
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling substituent effects on frontier orbitals of conjugated molecules using an absolutely localized molecular orbital based analysis†
Yuezhi
Mao
 a,
Martin
Head-Gordon
a and
Yihan
Shao
*b
a,
Martin
Head-Gordon
a and
Yihan
Shao
*b
aKenneth S. Pitzer Center for Theoretical Chemistry, Department of Chemistry, University of California at Berkeley, Berkeley, CA 94720, USA
bDepartment of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73019, USA. E-mail: yihan.shao@ou.edu; Tel: +1 405 325 0442
First published on 18th September 2018
Abstract
It is common to introduce electron-donating or electron-withdrawing substituent groups into functional conjugated molecules (such as dyes) to tune their electronic structure properties (such as frontier orbital energy levels) and photophysical properties (such as absorption and emission wavelengths). However, there lacks a generally applicable tool that can unravel the underlying interactions between orbitals from a substrate molecule and those from its substituents in modern electronic structure calculations, despite the long history of qualitative molecular orbital theory. In this work, the absolutely localized molecular orbitals (ALMO) based analysis is extended to analyze the effects of substituent groups on the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of a given system. This provides a bottom-up avenue towards quantification of effects from distinct physical origins (e.g. permanent electrostatics/Pauli repulsion, mutual polarization, inter-fragment orbital mixing). For the example case of prodan (a typical dye molecule), it is found that inter-fragment orbital mixing plays a key role in narrowing the HOMO–LUMO gap of the naphthalene core. Specifically, an out-of-phase mixing of high-lying occupied orbitals on the naphthalene core and the dimethylamino group leads to an elevated HOMO, whereas an in-phase combination of LUMOs on the naphthalene core and the propionyl group lowers the LUMO energy of the entire molecule. We expect this ALMO-based analysis to bridge the gap between concepts from qualitative orbital interaction analysis and quantitative electronic structure calculations.
1 Introduction
Substituent effects on conjugated molecules have been extensively explored in chemical, biochemical, and materials research. In the design of organic light-emitting/solar-cell materials and fluorescent probe molecules,1–5 it is routine to attach electron-donating groups (EDGs) and electron-withdrawing groups (EWGs) to dye molecules to tune their frontier orbital energies and absorption/emission wavelengths. In synthetic chemistry, it has been common practice over the last several decades to introduce EDGs/EWGs onto conjugated organic molecules to modulate their chemical reactivity.6Empirically, the substituent effects on conjugated systems have been explained using classical descriptors, especially Hammett constants (e.g. σp, σm, σp−, and σp+).6,7 These constants were developed originally to quantify the electron-donating and electron-withdrawing capabilities of different substituent groups in affecting the dissociation of benzoic acid and other reactions. Ever since, Hammett constants have also been employed in many other structure–property analyses.8–13 Most recently, researchers have observed some correlation between the Hammett constants and photophysical properties of oxyluciferin analogs,14,15 curcumin derivatives,5 and other chromophores/fluorophores. Despite numerous efforts,16–24 Hammett constants still lack a rigorous theoretical basis, especially in the context of analyzing substituent effects on the photophysical properties of conjugated molecules. Also noteworthy are several other empirical constants, including analogs of the Hammett parameter25,26 and quantities defined based on charge population analysis,27,28 as descriptors exclusively for excited-state properties, while their general applicability and usefulness remain unclear.
Quantum mechanically, the substituent effect on the photophysical properties of conjugated molecules can be investigated using either qualitative molecular orbital (MO) theory or quantitative quantum chemistry calculations. Within the qualitative MO theory,29 which dates back to the early days of quantum chemistry, one performs perturbational MO (PMO) analysis30–34 and studies constructive/destructive interference via mixing degenerate (such as the H2 molecule) or non-degenerate (such as the HF molecule) orbitals. Qualitative MO analysis has contributed greatly to our understanding of key chemical concepts, such as covalent bonding, metal–ligand binding, conjugation, hyperconjugation, hypervalency, and substituent effects in small-size molecular systems (see ref. 29 for detailed discussions).
In quantitative quantum chemistry calculations for excited states, one goes directly after a quantitative prediction of the electronic and photophysical properties, such as frontier orbital energies and excitation wavelengths/intensities. These calculations are usually based on modern wavefunction theory [such as configuration interaction singles (CIS),35 equation-of-motion coupled-cluster theory (EOM-CC),36,37 and the algebraic diagrammatic construction (ADC) approach38] or time-dependent density functional theory (TDDFT).39–42 In quantitative electronic structure theory, less and less emphasis has been placed on the orbital interaction pictures, despite the effort to develop new areas such as conceptual density functional theory.43 This, in our view, is mainly because only canonical MOs (CMOs) of the entire substituted system emerge naturally from such calculations. However, without resorting to the orbital interaction picture, one often finds it not straightforward to interpret substituent effects on molecular properties.
