
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile and systematic access to the least-coordinating WCA [(RFO)3Al–F–Al(ORF)3]− and its more Lewis-basic brother [F–Al(ORF)3]− (RF = C(CF3)3)†
Arthur
Martens
 ,
Philippe
Weis
,
Michael Christian
Krummer
,
Marvin
Kreuzer
,
Andreas
Meierhöfer
,
Stefan C.
Meier
,
Jan
Bohnenberger
,
Harald
Scherer
,
Ian
Riddlestone
and
Ingo
Krossing
*
,
Philippe
Weis
,
Michael Christian
Krummer
,
Marvin
Kreuzer
,
Andreas
Meierhöfer
,
Stefan C.
Meier
,
Jan
Bohnenberger
,
Harald
Scherer
,
Ian
Riddlestone
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
First published on 1st August 2018
Abstract
By reaction of the Lewis acid Me3Si–F–Al(ORF)3 with a series of [PF6]− salts, gaseous PF5 and Me3Si–F are liberated and salts of the anion [F–Al(ORF)3]− ([f–al]−; RF = C(CF3)3) can be obtained. By addition of another equivalent of Me3Si–F–Al(ORF)3 to [f–al]−, gaseous Me3Si–F is released and salts of the least coordinating anion [(RFO)3Al–F–Al(ORF)3]− ([al–f–al]−) are formed. Both procedures work for a series of synthetically useful cations including Ag+, [NO]+, [Ph3C]+ and in very clean reactions with 5 g batch sizes giving excellent yields typically exceeding 90%. In addition, the synthesis of Me3Si–F–Al(ORF)3 has been optimized and scaled up to 85 g batches in an one-pot procedure. These anions could previously only be obtained by difficult to control decomposition reactions of [Al(ORF)4]− or by halide abstraction reactions with Me3Si–F–Al(ORF)3, generating relatively large countercations that are unsuited for further use as universal starting materials. Especially [al–f–al]− is of interest for the stabilization of reactive cations, since it is even weaker coordinating than [Al(ORF)4]− and more stable against strong electrophiles. This bridged anion can be seen as an adduct of [f–al]− and Al(ORF)3. Thus, it is similarly Lewis acidic as BF3 and eventually reacts with nucleophiles (Nu) from the reaction environment to yield Nu–Al(ORF)3 and [f–al]−. This prevents working with [al–f–al]− salts in ethereal or other donor solvents. By contrast, the [f–al]− anion is no longer Lewis acidic and may therefore be used for reactions involving stronger nucleophiles than the [al–f–al]− anion can withstand. Subsequently it may be transformed into the [al–f–al]− salt by simple addition of one equivalent of Me3Si–F–Al(ORF)3.
Introduction
For many years our group has been interested in the stabilization of reactive cations with the weakly coordinating anion [Al(ORF)4]− (WCA, RF = C(CF3)3). With this anion shown in Scheme 1, a large variety of reactive cations being electrophilic like [CX3]+, RZn+ (R = Me, Et), [Sn(Cp)]+, [P2X5]+ (X = Cl, Br, I) and [P9]+, oxidizing like [NO]+, [NO2]+ or [Ag(X2)n]+-complexes, or very weakly bound to a central atom like the huge number of [M(L)n]+ complexes with M = Cu, Ag, Au and L = CO, C2H2, C2H4, P4, S8, S12, Se6, Se12 or Se19 could be stabilized.1–5 One of the biggest advantages of this [Al(ORF)4]− anion is its facile and fast synthesis in high yields.6 Other commonly used WCAs are [B(C6F5)4]−, perhalogenated carborane anions [R–CB11X12]− (X = F, Cl, Br) and the teflates [E(OTeF5)x]− (E = B, Al, Sb, As, Bi, Nb; x = 4, 6).7,8 However, the synthesis of these anions either require handling of explosive LiC6F5,9 elaborate syntheses in small scales,10 or handling of the toxic and hydrolysis sensitive HOTeF5.8,11 Yet, especially the carborane anions are chemically more stable against fierce electrophiles than [Al(ORF)4]−, e.g. against small silylium ions.12 As shown in eqn (1a), [Al(ORF)4]− decomposes in the presence of small silylium ions [R3Si]+ above −30 °C to give R3Si–F–Al(ORF)3.13,14 However, it is compatible with the bulky [Si(C6Me5)3]+ silylium ion.15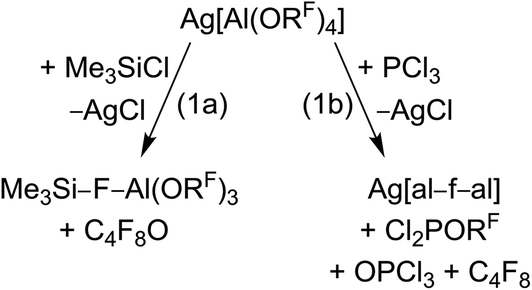
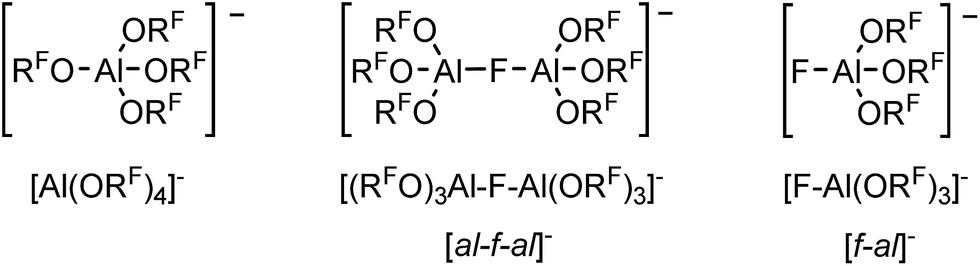 | ||
| Scheme 1 Overview to the anions presented in this paper and their abbreviations. | ||
More general, very small and very potent electrophiles, like “[PCl2]+” or “[SiCl3]+” induce [Al(ORF)4]− decomposition, very often under formation of [(RFO)3Al–F–Al(ORF)3]− ([al–f–al]−, eqn (1b)), which is even less coordinating than [Al(ORF)4]−.16 For more than a decade, access to a starting material with this anion was only accessible by decomposition of Ag[Al(ORF)4]. However, this synthesis proved to be delicate to reproduce, requires the two-step synthesis of Ag[Al(ORF)4], and is combined with loss of Ag+ and ORF moieties. The bridging motif of [al–f–al]− may be seen in analogy to related bridged anions known to the literature,17e.g. as in [(C6F5)3B–CN–B(C6F5)3]−, [(C6F5)3B–NH2–B(C6F5)3]− and [(C6F5)3E–F–E(C6F5)3]− (E = B, Al, Ga).18
In some cases, where decomposition of [Al(ORF)4]− occurred, formation of [al–f–al]− could not be observed. Instead the formal [F–Al(ORF)3]− anion ([f–al]−) was obtained, often as part of a neutral compound, like in Me3Si–F–Al(ORF)3 or Cp*Be–F–Al(ORF)3.13,19 Therefore, both were rationalized as being ion-like compounds that behave intermediate between being covalently bound or separated ions. Nevertheless also compounds, where [f–al]− and the cation are separated, were isolated.20
Me3Si–F–Al(ORF)3 has already shown its usefulness for silylation reactions,13,21 the polymerization of isobutene,13 halide abstraction reactions,21 and abstraction of [Cp]−.3 It is available in an one pot synthesis that was optimized here to 85 g scale. Upon investigation of its reactivity, formation of [al–f–al]− and [f–al]− could often be observed and proved to be easily controllable.13,21 Therefore we investigated here, if it is possible to use Me3Si–F–Al(ORF)3 to synthesize both anions with cations that would make them good starting materials for further chemistry.
