kJ mol−1). The terminal O-site acts then as the oxidizing centre to convert CO to CO2 (12) (E‡(TS13) = 39 kJ mol−1).
These reactions in terms of proton and electron transfer can be classified into (1) 2 H atoms transfer from the molecule to the active site (CH3OH → CH2O, CH2O → CO, HCOOH → CO2), (2) 1 H atom transfers to the active site and an OH group transfers from the active site to the reactant (CH4 → CH3OH, CH2O → HCOOH) or (3) an O atom transfers from the active site (CO → CO2). The formal oxidation state of C increases by two with each step from −4 of CH4 to +4 of CO2. This requires the reduction of 4 H2O2 molecules. The reduction of the H2O2 and the oxidation of the (oxygenated) hydrocarbons are decoupled with the help of the Fe site. Fe(III) is oxidized by H2O2 to Fe(IV) which is reduced again by the hydrocarbon to Fe(III). With the oxidation state of the Fe cluster the HS state also changes: the Fe(III) dimer is in the S = 5 state, while the Fe(IV) dimer is in the S = 4 HS state.
The calculations indicate that it is not likely that methane is overoxidized while being adsorbed to the active site, as the release of methanol to the MOF pore and the regeneration of the initial Fe cluster is strongly favourable thermodynamically. Therefore, we propose that the further transformation of methanol should take place on an active site different from that where it was originally formed. Furthermore, the comparison of the computed energetics for the overoxidation steps suggests that the oxidation of CH3OH and HCOOH proceeds with activation barriers similar to that of the initial methane activation despite the considerably lower C–H bond energies in the former molecules. This provides a fundamental possibility to obtain methanol as the kinetic product with a good selectivity. The low activation energies predicted for CH2O and CO conversions indicate a very short life-time of these intermediates in line with the experimental observations.52
H2O2 decomposition (cycle II) vs. CH4 oxidation
When H2O2 is used as the oxidant, the target methane oxidation process is inevitably accompanied by the direct decomposition of H2O2. The computed reaction energy diagrams for this process are shown in Fig. 5A.After the formation of the active site (H2O/3) a second H2O2 molecule is adsorbed in the pore of the MOF catalyst (H2O2 + H2O/3). This is followed by the deprotonation of H2O2 by the bridging O site to form an OOH radical intermediate (16) (ΔE = −43 kJ mol−1, E‡(TS14) = 14 kJ mol−1). The radical reacts then with an Fe–OH moiety to form molecular O2 (O2 + H2O/7) (ΔE = −83 kJ mol−1, E‡(TS15) = 1 kJ mol−1). The active site is regenerated by the decoordination of an H2O ligand and the formation of the Fe-OCRO bond (O2 + H2O/1) (ΔE = −16 kJ mol−1, E‡(TS16) = 45 kJ mol−1). During the course of the reaction the oxidation state of Fe and O atoms changes in two steps. Each step has a different HS state. The HS of the oxidized active site 3 is S = 4. After the TS14 Fe(III)–Fe(IV) cluster and a radical are formed, the HS state of 16 becomes S = 5. In the next step O2 is formed, and Fe is further reduced to Fe(III)–Fe(III) (O2 + H2O/7). The HS state of the Fe(III)–Fe(III) cluster is S = 5, however the total spin of the system will be S = 6 due to the triplet state of the molecular oxygen product.
The same active site (3) promotes the oxidation of both CH4 and H2O2. To facilitate the comparison of the two paths, both energy diagrams are shown in Fig. 5A. Although thermodynamically the formation of CH3OH is preferred, the undesirable H2O2 decomposition proceeds with a barrier of more than 60 kJ mol−1 lower than the C–H bond activation. This implies that the conversion of H2O2 is strongly kinetically favoured over the methane activation. The latter path can in principle be promoted at a lower concentration of hydrogen peroxide. However, H2O2 is necessary to generate the active site for methane oxidation, and the barrier for the active site formation is similar to that determined for CH4 activation, suggesting that this selectivity enhancement will always be achieved at the expense of the decreased reaction rate.