To bridge the gap between qualitative MO analysis and quantitative quantum chemistry calculations for substituted conjugated molecules, we will revisit the orbital interaction concepts and reformulate such analyses via a procedure similar to a variational energy decomposition analysis (vEDA). Dating back to the pioneering work of Kitaura and Morokuma,44,45 the vEDA family of methods46–56 have become a powerful tool for analyzing intermolecular interactions (see ref. 57 for a review). These methods connect non-interacting fragments (the initial state) and the fully interacting complex (the final state) via a series of intermediate states in which certain components of the interaction are “switched on/off”. Therefore, the energies of these intermediate states are usually variational upper bounds to that of the fully interacting state as obtained from quantitative quantum chemistry calculations based on Hartree–Fock or density functional theory (DFT). The energy differences between these (including the initial, intermediate, and final) states are then taken as the components of total interaction energy. In modern variants of vEDA for intermolecular interactions, the construction of intermediate states can be based on localized fragment orbitals [such as the absolutely localized molecular orbital (ALMO)-EDA46–49 and the block-localized wavefunction (BLW)-EDA50–52], natural bond orbitals [such as the natural energy decomposition analysis (NEDA)53,54], or fragment partition of electronic density [such as the density-based energy decomposition analysis (DEDA)55,56].
In this article, we extend the ALMO-based vEDA procedure46–49 to systems where a substituent is covalently bound to a conjugated molecule, giving rise to a bottom–up analysis of substituent effects on frontier orbitals. While many vEDA approaches for intermolecular interactions can be directly applied or extended to covalently bound systems,19,56,58–64 these methods mainly focus on the bonded interaction itself and usually employ a “bond-breaking” initial state (open-shell fragments). For example, another recent extension of ALMO-EDA for probing the “fingerprints” of single chemical bonds (e.g. ionicity, covalency, polarity, etc.)63,64 employed two spin-pure doublet fragments to construct the frozen wavefunction (which is a high-spin triplet). Our vEDA-like analysis (the word “vEDA-like” indicates that the quantity to be decomposed is not necessarily the interaction energy) differs from the above-referenced methods in not only the objective but also the preparation of the initial “fragment” state: instead of starting with radicals, we will adopt two separately prepared closed-shell fragments bridged via a “spectator” σ bond as the initial state (vide infra). On the other hand, our approach is inspired by EDA schemes for analyzing interactions between moieties belonging to the same molecule65–69 or intramolecular conjugation/hyperconjugation effects,70–74 which do not require cleavage of covalent bonds to prepare the initial state either but are for rather different purposes.
We should also mention that orbital interaction pictures can also be generated in a top–down manner, i.e., MOs belonging to different moieties can be constructed from fully converged self-consistent field (SCF) solutions.33,75–77 These approaches, however, do not facilitate a complete separation of individual contributions to the substituent effect. As a bottom–up approach, our ALMO-based analysis resembles the BLW analysis. Both analyses construct a mutually polarized but fragment-orbital-mixing-quenched state (referred to as the “POL” state below) by variationally optimizing its energy subject to the constraint that MOs are absolutely localized on fragments. The BLW approach was utilized by Mo and co-workers74 to probe the intramolecular interactions between the π-orbitals of the benzene ring and the amino/nitro groups in aniline/nitrobenzene, where similar “in situ” orbital interaction diagrams were produced. Instead of focusing solely on the π-substituent orbital interaction (during the transition from “POL” to the final state),74 our scheme is designed to provide well-defined “isolated fragments” (FRAG) and “frozen fragments” (FRZ) states (in addition to the POL state) in order to unravel other key perturbations (permanent electrostatics/Pauli repulsion, mutual polarization) to the frontier orbitals as well.