Results and discussion
Large-scale optimized synthesis of Me3Si–F–Al(ORF)3
In order to establish optimized routes to the anions [al–f–al]− and [f–al]−, we needed access to larger amounts of Me3Si–F–Al(ORF)3. Therefore, we optimized this synthesis and performed an upscaling from 40 g (ref. 13) up to 85 g. Although we believe that further upscaling is possible, we do not recommend it. The vigorous stirring of the reaction solution is crucial and becomes complicated in larger scales, when using a Dewar vessel for the cooling bath. Additionally, since 25 g of Me3SiF cost 364€ (ABCR), we elaborated a synthesis starting from Me3SiCl and [NH4]2[SiF6] on the basis of ref. 22, yielding Me3SiF in high purities at very low cost (see ESI† for details).In order to maximize the yield and purity of Me3Si–F–Al(ORF)3 it is highly important to dry all glassware with a gas burner in vacuo until the flame turns orange. When opening the reaction vessel containing the AlEt3, the argon flow must be carefully controlled. If the argon flow is too high, the AlEt3 may start to smoke due to swirling of ambient air. Additionally, the HORF has to be dried to a water content of less than 1 ppm, which can be achieved by stirring with P4O10 for two weeks. It is also highly advisable to use as large of a stir bar as possible, since vigorous stirring is crucial for this synthesis and during the reaction, the viscosity of the reaction solution increases.
The Me3Si–F–Al(ORF)3 used for all syntheses in this paper was prepared according to the following optimized procedure: AlEt3 (15.0 mL, 109 mmol, 93% purity) was dissolved in heptane (120 mL) and cooled to −40 °C. Under vigorous (!) stirring, the first two equivalents of HORF (30.0 mL, 215 mmol, 2.0 eq.) were added dropwise to the reaction mixture, while keeping the cooling bath between −40 and −20 °C. During the addition of HORF, gas evolution (C2H6) was observed, the viscosity of the solution increased and small amounts of precipitate formed. After complete addition of the first HORF portion (typically within 1 hour), Me3SiF (12.5 g, 136 mmol, 1.2 eq.) was condensed onto the reaction mixture at −40 °C. The solution was stirred for 10 min, and then the third equivalent of HORF (20.0 mL, 143 mmol, 1.3 eq.) was added within 5 min. The cooling bath was removed and the reaction mixture was allowed to reach rt, which led to dissolution of the precipitate. After 30 min, more HORF (5.0 mL, 36 mmol, 0.3 eq.) was added and the solution was stirred overnight. From the clear solution, slowly a white powder crystallized. For product isolation, simply the solvent was removed in vacuo yielding a white crystalline powder (85.4 g, 103 mmol, 94%). Note: one needs the excess of the fluorinated alcohol, as this is very volatile (b.p. = +45 °C) and the continuous stream of evolving ethane removes the excess alcohol through the bubbler.
The quality of the Me3Si–F–Al(ORF)3 can be verified by different methods: if the obtained powder seems to be moist, but cannot be completely dried, there are probably residual Al–Et groups present in the product. In this case, more HORF should be added. Another qualitative test for the purity of the product is by dissolving it in 1,2-difluorobenzene (o-DFB). A solution of pure Me3Si–F–Al(ORF)3 is nearly colorless, while impurities (mainly residual Al–Cl groups stemming from the AlEt3) induce a light yellow color. These impurities usually enrich at the top of the crystalized product and stick to the glass of the reaction vessel. These crystals are yellow colored and isolable. Therefore they should not be scratched out of the reaction vessel. Purification of the white powder can be achieved either by sublimation at 60 °C in dynamic vacuum (10−3 mbar) or by washing with warm CH2Cl2, let it cool and then filter it. Additionally, NMR spectra of the product should be taken in both, o-DFB and CH2Cl2. Me3Si–F–Al(ORF)3 is only poorly soluble in CH2Cl2, but not its impurities. Therefore, if a NMR spectrum in CH2Cl2 shows only negligible amounts of impurities, the product can be considered pure. In o-DFB Me3Si–F–Al(ORF)3 is highly soluble and here the signal intensity is increased compared to the impurities. Therefore, NMR spectra in CH2Cl2 may show a purity of only 60%, while the real purity is >90%, as can be seen from NMR spectra in o-DFB. Pure Me3Si–F–Al(ORF)3 can be stored in a glove box for at least a year without decomposition.
Synthesis of [f–al]− and [al–f–al]− salts
In a general procedure [Cat]+[f–al]− and [Cat]+[al–f–al]− (Cat = Li, K, Ag, NO, Ph3C) can be obtained by reaction of 1 and 2 equiv. Me3Si–F–Al(ORF)3, respectively, with [Cat][PF6] at rt or 60 °C, depending on the solubility of the [PF6]− salt (eqn (2)). The calculated rather exergonic reaction energies are included with eqn (2) and are given in kJ mol−1 (BP86-D3(BJ)/def-SV(P)).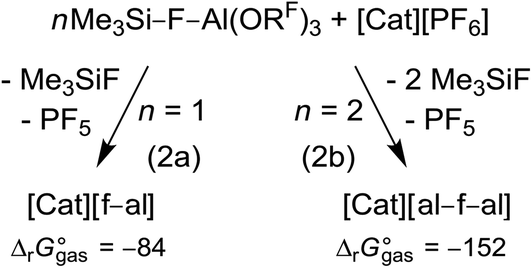
The preferred reaction conditions for all investigated compounds are listed in Table 1. The formation of the respective anion is controlled by the stoichiometry of Me3Si–F–Al(ORF)3 used (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1) – the very Pearson hard Li+ case presents the only exception and stops at the [f–al]− stage. The reactions performed at rt are usually finished within a few minutes and are accompanied by a vigorous evolution of PF5 after addition of the solvent. In the following, we briefly describe the reactions for each cation separately.
:1 or 2:1) – the very Pearson hard Li+ case presents the only exception and stops at the [f–al]− stage. The reactions performed at rt are usually finished within a few minutes and are accompanied by a vigorous evolution of PF5 after addition of the solvent. In the following, we briefly describe the reactions for each cation separately.
| Cation | Solvent | Reaction time | Temperature | Yield | Cation (bulk) | Cation (single crystal) | CCDC number |
|---|---|---|---|---|---|---|---|
| a Only the [f–al]− anion is obtained. b K[f–al] can only be obtained with significant amounts of impurities, therefore no yield is given. | |||||||
| Li+a | o-DFB/Cl3CCN | 4 h | 60 °C | — (84%) | — | ([Li(NCCCl3)]+) | (1845808) |
| Li+a | DMC ((MeO)2CO) | 2 h | 70 °C | — (86%) | ([Li(DMC)2]+) | ([Li(DMC)3]+) | (1845810) |
| K+b | o-DFB | 2 h | 60 °C | 90%b | K+ | — | |
| Ag+ | CH2Cl2 | 1 h | rt | 97% (98%) | [Ag(CH2Cl2)3]+ ([Ag(CH2Cl2)]+) | — | |
| Ag+ | SO2 | 2 h | −20–0 °C | 95% (98%) | Ag+ (Ag+) | — | |
| Ag+ | o-DFB | 30 min | rt | 96% (70%) | [Ag(o-DFB)2]+, ([Ag(o-DFB)x]+; x = 0–2) | [Ag(o-DFB)2]+, [Ag(o-DFB)3]+ ([Ag(o-DFB)3]+) | 1845812, 1845817 (1845811) |
| [NO]+ | SO2 | 1 h | −35–0 °C | 88% (90%) | [NO]+ | [NO]+ | 18458114 |
| [Ph3C]+ | o-DFB | 1 h | rt | 88% (95%) | [Ph3C]+ | — | |
The synthesis of K[f–al] proved to be more problematic. Compared to Me3Si–F–Al(ORF)3, only little K[PF6] in solution, and thus only K[al–f–al] is formed at the beginning of the reaction. The [f–al]− anion is therefore mainly formed by reaction of [PF6]− and [al–f–al]−, which is hindered by coulombic repulsion. After heating the reaction mixture to 60 °C for 9 h with an excess of finely ground K[PF6] (2 eq.) the reaction mixture still contained 4% K[al–f–al]. Additionally, NMR spectra show signals of K[ORF] (6%) and [F1+xAl(ORF)3−x]− (12%). Therefore we do not recommend to synthesize K[f–al] for the use as starting material, as it can only be obtained with significant amounts of impurities by this route. The K[f–al] obtained this way (∼80% purity), however, is sufficiently pure for IR and Raman spectroscopy.