Mononuclear Fe sites
Characterization of the catalyst suggests that a considerable portion of the Fe sites in the mixed-metal MIL-53(Fe,AL) catalyst are monomeric complexes. The main difference between the monomeric and dimeric Fe sites is in the formal oxidation state. The reduction of H2O2 requires two electrons from the Fe centre. This implies that such a reaction over the dimeric species will oxidise both Fe sites to the +4 state, while for the monomeric site, the oxidized Fe is in the +5 state (light pink line of Fig. 3B). The formation of the mononuclear +5 species is thermodynamically less favourable than the oxidation of the binuclear cluster (ΔE(3-monomer)–ΔE(3-dimer) = 27 kJ mol−1). Nevertheless, the mononuclear Fe site still provides favourable reaction channels for the active site formation and the oxidation of methane to methanol (energy diagram in Fig. 3B, and 5C). Importantly, we find here that this mononuclear site also promotes the H2O2 decomposition. The associated two H-abstraction process in this case proceeds with a less than 4 kJ mol−1 activation barrier each, following the same trend as that described for the binuclear active site above.These results indicate that the behaviour of the mononuclear Fe site in the MIL-53 structure is similar to that of the binuclear Fe clusters. This implies that the coordination sphere (geometry and quality of the ligands) of the Fe has a more significant influence on the reactivity than the nuclearity of the complex.
Conclusions
The conversion of methane to methanol over a mixed metal Fe-MIL-53(Al) catalyst was investigated by periodic DFT calculations. Other than the most important C–H bond activation step the whole catalytic cycle was explored and different reaction paths were identified for the formation of the active site and the overoxidation of methane towards CO2. An important side reaction, the decomposition of H2O2 to O2 and H2O was investigated. The reactivities of mononuclear and binuclear Fe species were compared. As expected the calculations reveal a highly complex reaction network with many possibilities. Our main findings can be outlined as follows.(i) Methanol can be yielded with good selectivity as the kinetic product of the reaction, as the consecutive oxidation of methane has a high activation barrier, and the desorption of methanol and the regeneration of the initial Fe site is thermodynamically favourable.
(ii) The first C–H bond dissociation of methane is only one of the rate determining steps. The reaction barrier that leads to the active site formation, the O–H bond dissociation of CH3OH and the conversion of HCOOH is in the same order of magnitude. The oxidation of H2CO and CO has a low reaction barrier indicating that these intermediates have a low concentration in the reaction mixture in line with experiments.
(iii) The same active site (3) promotes the oxidation of both CH4 and H2O2. The conversion of H2O2 is favoured over CH4 as the reaction barrier of the former reaction is over 60 kJ mol−1 lower. This renders H2O2 unsuitable for methane oxidation in combination with Fe complexes.
(iv) The activities of mononuclear and binuclear sites were compared. The calculations indicate that despite monomeric species go through a formal oxidation state of +5 in the reaction, while dimeric species are only oxidized until the formal oxidation state +4, the activities of the species are comparable, and they promote the same reaction steps.
Conflicts of interest
We have no conflicts of interest to declare.Acknowledgements
The Dutch Science Foundation (NWO) is gratefully acknowledged for financial support through the VIDI personal grant “MetMOFCat”. Dr G. Li acknowledges the financial support from NWO for her personal VENI grant (no. 016.Veni.172.034). SurfSARA and NWO (The Netherlands Organisation for Scientific Research) are acknowledged for providing access to supercomputer resources.References
- Q. Zhang, D. He and Q. Zhu, J. Nat. Gas Chem., 2003, 12, 81–89 Search PubMed.
- J. H. Edwards and N. R. Foster, Fuel Sci. Technol. Int., 1986, 4, 365–390 CrossRef.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef PubMed.
- A. I. Olivos-Suarez, A. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef.
- C. A. Moulder and T. R. Cundari, Isr. J. Chem., 2017, 57, 1023–1031 CrossRef.
- S.-P. Huang, Y. Shiota and K. Yoshizawa, Dalton Trans., 2013, 42, 1011–1023 RSC.
- T. Quinn and P. Choudhury, Mol. Catal., 2017, 431, 9–14 CrossRef.
- A. Trinchero, A. Hellman and H. Grönbeck, Surf. Sci., 2013, 616, 206–213 CrossRef.
- H. Basch, K. Mogi, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 1999, 121, 7249–7256 CrossRef.
- S. Chempath and A. T. Bell, J. Am. Chem. Soc., 2006, 128, 4650–4657 CrossRef PubMed.
- A. A. Latimer, A. R. Kulkarni, H. Aljama, J. H. Montoya, J. S. Yoo, C. Tsai, F. Abild-Pedersen, F. Studt and J. K. Nørskov, Nat. Mater., 2017, 16, 225–229 CrossRef PubMed.