2 Methods
2.1 Notation
We shall follow the tensor formulation for electron structure calculations in a non-orthogonal basis.78 Throughout this paper, we use |ϕ〉 for atomic orbital (AO) basis functions, |ψ〉 for molecular orbitals (MOs), x, y for fragment indices, μ, ν for AO basis indices, i, j for occupied MO indices, a, b for unoccupied (virtual) MO indices, and p, q for generic (occupied or virtual) MO indices. Einstein summation convention is applied to a pair of identical covariant and contravariant indices (except for the orbital energy definitions in eqn (7) and (8)). Using this notation, occupied and virtual orbitals on the x-th fragment can be written as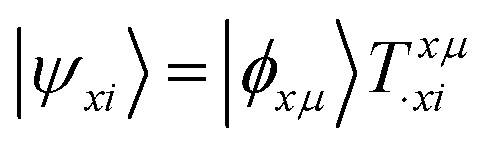 | (1) |
 | (2) |
 | (3) |
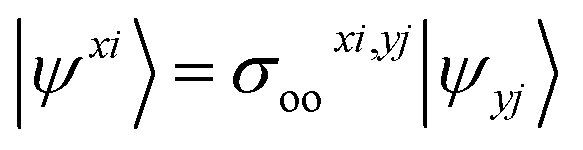 | (4) |
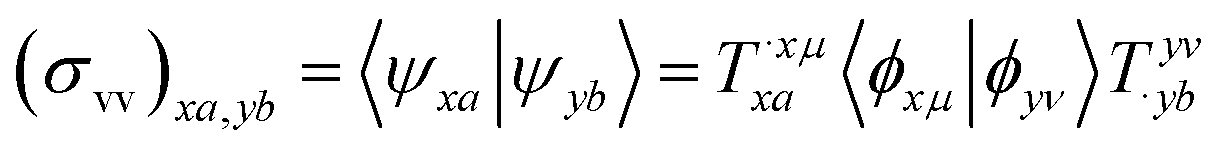 | (5) |
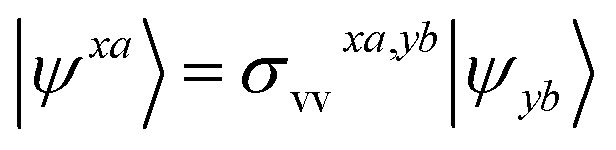 | (6) |
2.2 ALMO analysis procedure
Before introducing our method, we first briefly recapitulate the regular ALMO-EDA scheme46 for intermolecular interactions, which is schematically summarized in Fig. 1(a). The procedure begins with separately converged SCF solutions for isolated fragments, which correspond to the “FRAG” stage. The isolated fragments are then brought together in the complex with their orbitals remaining unrelaxed, giving rise to the frozen (FRZ) state of the supersystem. The total energy of the supersystem is then minimized subject to the “absolute localization” constraint, i.e., the fragment MOs are only allowed to be spanned by the AO basis functions that belong to the same fragment. The SCF solution converged therefrom represents the mutually polarized (POL) state. Further relaxations involve interfragment ov-mixings, and thus the associated energy lowering is interpreted as charge transfer (CT). It can be further split into two parts: (i) the perturbative CT energy calculated by performing a “Roothaan step” (RS) on top of the POL state,46,79 which corresponds to a single diagonalization of the supersystem Fock matrix associated with the POL state; and (ii) the high-order CT energy defined as the energy difference between the RS state and the fully converged SCF solution (the “FULL” state).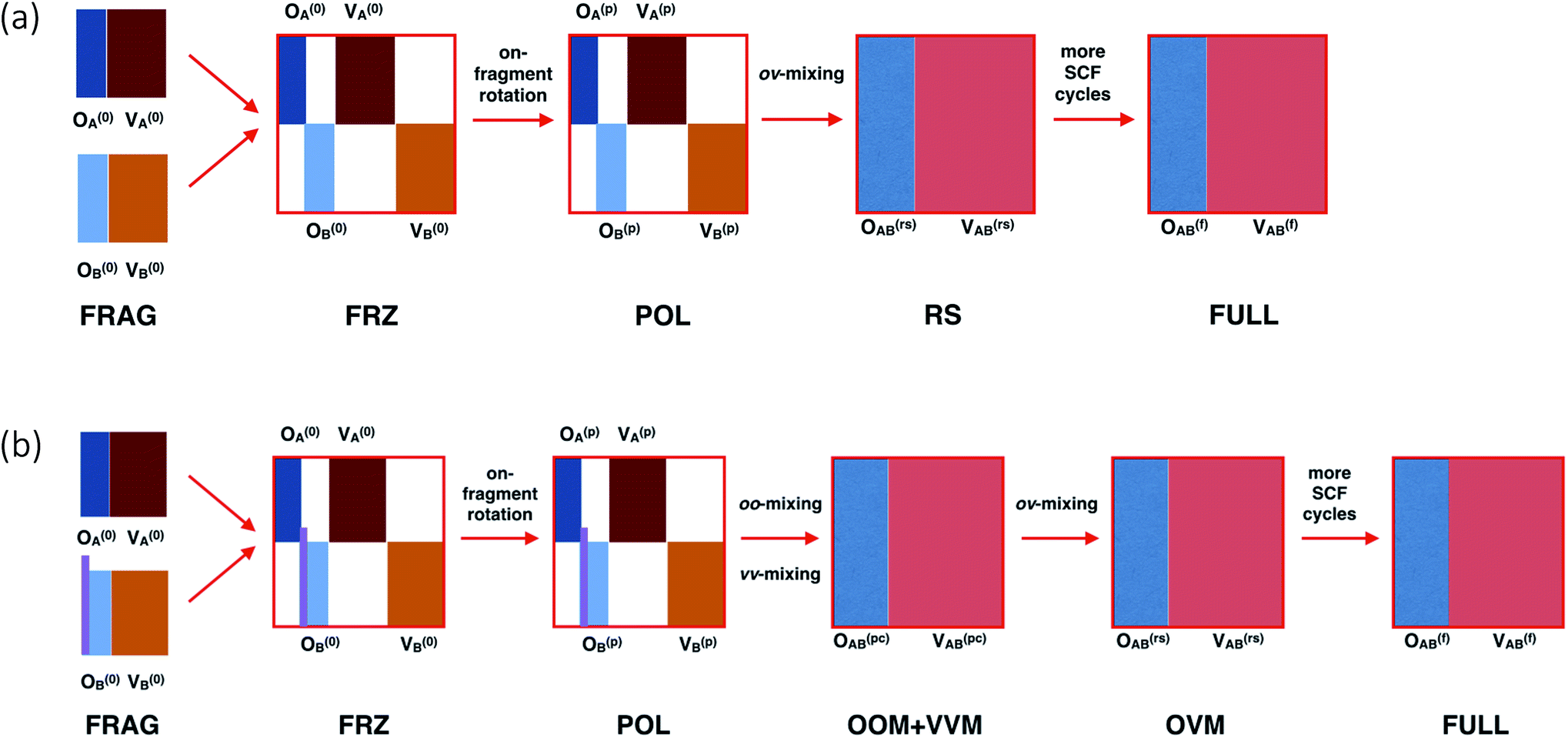 | ||
| Fig. 1 Schematic illustration of the procedure of (a) the regular ALMO-EDA for intermolecular interactions and (b) our ALMO-based analysis for substituent effects on frontier orbitals. “A” and “B” denote two fragments, and “O” and “V” refer to occupied and virtual orbitals, respectively. The vertical bars in light purple in panel (b) refer to the link orbital between two fragments (corresponding to the C–X single bond in Fig. 2). | ||
In this work, we focus on molecules with two covalently linked fragments: a conjugated molecule (Ar) and its substituent (X). As shown in Fig. 1(b), there are six states in our frontier orbital analysis. Compared to the regular ALMO-EDA procedure for intermolecular complexes, the preparation of the FRAG state here is more complicated due to the covalent linking, and the RS state in a regular ALMO analysis has been divided into two consecutive states “OOM + VVM” and “OVM” (defined below) because they have distinct effects on the energy of frontier orbitals. The details associated with these steps will be elaborated in the rest of this section.