The reaction in SO2 has to be performed at −20 °C in order to prevent loss of the solvent, while at the same time the evolving PF5 is allowed to evaporate from the solution. The turbid reaction solution is then stirred at −20 °C until the gas evolution has completely stopped. Subsequently, the reaction solution is allowed to reach 0 °C, followed by removal of the solvent. The solvent free product is obtained as an off-white powder.
Spectroscopic differentiation of the anions
Both anions can be differentiated by standard spectroscopic methods (IR, Raman, NMR). However, in all cases spectra of [f–al]− and [al–f–al]− are very similar and therefore we highlight key differences in the following.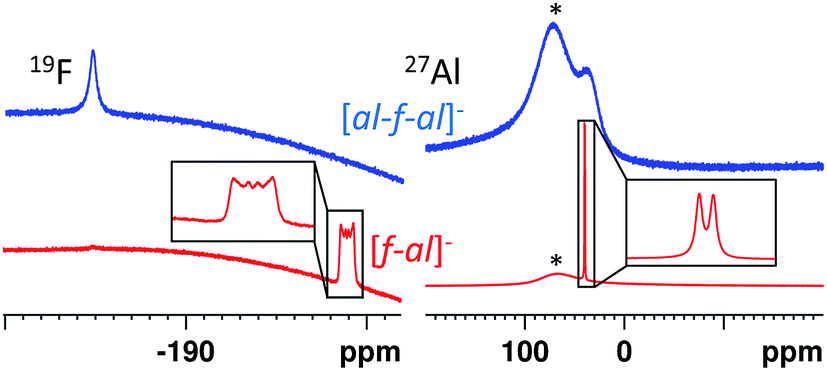 | ||
| Fig. 1 Characteristic 19F (Al–F range only) and 27Al NMR signals of [f–al]− and [al–f–al]−. The broad resonance at ∼60 ppm in the 27Al NMR spectra is caused by the probe head; red: Ag[f–al] in o-DFB, blue: Ag[al–f–al] in o-DFB, the signals marked by the asterisk are caused by the NMR spectrometer. | ||
By contrast, for [f–al]− the Al–F signal in the 19F NMR spectrum generates a sextet, due to coupling to the aluminum atom. This signal is usually found between −185 and −200 ppm, but may also be found at −145 ppm in case of the [NO]+ and [SeCl3]+ salts. The signal of the alkoxy groups is visible at −75.6 ppm with 5J(F–F) = 1.5–2 Hz due to coupling to the Al-bound fluorine atom. In the 27Al NMR spectrum a sharp doublet at 41 ppm with 1J(Al–F) = 40 Hz is observable due to an increased relaxation time of the Al atom compared to [al–f–al]−.
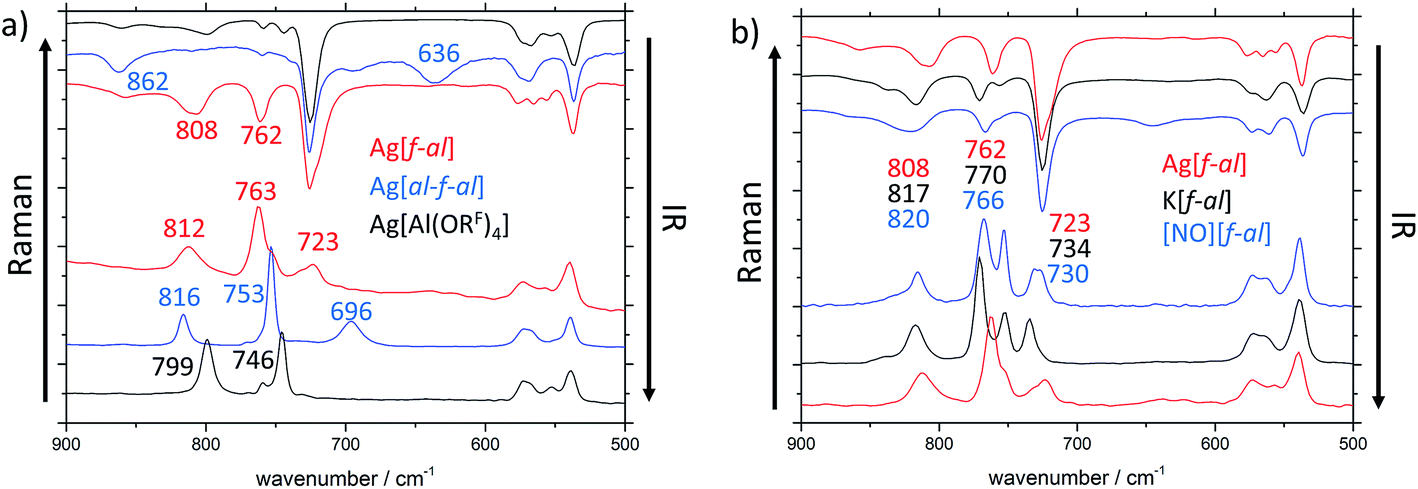 | ||
| Fig. 2 IR (top) and Raman (bottom) spectra in the region between 900 and 500 cm−1 and frequencies of characteristic bands in cm−1 of (a) Ag[Al(ORF)4] (black), Ag[al–f–al] (blue) and Ag[f–al] (red); (b) Ag[f–al] (red), K[f–al] (black) and [NO][f–al] (blue). | ||
The main differences between these anions can be found in the region between 900 and 500 cm−1 (Fig. 2a). In the IR spectra additional bands at 636 (νas(Al–F–Al)) and 862 cm−1 are visible for [al–f–al]−, and at 762 (ν(Al–F)) and 808 cm−1 for [f–al]−, when compared to [Al(ORF)4]−. The Raman spectra of these anions also show new bands at 636 and 723 cm−1, respectively. One of the most characteristic differences between the three discussed anions in the Raman spectra is the frequency of the vibrational bands at ∼750 and ∼800 cm−1 and their intensities. These bands are approximately equally intense for [Al(ORF)4]−. When looking at [al–f–al]− and [f–al]−, the vibrational band at ∼750 cm−1 is about twice as intense as the band at ∼800 cm−1.
Especially for [f–al]−, the coordination of the cation to the (Al–)F atom may have an impact on the vibrational bands in the vibrational spectra. Therefore, we compared the IR and Raman spectra of Ag[f–al], K[f–al] and [NO][f–al] (Fig. 2b). Among these cations, [NO]+ is considered to be the least and Ag+ (donor-free) to be the strongest coordinating. Most of the vibrational bands in these compounds are identical. Only the molecular vibrations containing partial Al–F contributions (∼810, ∼760 and ∼730 cm−1) are influenced by the coordination to the cation. These bands appear at higher wave numbers, the weaker coordinating the cation is. For [NO][f–al] and K[f–al] these bands are blue shifted by up to 12 cm−1 compared to Ag[f–al]. Although a comparison of these compounds with the analogous [Ph3C]+ and [Li(L)x]+ salts would be interesting, it proved to be rather complicated, since the vibrational bands of these cations overlap with the ones of the anion.