- M. H. Mahyuddin, A. Staykov, Y. Shiota, M. Miyanishi and K. Yoshizawa, ACS Catal., 2017, 7, 3741–3751 CrossRef.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 344, 570–577 CrossRef.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef PubMed.
- H. Aljama, J. K. Nørskov and F. Abild-Pedersen, J. Phys. Chem. C, 2017, 121, 16440–16446 CrossRef.
- M. H. Mahyuddin, T. Tanaka, Y. Shiota, A. Staykov and K. Yoshizawa, ACS Catal., 2018, 8, 1500–1509 CrossRef.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef.
- D. Park and J. Lee, Korean J. Chem. Eng., 2013, 30, 977–987 CrossRef.
- X. Liu, Y. Ryabenkova and M. Conte, Phys. Chem. Chem. Phys., 2015, 17, 715–731 RSC.
- K. A. Dubkov, V. I. Sobolev and G. I. Panov, Kinet. Catal., 1998, 39, 72–79 Search PubMed.
- G. I. Panov, V. I. Sobolev, K. A. Dubkov, V. N. Parmon, N. S. Ovanesyan, A. E. Shilov and A. A. Shteinman, React. Kinet. Catal. Lett., 1997, 61, 251–258 CrossRef.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef PubMed.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 136, 52–56 CrossRef.
- J. Xu, A. Zheng, X. Wang, G. Qi, J. Su, J. Du, Z. Gan, J. Wu, W. Wang and F. Deng, Chem. Sci., 2012, 3, 2932–2940 RSC.
- A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi and Y. Kuroda, J. Phys. Chem. C, 2013, 117, 19525–19534 Search PubMed.
- J. Shan, W. Huang, L. Nguyen, Y. Yu, S. Zhang, Y. Li, A. I. Frenkel and F. Tao, Langmuir, 2014, 30, 8558–8569 CrossRef PubMed.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC.
- V. I. Sobolev, K. A. Dubkov, O. V. Panna and G. I. Panov, Catal. Today, 1995, 24, 251–252 CrossRef.
- M. V. Parfenov, E. V. Starokon, L. V. Pirutko and G. I. Panov, J. Catal., 2014, 318, 14–21 CrossRef.
- T. Z. H. Gani and H. J. Kulik, ACS Catal., 2018, 8, 975–986 CrossRef.
- K. Otsuka and Y. Wang, Appl. Catal., A, 2001, 222, 145–161 CrossRef.
- G. B. Shulpin and G. V. Nizova, React. Kinet. Catal. Lett., 1992, 48, 333–338 CrossRef.
- J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef PubMed.
- J. Weitkamp, Solid State Ionics, 2000, 131, 175–188 CrossRef.
- G. Li, E. A. Pidko, R. A. Van Santen, Z. Feng, C. Li and E. J. M. Hensen, J. Catal., 2011, 284, 194–206 CrossRef.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef PubMed.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Appl. Catal., A, 2010, 373, 168–175 CrossRef.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef PubMed.
- A. Corma, H. García, F. X. Llabrés and I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef PubMed.
- B. Li, M. Chrzanowski, Y. Zhang and S. Ma, Coord. Chem. Rev., 2016, 307, 106–129 CrossRef.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef PubMed.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef PubMed.
- D. Osadchii, A. Olivos Suarez, A. Szécsényi, G. Li, M. Nasalevich, A. Dugulan, P. Serra-Crespo, E. Hensen, S. Veber, M. Fedin, G. Sankar, E. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef.
- A. Kazaryan and E. Baerends, ACS Catal., 2015, 5, 1475–1488 CrossRef.
- G. Gopakumar, P. Belanzoni and E. Baerends, Inorg. Chem., 2012, 51, 63–75 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B, 1994, 49, 14251–14269 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- P. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef.
- P. Belanzoni, L. Bernasconi and E. J. Baerends, J. Phys. Chem. A, 2009, 113, 11926–11937 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- J. Stone, M. Sc. thesis, Computer Science Department, University of Missouri-Rolla, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates of calculated structures, relative energies of different spin states of structure 1, spin densities and partial charges of structure H2O/3. See DOI: 10.1039/c8sc02376j |
| This journal is © The Royal Society of Chemistry 2018 |