From the optimized geometry of the entire Ar–X molecule, the construction of two “tailored-then-capped” fragments (Ar–H and Ph–X) is automated using a python script. Subsequently, from the full SCF solutions of these two systems, two sets of initial fragment orbitals can be prepared by truncating the Pipek–Mezey80 localized orbitals in the AO space and then re-optimizing them with a generalized ALMO-SCF scheme: one set for the aromatic ring (Ar) from Ar–H (excluding the C–H bond), and the other set for the substituent group (–X) from Ph–X (including the C–X single bond). The fragment orbital preparation scheme is illustrated in Fig. 2, and a detailed description of all steps here can be found in Section S1 of the ESI.†
 | ||
| Fig. 2 The “fragmentation” scheme for a substituted naphthalene. Orbitals belonging to Ar and –X are prepared through ALMO calculations for Ar–H and Ph–X, respectively. The combination of converged orbitals for Ar and –X yields the frozen state of Ar–X. | ||
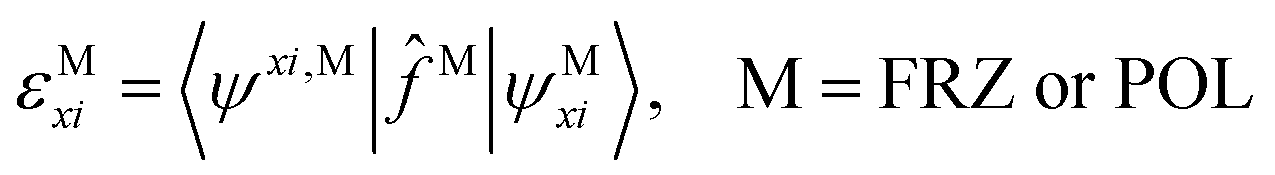 | (7) |
 | (8) |
The change in these orbital energies relative to isolated fragments contains both permanent electrostatics and Pauli repulsion contributions. It is possible to further separate them by introducing an extra intermediate state where the fragment wavefunctions are not yet antisymmetrized against each other.
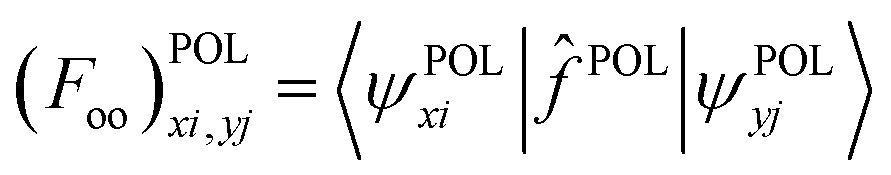 | (9) |
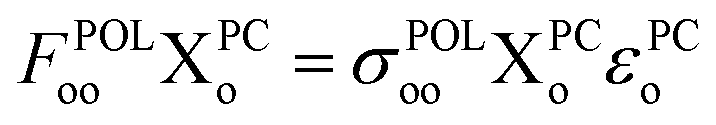 | (10) |
Such an oo-mixing does not change the 1PDM of the supersystem. Thus, it has no impact on the total energy or any density-dependent fragment charge population for the electronic ground state. However, as known in PMO theory (see, for example, Fig. 10.17 in ref. 29), oo-mixing does affect frontier orbital energies. Specifically, it can increase the HOMO energy, because the highest-energy pseudocanonical occupied orbital is expected to be an out-of-phase (i.e. destructive) combination of fragment occupied orbitals.