Molecular structures of [f–al]− and [al–f–al]− salts
Although our commonly used [Al(ORF)4]− anion usually does not show significant coordination, very small cations, like Li+ and [Ag(L)]+ are able to be coordinated by its oxygen atoms.4,5,23 As can be seen from Fig. 3, [f–al]− usually coordinates to the cation with the (Al–)F atom. In case of the dimeric {[Li(NCCCl3)][f–al]}2 each (Al–)F atom is coordinated to two Li atoms and also the alkoxy moieties are involved in the coordination, forming a nearly planar Al–O–Li–F plane. This coordination leads to an elongation of the Al–F and Al–Ocoordinated bonds compared to [Li(DMC)3][f–al] (Table 2). A similar coordination of the oxygen atoms of [al–f–al]− to a cation is highly unlikely for sterical reasons and was not observed in the compounds presented in this work. However, the [ZnEt]+ cation is small enough to co-ordinate to these alkoxy groups.2 Here, both Zn–O distances are nearly equal (209.8 pm on average), while in [ZnEt][Al(ORF)4] they differ by ∼5 pm (211.2 and 205.8 pm on average). The non-existing difference between the Zn–O distances in [ZnEt][al–f–al] not only is evidence for the weaker coordination of [al–f–al]− compared to [Al(ORF)4]−, but also for the inferior accessibility of the oxygen atoms.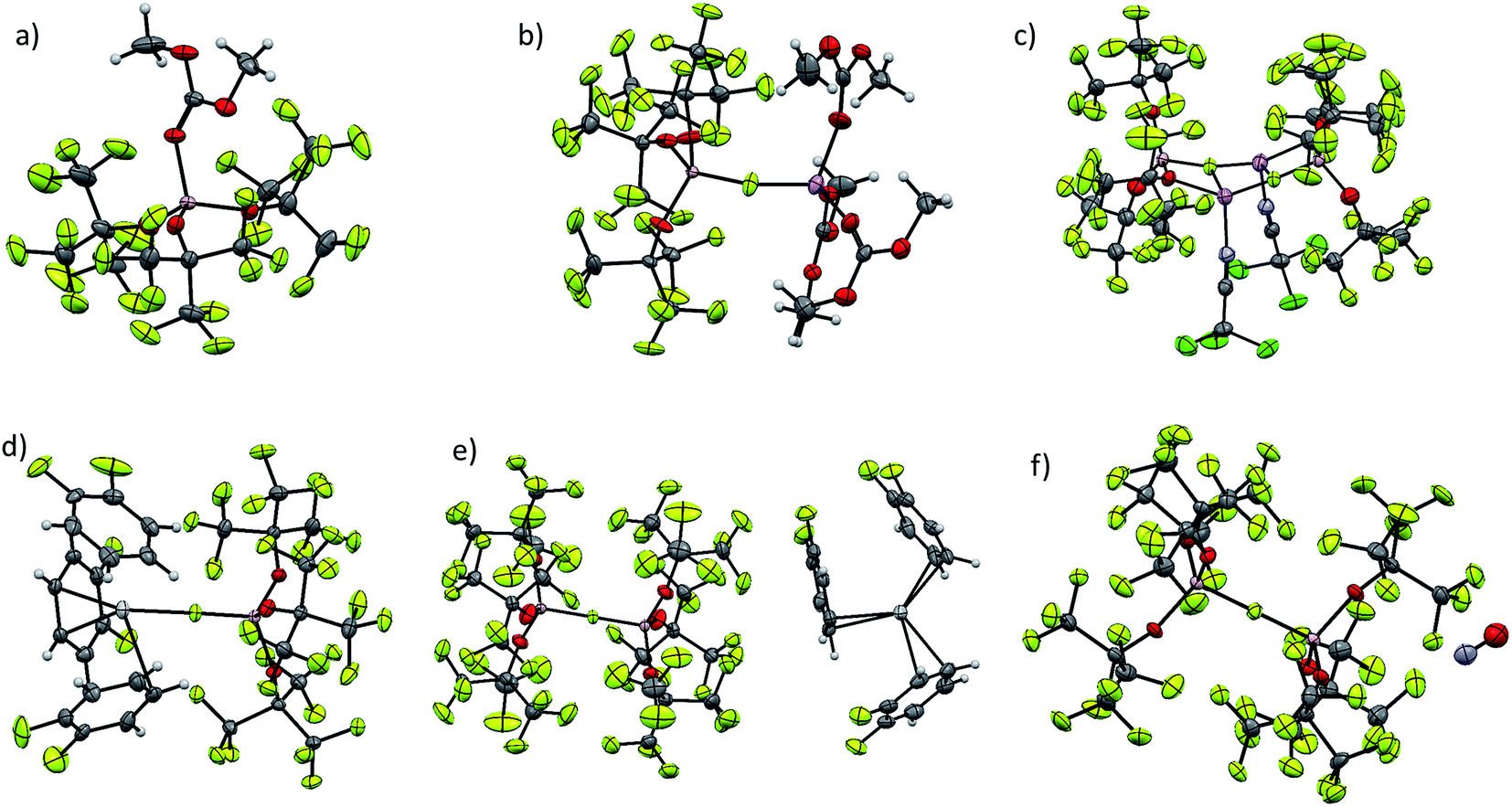 | ||
| Fig. 3 Overview of molecular structures of some starting materials presented in this work. Disorder was omitted in all cases, for the sake of clarity. (a) DMC–Al(ORF)3; (b) [Li(DMC)3][f–al]; (c) [Li(NCCCl3)][f–al]; (d) [Ag(o-DFB)3][f–al]; (e) [Ag(o-DFB)3][al–f–al]; (f) [NO][al–f–al]. | ||
| Anion | Cation | d(Al–F) | d(Al–O) | d(A–Cat) |
|---|---|---|---|---|
| a Li–F(1). b Li–O. c Li–F(2). d Ag–F. e H–F. f N–F. | ||||
| [f–al]− | [Li(NCCCl3)]+ | 171.2(2) | 169.9(5)/177.4(4) | 185.7(7)a/206.0(9)b/220(1)c |
| [f–al]− | [Li(DMC)3]+ | 167.4(2) | 171.9(2) | 184.1(5)a |
| [f–al]− | [Ag(o-DFB)3]+ | 168.7(1) | 172.7(1) | 242.0(1)d |
| [al–f–al]− | [Ag(o-DFB)3]+ | 176.1(2) | 169.3(2) | 348.1(2)e |
| [al–f–al]− | [NO]+ | 176.7(2) | 170.2(13) | 273.0(3)f |
Some aspects for single crystal X-ray diffraction refinements
For [Al(ORF)4]− compounds, the structure determination by single crystal X-ray diffraction frequently tends to be problematic. Due to the very weak interactions of the perfluorinated residues with the cation, but also with each other, these residues are able to rotate around the C–O and the Al–O bonds. As a result, the OC(CF3)3 moieties in this anion are often heavily disordered and/or feature rather large anisotropic displacement ellipsoids. This is one of the reasons, why we introduced the DSR program code, nowadays included with OLEX2 (ref. 24) and ShelXLe,25 to facilitate the description of these often very complicated disorders.26 These problems are typically less pronounced in salts containing [al–f–al]−. This is due to the further shortened Al–O bonds (169.7 pm vs. 172.5 pm on average)6,16 that force the six OC(CF3)3 groups to be distributed over the rather small Al–F–Al core and increase the steric strain. For these reasons, the OC(CF3)3 groups are not able to rotate as freely as in [Al(ORF)4]− and are usually locked in one or two orientations. Therefore in many cases there is much less disorder and also smaller displacement ellipsoids were observed for [al–f–al]− structures compared to [Al(ORF)4]−. Nevertheless, there are also cases, where the entire anion is disordered around the bridging fluorine atom (see ESI of ref. 21†).Attempts to exchange Cat+[PF6]− for Cat+[BF4]−
[PF6]− salts are usually more expensive than the analogous [BF4]− salts (the price per mole of Ag[PF6] is more than 3 times as high as that of Ag[BF4]). Therefore we tried to use [Cat][BF4] for the synthesis of [f–al]− and [al–f–al]−. NMR spectra of the reaction solution taken in C6H5F/CDCl3 showed several signals for the ORF and Al–F moieties in the 19F NMR spectrum. This suggests a ligand scrambling reaction and formation of [AlFx(ORF)4−x]− (x = 1–3) and their F-bridged analogues. From the 11B NMR spectrum only formation of BF(ORF)2 was visible. Since this solution contained lots of precipitate, another sample was prepared in C6H5F/acetone-d6 in order to dissolve this precipitate. Here NMR spectra showed mainly the formation of unbridged [AlFx(ORF)4−x]− and several boron containing species. The formation of [AlFx(ORF)4−x]− occurs due to the Lewis basicity of acetone and will be discussed later. These NMR spectra suggested a complex mixture of different boron and aluminum containing species. Inter alia, [Ag(PhF)][F2B(ORF)2] (see ESI† for details) crystallized from one of the mixtures. Interestingly, in this structure the Ag+ is coordinated to the oxygen atoms of the anion, since the more Lewis basic (B–)F atoms form good hydrogen bonds with the coordinated solvent molecules.Due to the lower Lewis acidity of the evolving BF3 compared to PF5, the ligand scrambling reaction would rather be expected when using [PF6]− salts for these reactions (FIA = 346 vs. 380 kJ mol−1; the CIA, HIA and MIA are also higher for PF5).27 The same trend holds, when comparing the calculated [ORF]− ion affinities of BF3 (208 kJ mol−1) and PF5 (239 kJ mol−1) at BP86-D3(BJ)/def-SV(P) level. Therefore, a kinetic reason for this ligand scrambling has to be considered. The according reaction intermediate very likely involves Al–O(RF)–B/P and Al–F–B/P bridging. In case of PF5, this transition state is energetically less accessible due to its increased size and decreased tendency to undergo bridging.