Moving next to the virtual orbitals, one first needs to project out the components in the occupied space from them:
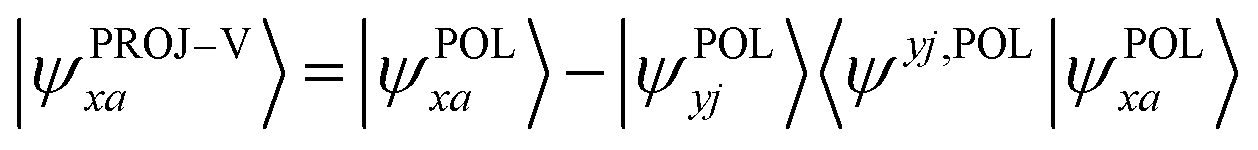 | (11) |
For virtual–virtual orbital mixing (VVM), the Fock matrix represented in the subspace of projected virtual orbitals
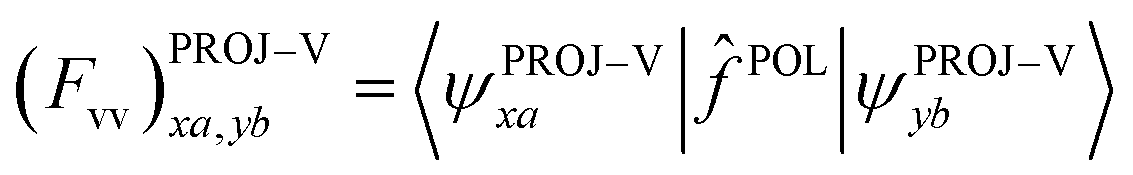 | (12) |
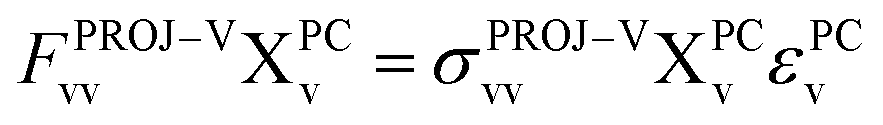 | (13) |
As is well known in PMO theory, when two orbitals with significantly different energies interact with each other, the lower-energy orbital acquires an even lower energy, and the higher-energy orbital is going to further increase its energy. Consequently, ov-mixing is expected to lower the HOMO energy while raising the LUMO energy, thus enlarging the HOMO–LUMO gap. This is exactly the opposite to oo- and vv-mixing.
 | (14) |
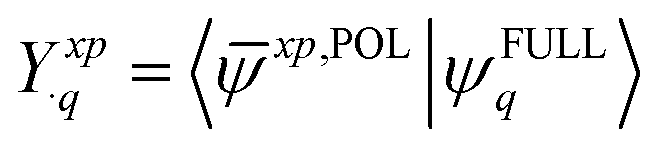 | (15) |
![[small psi, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d9.gif) xp,POL〉, we utilize a full overlap metric that contains not only the occupied–occupied (eqn (3)) and virtual–virtual blocks (eqn (5)) but also the occupied-virtual and virtual-occupied blocks.
xp,POL〉, we utilize a full overlap metric that contains not only the occupied–occupied (eqn (3)) and virtual–virtual blocks (eqn (5)) but also the occupied-virtual and virtual-occupied blocks.
3 Results and discussion
3.1 Test case—prodan molecule
Prodan [6-propionyl-2-(N,N-dimethylamino)naphthalene], a widely-used fluorescent probe molecule in biology,81,82 is employed in this work as an example to illustrate our ALMO-based analysis of substituent effects. With an absorption peak ranging from 342 nm in cyclohexane to 362 nm in water,83 its vacuum absorption wavelength is estimated to be around 340 nm. This is 65 nm longer than the 275 nm absorption wavelength of naphthalene vapor,84 which amounts to a reduction of the vertical excitation energy (VEE) from 4.50 eV (naphthalene) to 3.64 eV (prodan).As shown in Fig. 3, the absorption energies of both molecules can be well reproduced from TDDFT calculations. Using the B3LYP functional85,86 and the 6-31G(d) basis set for gas-phase ground-state geometry optimization and subsequent TDDFT calculations, the lowest VEE was computed to be 4.46 eV for naphthalene, and 3.55 eV for prodan, both within 0.1 eV from the experimental values. These calculations also indicated that, for both molecules, the lowest-energy electronic excitation is dominated by a HOMO → LUMO transition (amplitude > 0.94). Therefore, the reduction in the lowest VEE is mainly caused by the narrowing of the HOMO–LUMO gap, whose computed values as shown in Fig. 3 are 4.83 eV (naphthalene) and 3.87 eV (prodan).
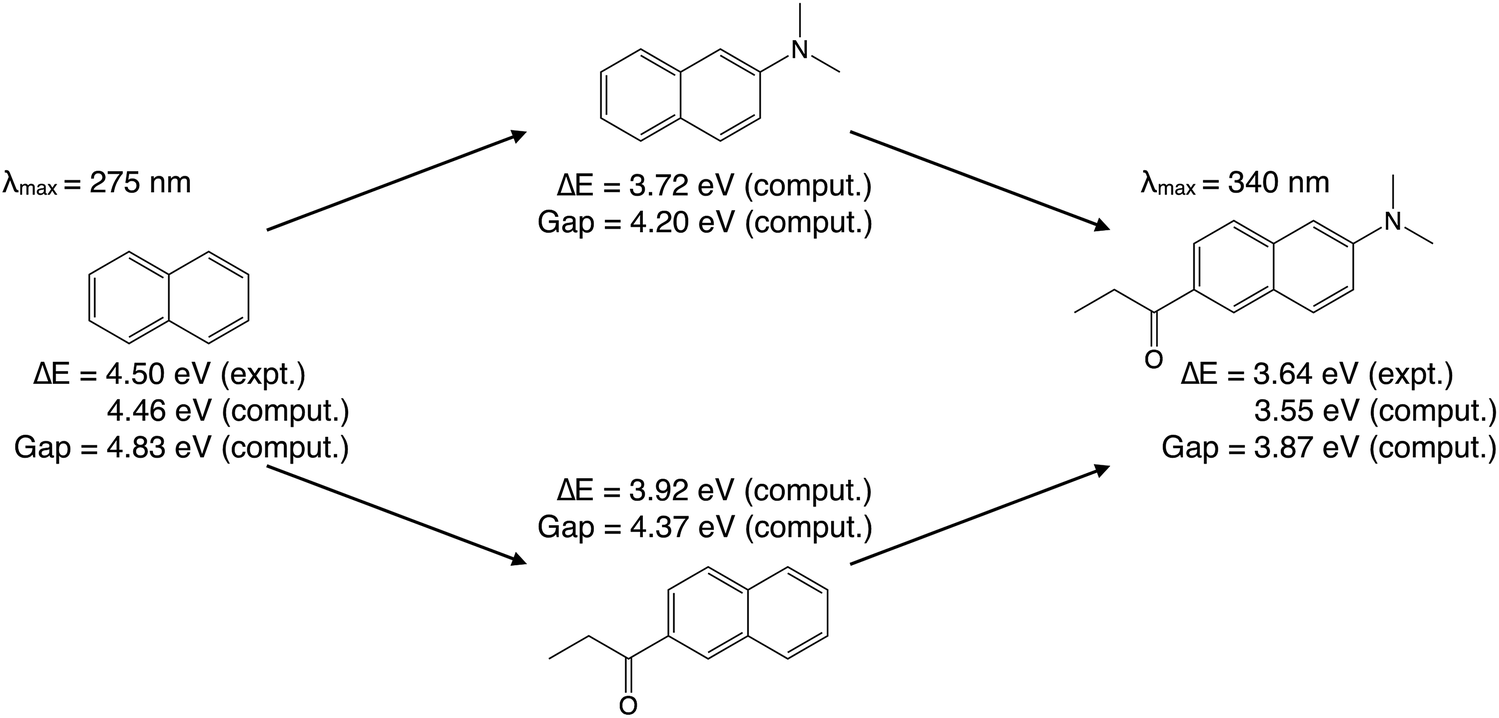 | ||
| Fig. 3 Experimental and computed gas-phase vertical excitation energies of naphthalene, prodan, and two intermediates. Computed absorption energies and HOMO–LUMO gaps were obtained from TDDFT calculations using B3LYP/6-31G(d). | ||
In computational studies, one can choose to attach only the electron-donating dimethylamino group or the electron-withdrawing propionyl group (of the prodan molecule) to the naphthalene core first, leading to the two intermediates in Fig. 3. Both 2-(dimethylamino)naphthalene and 6-(propionyl)naphthalene have smaller computed HOMO–LUMO gaps (4.20 and 4.37 eV, respectively) than that of naphthalene (4.83 eV). As a result, their computed VEEs (based on TDDFT) are 3.72 eV and 3.92 eV, respectively, which are more than 0.5 eV lower than the computed VEE of naphthalene. That raises an intriguing question: how can an EDG and an EWG both reduce the HOMO–LUMO gap and thus the lowest electronic excitation energy? This question will be addressed below by performing the ALMO-based analysis.
All calculations below were carried out in gas-phase with the B3LYP functional and the 6-31G(d) basis set using a development version of Q-Chem 5.0.87 To test the applicability to different basis sets, the ALMO-based analysis was also performed using the 6-31+G(d) basis and yielded very similar results for the prodan molecule (see Fig. S1 and S2 in the ESI†). Python scripts were employed to automate the visualization of orbital energy changes among different states (such as Fig. 4 and 6) and the construction of orbital interaction diagrams (such as Fig. 5 and 7).