Additionally, the stabilities of the theoretical reaction products, i.e. BF2(ORF) and PF4(ORF), also should be considered, but this is rather a secondary effect. BF2(ORF) features a tricoordinated, planar boron atom, which is able to undergo π back bonding with the lone pairs of the alkoxy moiety. This back bonding is stronger than that of fluoride and leads to a considerable stabilization of this molecule. The BF2(ORF) is able to undergo a second ligand scrambling to form the observed BF(ORF)2. In PF4(ORF) this π back bonding is not present, presumably resulting in less favorable thermodynamics in matters of the discussed ligand scrambling reaction. Therefore, the isolation of [f–al]− and [al–f–al]− is best possible when using [PF6]− salts, although the Lewis acidities of PF5 and BF3 would suggest otherwise.
Improved WCA performance of [al–f–al]−
When considering the calculated electrostatic potentials of [Al(ORF)4]−, [al–f–al]− and [f–al]− (Fig. 4) it can be concluded that [al–f–al]− should be the least coordinating of these anions and about as coordinating as [(C6F5)3Al–F–Al(C6F5)3]−. This is mainly due to the delocalization of the negative charge over 55 fluorine atoms, but also because the negative charge is partially localized at the bridging fluorine atom with an NPA charge of −0.67 vs. −0.34 for the terminal F atoms, similar to [f–al]− (NPA charge = −0.69 vs. −0.35; BP86/def-SV(P)). This makes [al–f–al]− more stable towards F− and [RFO]− abstraction than [Al(ORF)4]− (see later).16,28,29 | ||
| Fig. 4 Projection of the calculated electrostatic potential onto a 0.025 e− Bohr−3 isodensity surface of commonly used WCAs and the anions presented in this work; BP86/def-SV(P). | ||
The reactions of Ag[Al(ORF)4] with halosilanes R3SiX (R = Me, tBu, Ph; X = Cl, Br, I) in CH2Cl2 were already described.13,14 At rt these reactions yield R3Si–F–Al(ORF)3, while at −50 °C the halide bridged bis-silylium ions [R3Si–X–SiR3]+ were obtained. When we tried to reproduce these reactions with Ag[al–f–al] we noticed several curiosities. First of all, while Ag[Al(ORF)4] is highly soluble in CH2Cl2 at low temperatures, Ag[al–f–al] is only poorly soluble in cold CH2Cl2, although a higher solubility would have been expected with the weaker interactions between cation and anion. Secondly, when Me3SiCl, Ph3SiCl or tBu3SiBr were added to the reaction solution, no silver halide formed. NMR spectra also revealed almost quantitative retention of the anion and the halosilane. The 29Si NMR chemical shifts of the silanes, however, were slightly shifted to lower field (0.5–2 ppm), probably due to coordination to the silver cation. From the reaction with tBu3SiBr single crystals could be obtained and were identified as [Ag(tBu3SiBr)2(CH2Cl2)2][al–f–al] (Fig. 5). It is possible that related products may form with Me3SiCl and Ph3SiCl.
 | ||
| Fig. 5 Molecular structure of [Ag(tBu3SiBr)2(CH2Cl2)2]+[al–f–al]− at 100 K with thermal ellipsoids at 50% probability level. The C(CH3)3 moieties are shown in wireframe. Scheme: Ag (light gray), Br (brown), Si (yellow), Cl (green), Al (pink), O (red), F (light green), C (grey), H (white). Selected distances (pm) and bond angles (deg): Si1–Br1 231.87(7), Si2–Br2 231.88(6), Ag–Br1 265.68(5), Ag–Br2 268.33(5), Ag–Cl1 262.22(8), Ag–Cl2 276.0(1), Al1–F1 176.6(1), Al2–F1 176.9(1), Al1–F1–Al2 159.51(8). | ||
Although the formation of this cation is in agreement with the findings of Reed et al. that silylium ions are able to dissolve AgBr,30 at the same time it is contradictory to the reactions of Ag[Al(ORF)4], where halide abstraction from halosilanes was observed.13,14 This leads to the conclusion that [Al(ORF)4]− cannot be innocent in these reactions as similar halide abstractions are observed with Ag[ClO4]. Only when Me3SiI was added to Ag[al–f–al] in toluene/1,2,3-C6H3F3 in a 1:1 stoichiometry, the formation of AgI could be observed. NMR spectra of this solution predominantly showed decomposition of the anion and formation of Me3Si–F–Al(ORF)3 next to other unidentified products.
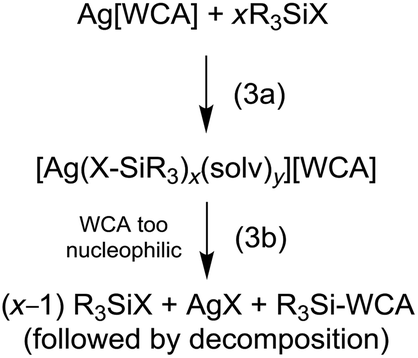
Judged from these reactions and their solution NMR spectra, we propose that upon reaction of Ag[WCA] and halosilanes, R3SiX, always the according silver complexes [Ag(X–SiR3)x(solv)y]+ are formed (eqn (3a)). These complexes contain an activated silicon and are strong silylating reagents. They react with nucleophiles (Nu) under formation of Nu–SiR3 and AgX. It appears, that for anions like [ClO4]− or even [Al(ORF)4]− this nucleophile can also be the anion (!), which subsequently may decompose (eqn (3b)). However, apparently the nucleophilicity of the [al–f–al]− WCA is too low, to induce AgX separation. In addition, the silylation strengths of the [Ag(X–SiR3)x(solv)y]+ cation increases, when going from X = Cl to I, mainly due to weakening of the Si–X bond. Overall, the lacking formation of AgX by reaction of Ag[al–f–al] with R3SiX (X = Cl, Br) can be seen as first experimental proof for the less coordinating nature of [al–f–al]− with respect to [Al(ORF)4]− or other WCAs that react with halosilanes under separation of AgX.
Another evidence for the weaker coordination and increased stability of [al–f–al]− compared to [Al(ORF)4]− is the intermediate synthesis of silylium ions by reaction of [Ph3C][WCA] with Me3SiH in CH2Cl2. In case of [WCA]− = [Al(ORF)4]− only Me3Si–F–Al(ORF)3 is formed at rt within 30 min. In contrast to this, in case of [WCA]− = [al–f–al]− the solvent is attacked under formation of Me3SiCl. In the NMR spectra of the latter reaction, the anion is mostly intact but decomposes within two days at room temperature. We were not able to identify the according cation, but judged by the formation of Me3SiCl, it might be a chlorinated carbocation resulting from the solvent CH2Cl2. Exchanging the CH2Cl2 for o-DFB, we were not able to isolate silylium ions at rt by this route when using Me3SiH or iPr3SiH. Again, we mainly observed formation of R3Si–F–Al(ORF)3 and other decomposition products, similar to the reaction of Ag[al–f–al] with Me3SiI.
Thermodynamic stability of [al–f–al]−
[al–f–al]− is stable against very strong silylating agents, such as [Ag(X–SiR3)x(solv)y]+ (X = Cl, Br), but not against small silylium ions. Therefore, we were interested in evaluating its general stability towards fluoride abstraction. This was done by calculating the decomposition reaction energies of [al–f–al]− and [Al(ORF)4]− using to the FIA procedure (Table 3).31 Although this was already done in the literature,28 we repeated these calculations with inclusion of the D3(BJ) dispersion correction (BP86-D3(BJ)/def-SV(P)). This seemed reasonable, since the strength of the Al–F–Al bond in [al–f–al]− is underestimated by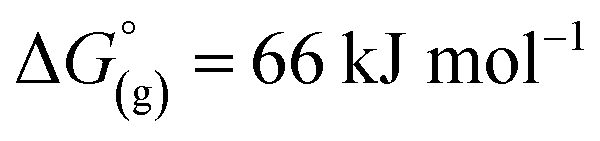 at BP86/def-SV(P) level. This also holds for the weak interaction between the evolving epoxide C4F8O and the corresponding Lewis acids. Yet, these calculations show only minor discrepancies to the literature.
at BP86/def-SV(P) level. This also holds for the weak interaction between the evolving epoxide C4F8O and the corresponding Lewis acids. Yet, these calculations show only minor discrepancies to the literature.
| Decomposition reaction |
|
|
ΔG°(CH2Cl2) |
|---|---|---|---|
| a Thermodynamic values for F− were calculated using the Sackur–Tetrode equation. Only the solvation free energy was calculated at DFT level. | |||
| [Al(ORF)4]− → F− + C4F8O–Al(ORF)3a | 605 | 561 | 324 |
| [al–f–al]− → F− + C4F8O–Al(ORF)2–F–Al(ORF)3a | 676 | 639 | 383 |
| [Al(ORF)4]− + [Me3Si]+ → Me3SiF + C4F8O–Al(ORF)3 | −349 | −336 | −49 |
| [al–f–al]− + [Me3Si]+ → Me3SiF + C4F8O–Al(ORF)2–F–Al(ORF)3 | −277 | −257 | 10 |
| 2CH2Cl2 + [Me3Si]+ → Me3SiCl + [(H2)(Cl)C–Cl–CH2Cl]+ | −60 | −4 | 0 |
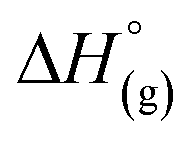
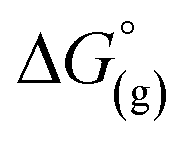
Again, these calculations support the previous experiments and show that [al–f–al]− is more stable against fluoride abstraction than [Al(ORF)4]−. Additionally, the decomposition of [al–f–al]− by [Me3Si]+ was calculated to be endergonic in CH2Cl2 by ΔG° = 10 kJ mol−1 and to be less favored than the decomposition of CH2Cl2. This is also in accordance to our reactions of [Ph3C][WCA] with Me3SiH in CH2Cl2. It should be noted, that the calculated reactions are only the first step in the decomposition of CH2Cl2, [Al(ORF)4]− and [al–f–al]− and are not the final reaction products. As a result, [al–f–al]− is not stable against small silylium ions at rt, although these calculations suggest so.
The Lewis acidity of [al–f–al]−: implications for solvent compatibility
The increased stability of [al–f–al]− against electrophiles compared to [Al(ORF)4]− seems to make it the anion of choice for the synthesis of reactive cations.28 However, this increased stability comes for a price: it's clearly noticeable Lewis acidity. Thus, when investigating the NMR spectra of K+[al–f–al]− we learned that as soon as we added Et2O to the NMR sample in order to completely dissolve the K[al–f–al], the NMR signals of the anion disappeared, and only the signals of Et2O–Al(ORF)3 and [f–al]− remained. The fluoride bridged anion [al–f–al]− can thus be seen as an adduct of [f–al]− and the Lewis superacid Al(ORF)3.32 Obviously, a large part of the Lewis acidity is retained in the final product leading to the cleavage of the anion in presence of Lewis bases (Scheme 2).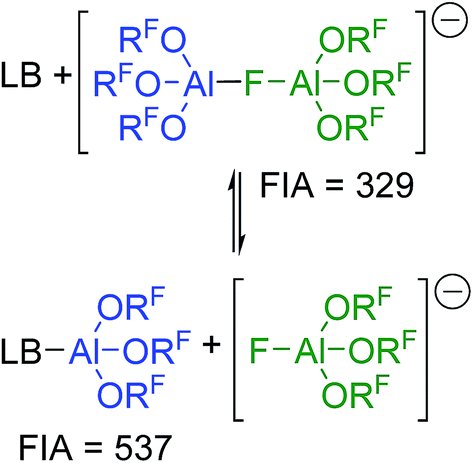 | ||
| Scheme 2 Equilibrium between LB/[al–f–al]− and LB–Al(ORF)3/[f–al]−; LB = Lewis base. Calculated FIAs refer to the free Lewis acids and are given in kJ mol−1; BP86-D3(BJ)/def-SV(P). | ||
Using the Fluoride Ion Affinity (FIA) as a measure, the Lewis acidity of [al–f–al]− is with a FIA-value of 329 kJ mol−1 in the region of BF3 (346 kJ mol−1). This limits the choice of solvents: in basic solvents such as Et2O and MeCN the equilibrium shown in Scheme 2 is on the side of LB–Al(ORF)3 and [f–al]−. In SO2, PhF, o-DFB, CH2Cl2 and CHCl2F the fluoride bridged anion [al–f–al]− was found to be completely intact by NMR spectroscopy. However, since SO2–Al(ORF)3 and Ph–F–Al(ORF)3 are known to be relatively stable,33 an equilibrium between [al–f–al]− and LB–Al(ORF)3/[f–al]− (here LB = SO2, PhF) may be possible. For SO2 this equilibrium was calculated to be on the side of [al–f–al]− by  using the COSMO model (ε = 16.3) at BP86-D3(BJ)/def-SV(P) level. This is in agreement with our experimental data, since we were able to synthesize [al–f–al]− salts free of [f–al]− in SO2. Theoretically, the [f–al]− anion may also act as Lewis base here, i.e. when the [al–f–al]− salt is contaminated with [f–al]−. In this case, an exchange of Al(ORF)3 moieties between [f–al]− and [al–f–al]− is expected. However, the presence of the 1JAl–F and 5JF–F coupling for both anions in this mixture definitely excludes this exchange. We did also not observe such an exchange in the 19F,19F-EXSY NMR spectrum or by line shape analysis. In contrast to this, the analogous chloride bridged anion [al–cl–al]− dissociates in PhF.34 Therefore, we suggest using CHCl2F, CH2Cl2, o-DFB, PhF and SO2 as solvents of choice, as they are polar and feature low Lewis basicity.
using the COSMO model (ε = 16.3) at BP86-D3(BJ)/def-SV(P) level. This is in agreement with our experimental data, since we were able to synthesize [al–f–al]− salts free of [f–al]− in SO2. Theoretically, the [f–al]− anion may also act as Lewis base here, i.e. when the [al–f–al]− salt is contaminated with [f–al]−. In this case, an exchange of Al(ORF)3 moieties between [f–al]− and [al–f–al]− is expected. However, the presence of the 1JAl–F and 5JF–F coupling for both anions in this mixture definitely excludes this exchange. We did also not observe such an exchange in the 19F,19F-EXSY NMR spectrum or by line shape analysis. In contrast to this, the analogous chloride bridged anion [al–cl–al]− dissociates in PhF.34 Therefore, we suggest using CHCl2F, CH2Cl2, o-DFB, PhF and SO2 as solvents of choice, as they are polar and feature low Lewis basicity.