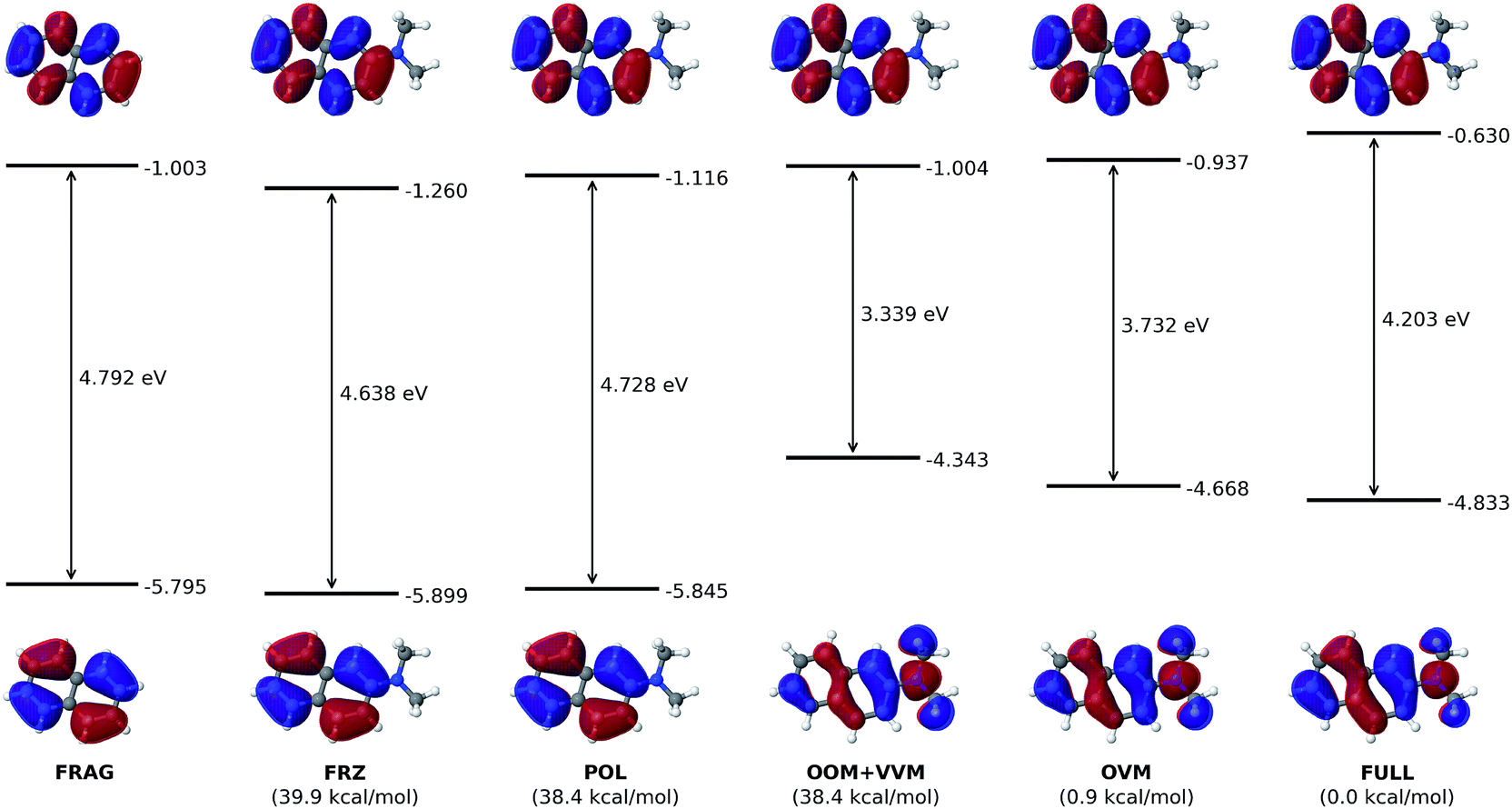 | ||
| Fig. 4 Effect of the electron-donating dimethylamino group on the HOMO and LUMO of the naphthalene chromophore. All orbital energies are in eV. Numbers in bottom parentheses show the total energy of each intermediate state relative to that of the fully converged state. | ||
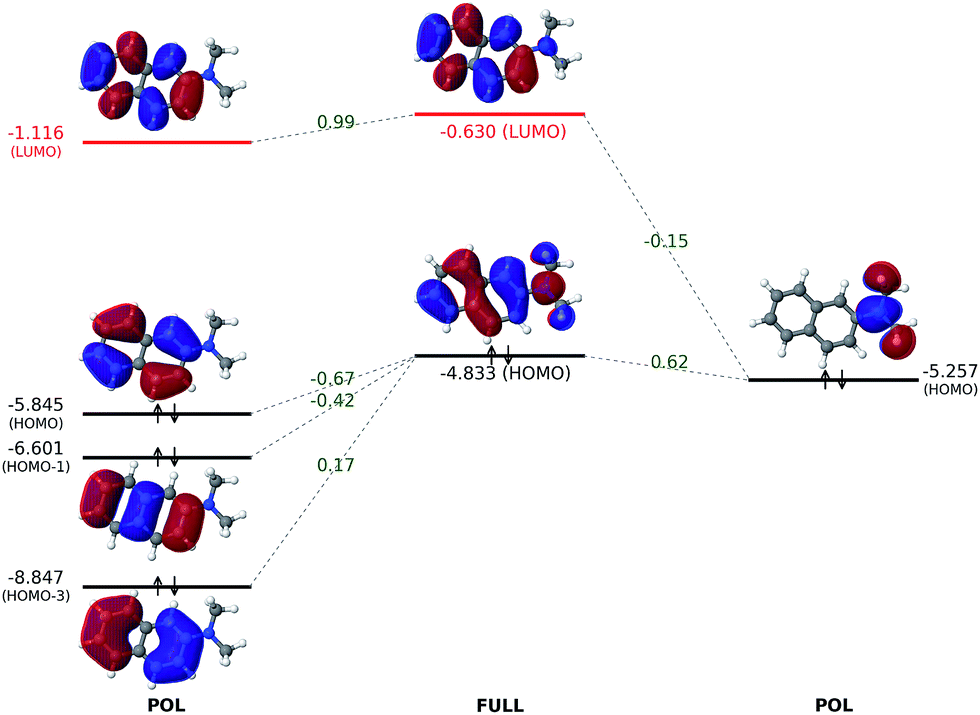 | ||
| Fig. 5 Interaction between polarized naphthalene and dimethylamino orbitals to produce the fully converged MOs of 2-(dimethylamino)naphthalene. All orbital energies are in eV. Numerical values in green refer to projection coefficients calculated based on eqn (15). Other superpositions of fragment occupied orbitals are not shown because they have lower energies. | ||
While we focus primarily on frontier orbital energies in this work, it would still be worthwhile to examine the total energy for various intermediate states in the ALMO analysis (which are shown in Fig. 4 and 6 as well as Fig. S3 and S5 from the ESI†). As expected, FRZ, POL, and OVM states (OOM + VVM shares the same total energy with POL) have successively lower energies and approach the FULL energy from above, which is consistent with the fact that they provide variational upper bounds to the full SCF energy. The polarization energy (i.e. the difference between the FRZ and POL states) is less than 2 kcal mol−1 in all cases, indicating that the fragment orbitals obtained at the FRAG stage closely resemble the mutually polarized ALMOs in the POL state. The dominant change in the total energy occurs upon ov-mixing (difference between the OOM + VVM and OVM states), where the total energy is lowered by as much as 20–40 kcal mol−1. After the ov-mixing, further SCF steps until full convergence only lower the total energy by less than 2 kcal mol−1. As shown below, the frontier orbital energies experience dramatically different changes than the total energy during the vEDA-like analysis procedure.