Taking advantage of [f–al]−
As already discussed, [al–f–al]− decomposes in presence of nucleophiles under formation of its more coordinating brother [f–al]−. This anion is less suited for the stabilization of reactive cations, since the anion is very likely to undergo ion pairing due to the strongly coordinating Al-bound fluorine atom. However, the characterization of reactive cations in combination with WCAs can be very demanding, e.g. because of bad crystallization behavior due to the very low Coulomb interactions between anion and cation. In these cases the use of [f–al]− as a coordinating anion may be very helpful. Due to the stronger interaction between the anion and the cation, the crystallization of the reaction product may be facilitated. In a different project to be published with all details separately,35 we were able to synthesize [SeCl3][WCA] by reaction of Ag[WCA] ([WCA]− = [Al(ORF)4]−, [al–f–al]−, [f–al]−) with SeCl4 and to crystallize all three compounds (Fig. 6).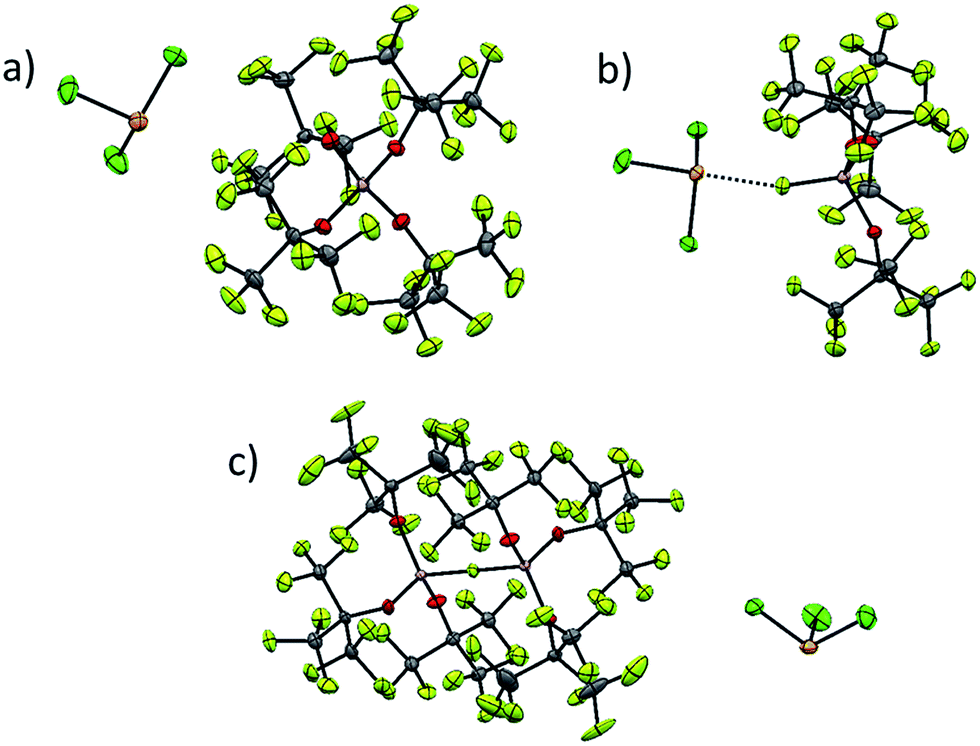 | ||
| Fig. 6 Complete ion pairs cut out of the solid state structures of the three [SeCl3]+[WCA]− salts at 100 K with thermal ellipsoids drawn at 50% probability level. Disorder was omitted for clarity (see ESI† for details). (a) [SeCl3]+[Al(ORF)4]−; (b) Cl3Se–[f–al]; (c) [SeCl3]+[al–f–al]−. Scheme: Se (orange), Cl (green), Al (pink), O (red), F (light green), C (grey). | ||
While single crystal X-ray diffraction measurements on these crystals yielded a superstructure at 100 K for [SeCl3][Al(ORF)4], a simpler solution was obtained, when using [f–al]− and [al–f–al]− as anions at 100 K. Especially the crystal structure of Cl3Se–[f–al], which crystallized as a contact ion pair, showed no disorder at all and only contains one molecule in the asymmetric unit. The bond valence36 (bv) of only 0.16 for the Se–F contact suggests a weak interaction between the cation and the anion (cf. bv(Al–F) = 0.68). Nevertheless, in the 19F NMR spectrum the (Al–)F signal is shifted from −187 to −145 ppm. A related effect was also observed for [NO][f–al], which may be caused by coordination to the cation.
This coordination of [f–al]− to [NO]+ can be evaluated by the Raman vibrational frequency of the N–O stretching vibration, since a strong coordination of the anion leads a redshift of this vibration.37 The N–O vibrational frequencies given in Table 4 suggest that [f–al]− (2313 cm−1) is stronger coordinating than [BF4]− and [B(CF3)4]− (2340 and 2337 cm−1), but less coordinating than [B(CF3)3CN]− and [GaCl4]− (2288 and 2226 cm−1).
| Compound | Vibrational frequency |
|---|---|
| [NO][GaCl4] | 2226 |
| [NO][B(CF3)3CN] | 2288 |
| [NO][f–al] | 2313 |
| [NO][B(CF3)4] | 2337 |
| [NO][BF4] | 2340 |
| [NO][Al(ORF)4] | 2340 |
| [NO][al–f–al] | 2340 (ref. 38) |
Additionally to crystallization, [f–al]− salts can be used as key intermediates towards [al–f–al]− salts. Since [f–al]− is compatible with nucleophiles, it can be used for the synthesis of cations, which require the presence of nucleophiles or coordinating solvents. The resulting product can then be transformed into the [al–f–al]− salt by addition of Me3Si–F–Al(ORF)3. The only prerequisite for this transformation is that the cation and [f–al]− have to be separated in solution to some extent, i.e. as solvent separated ion pair providing access to small amounts of free [f–al]− for the reaction with Me3Si–F–Al(ORF)3.
Limits of [f–al]−
To this point [f–al]− seemed to be rather stable against highly Lewis acidic cations, like [SeCl3]+ and [Me3Si]+ (FIA = 815 and 952 kJ mol−1, respectively; BP86/def-SV(P)), due to formation of stable adducts. A comparison of the Al–F bond valences of 0.68 (Cl3Se–[f–al]) and 0.50 (Me3Si–F–Al(ORF)3)13 already suggests, that extremely electrophilic cations might be able to abstract the Al-bound fluorine atom. Yet, the relatively high bv in Me3Si–F–Al(ORF)3 is astonishing, considering the FIA of [Me3Si]+ is higher by 415 kJ mol−1 compared to Al(ORF)3 (FIA = 537 kJ mol−1).32 Therefore, it seemed possible to stabilize an ion-like [PCl2]+ cation (FIA = 1001 kJ mol−1)39 with [f–al]−, although it is not possible with [Al(ORF)4]− (eqn (1b)). When we treated Ag[f–al] with PCl3 in o-DFB we were expecting the formation of Cl2P–F–Al(ORF)3 (eqn (4a)). As expected, after a few seconds a precipitate formed (AgCl). NMR spectra, however, did not show the formation of the expected product, but mainly signals of PF3, PCl3 and Ag[al–f–al], besides small signals of PClF2 and PCl2F. The presence of PFxCl3−x (x = 1, 2, 3) and [al–f–al]− is evidence that Cl2P–F–Al(ORF)3 was formed. But at the same time there was nucleophilic [f–al]− present in solution, which led to formation of [al–f–al]− and PCl2F (eqn (4b)), analogous to the previously described reactions (eqn (2b)). The PCl2F then further dismutated to PF3, as described in eqn (4b). The ratio of the products also suggests that the halide abstraction from PFxCl3−x is preferred over PCl3. Interestingly again, the Ag[al–f–al] formed did not participate in further halide abstraction reactions (see our comments above).
As a conclusion, [f–al]− may be well suited for the stabilization of reactive cations as neutral ion-like Cat–F–Al(ORF)3 compounds. However, the presence of another [f–al]− anion may also lead to formation of [al–f–al]− and release of Cat–F. In these cases the best way to obtain a desired Cat–F–Al(ORF)3 may be the reaction of Cat–F with Al(ORF)3 or its equivalent Me3Si–F–Al(ORF)3.
Conclusion
By reaction of Me3Si–F–Al(ORF)3 with different [PF6]− salts we were able to selectively synthesize the anions [(RFO)3Al–F–Al(ORF)3]− ([al–f–al]−) and [F–Al(ORF)3]− ([f–al]−). In the presence of nucleophiles or coordinating solvents, however, [al–f–al]− was shown to react under formation of [f–al]− and Nu–Al(ORF)3. By intermediate generation of small silylium ions and isolation of [Ag(tBu3SiBr)2(CH2Cl2)2]+ we were able to support the role of [al–f–al]− as “least-coordinating-anion”:16 thus, the elimination of AgX from the formed solvated [Ag(XSiR3)x]+ cation requires the back-side attack of a nucleophile (solvent, anion). It appears that in non-basic solvents and in contrast to [Al(ORF)4]−, the nucleophilicity of [al–f–al]− is too low to allow for AgX formation with X = Cl and Br. These reactions and quantum-chemical calculations also show that [al–f–al]− is more stable against electrophiles than [Al(ORF)4]− and may be similar or in part also better than the borate [B(C6F5)4]−. However, the synthetic access to [al–f–al]− is now greatly facilitated compared to that of the borate [B(C6F5)4]−, which requires handling of explosive LiC6F5. Compared to [Al(ORF)4]−, the refinement of single crystal X-ray diffraction data of [al–f–al]− and [f–al]− salts usually tends to be facilitated. This is due to less disorder of the OC(CF3)3 moieties for [al–f–al]− (increased bulk) and due to ion-pairing for [f–al]−. The stronger coordination of [f–al]− could be used for the synthesis and crystallization of Cl3Se–[f–al] without any disorder. Additionally, we were able to show that [f–al]− salts can be transformed into [al–f–al]− salts by addition of Me3Si–F–Al(ORF)3, if the salts exist as solvent separated ion pairs in solution.Due to the facile synthesis of Me3Si–F–Al(ORF)3 and the anions [f–al]− and [al–f–al]− in large scales and high yields, we believe that these anions will find wide-spread use for the generation and stabilization of reactive cations, but also suggest they may be suitable for catalytic processes. Here, the [f–al]− anion may even be helpful to stabilize in a hemilabile coordination scheme the resting state of the catalysis process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg, the ERC by the project UniChem, the BASF SE and the DFG. We would like to thank Daniel Himmel for the valuable discussion of the quantum-chemical calculations and reactions, the MagRes facility of the University of Freiburg and Fadime Bitgül for the measurement of the NMR spectra, Mario Schleep for the measurement and refinement of single crystal X-Ray diffraction data, Daniel Kratzert for the valuable discussions of the crystallographic data.References
- (a) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC; (b) I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201710782; (c) J. Schaefer, A. Kraft, S. Reininger, G. Santiso-Quinones, D. Himmel, N. Trapp, U. Gellrich, B. Breit and I. Krossing, Chem.–Eur. J., 2013, 19, 12468 CrossRef PubMed; (d) J. Schaefer, D. Himmel and I. Krossing, Eur. J. Inorg. Chem., 2013, 2013, 2712 CrossRef; (e) D. Aris, J. Beck, A. Decken, I. Dionne, J. Schmedt auf der Günne, W. Hoffbauer, T. Köchner, I. Krossing, J. Passmore, E. Rivard, F. Steden and X. Wang, Dalton Trans., 2011, 40, 5865 RSC; (f) T. Köchner, N. Trapp, T. A. Engesser, A. J. Lehner, C. Röhr, S. Riedel, C. Knapp, H. Scherer and I. Krossing, Angew. Chem., Int. Ed., 2011, 50, 11253 CrossRef PubMed; (g) J. Schaefer, A. Steffani, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6009 CrossRef PubMed.
- T. O. Petersen, E. Tausch, J. Schaefer, H. Scherer, P. W. Roesky and I. Krossing, Chem.–Eur. J., 2015, 21, 13696 CrossRef PubMed.
- M. Schleep, C. Hettich, J. Velázquez Rojas, D. Kratzert, T. Ludwig, K. Lieberth and I. Krossing, Angew. Chem., Int. Ed., 2017, 56, 2880 CrossRef PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9259 CrossRef PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9262 CrossRef PubMed.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490 CrossRef.
- (a) D. M. van Seggen, P. K. Hurlburt, O. P. Anderson and S. H. Strauss, Inorg. Chem., 1995, 34, 3453 CrossRef; (b) M. J. Collins and G. J. Schrobilgen, Inorg. Chem., 1985, 24, 2608 CrossRef; (c) H. P. A. Mercier, J. C. P. Sanders and G. J. Schrobilgen, J. Am. Chem. Soc., 1994, 116, 2921 CrossRef; (d) K. Moock and K. Seppelt, Z. Anorg. Allg. Chem., 1988, 561, 132 CrossRef.
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263 CrossRef PubMed.
- (a) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245 CrossRef; (b) E. Martin, D. L. Hughes and S. J. Lancaster, Inorg. Chim. Acta, 2010, 363, 275 CrossRef.
- M. Saleh, D. R. Powell and R. J. Wehmschulte, Inorg. Chem., 2016, 55, 10617 CrossRef PubMed.
- (a) H. Kropshofer, O. Leitzke, P. Peringer and F. Sladky, Chem. Ber., 1981, 114, 2644 CrossRef; (b) A. Engelbrecht and F. Sladky, Angew. Chem., 1964, 76, 379 CrossRef.
- (a) J. B. Lambert, S. Zhang, C. L. Stern and J. C. Huffman, Science, 1993, 260, 1917 CrossRef PubMed; (b) C. A. Reed, Acc. Chem. Res., 2010, 43, 121 CrossRef PubMed.
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem.–Eur. J., 2014, 20, 1218 CrossRef PubMed.
- A. Budanow, M. Bolte, M. Wagner and H.-W. Lerner, Eur. J. Inorg. Chem., 2015, 2015, 2524 CrossRef.
- H. Großekappenberg, M. Reißmann, M. Schmidtmann and T. Müller, Organometallics, 2015, 34, 4952 CrossRef.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem.–Eur. J., 2004, 10, 5041 CrossRef PubMed.
- L. Birckenbach and K. Kellermann, Ber. Dtsch. Chem. Ges., 1925, 58, 786 CrossRef.
- (a) S. J. Lancaster, D. A. Walker, M. Thornton-Pett and M. Bochmann, Chem. Commun., 1999, 1533 RSC; (b) A. J. Mountford, W. Clegg, S. J. Coles, R. W. Harrington, P. N. Horton, S. M. Humphrey, M. B. Hursthouse, J. A. Wright and S. J. Lancaster, Chem.–Eur. J., 2007, 13, 4535 CrossRef PubMed; (c) M.-C. Chen, J. A. S. Roberts, A. M. Seyam, L. Li, C. Zuccaccia, N. G. Stahl and T. J. Marks, Organometallics, 2006, 25, 2833 CrossRef.
- D. Himmel, H. Scherer, D. Kratzert and I. Krossing, Z. Anorg. Allg. Chem., 2015, 641, 655 CrossRef.
- (a) T. S. Cameron, A. Decken, I. Krossing, J. Passmore, J. M. Rautiainen, X. Wang and X. Zeng, Inorg. Chem., 2013, 52, 3113 CrossRef PubMed; (b) H. Poleschner and K. Seppelt, Angew. Chem., Int. Ed., 2013, 52, 12838 CrossRef PubMed.
- J. Possart, A. Martens, M. Schleep, A. Ripp, H. Scherer, D. Kratzert and I. Krossing, Chem.–Eur. J., 2017, 23, 12305 CrossRef PubMed.
- R. Damrauer, R. A. Simon and B. Kanner, Organometallics, 1988, 7, 1161 CrossRef.
- X. Zheng, Z. Zhang, G. Tan and X. Wang, Inorg. Chem., 2016, 55, 1008 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933 CrossRef PubMed.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC.
- I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721 CrossRef.
- I. Krossing and I. Raabe, Chem.–Eur. J., 2004, 10, 5017 CrossRef PubMed.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 325 CrossRef.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151 CrossRef.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659 CrossRef PubMed.
- A. Kraft, N. Trapp, D. Himmel, H. Böhrer, P. Schlüter, H. Scherer and I. Krossing, Chem.–Eur. J., 2012, 18, 9371 CrossRef PubMed.
- A. Kraft, J. Beck and I. Krossing, Chem.–Eur. J., 2011, 17, 12975 CrossRef.
- P. Weis and I. Krossing, manuscript in preparation.
- I. D. Brown, J. Appl. Crystallogr., 1996, 29, 479 CrossRef.
- T. A. Engesser, C. Friedmann, A. Martens, D. Kratzert, P. J. Malinowski and I. Krossing, Chem.–Eur. J., 2016, 22, 15085 CrossRef PubMed.
- J. Bohnenberger, W. Feuerstein, D. Himmel, F. Breher and I. Krossing, Nat. Chem., submitted Search PubMed.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, procedures, weights, 1D- and 2D-NMR spectra of the reactions are deposited. Details to the quantum chemical calculations are given together with crystallographic details. CCDC 1845808–1845817. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02591f |
| This journal is © The Royal Society of Chemistry 2018 |