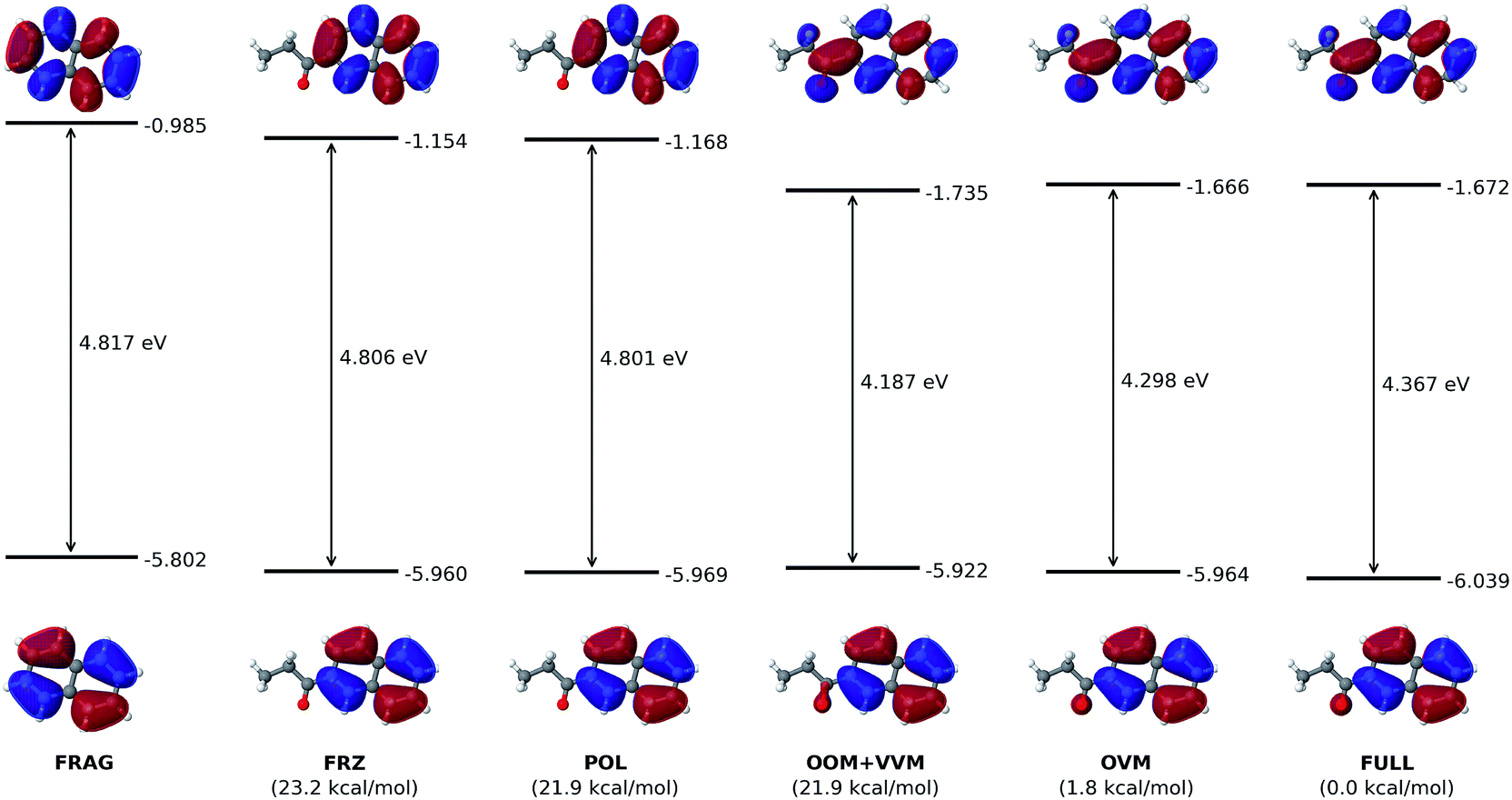 | ||
| Fig. 6 Effect of the electron-withdrawing propionyl group on the HOMO and LUMO of the naphthalene chromophore. All orbital energies are in eV. Numbers in bottom parentheses show the total energy of each intermediate state relative to that of the fully converged state. | ||
3.2 Effect of the dimethylamino group
According to the HOMO and LUMO energy levels shown in Fig. 4 for the initial (FRAG) and final (FULL) states of the analysis, both of which can be readily obtained from standard SCF calculations, there is clearly a significant reduction of the HOMO–LUMO gap from 4.792 eV (FRAG) to 4.203 eV (FULL). This arises mainly from an elevated HOMO level (from −5.795 to −4.833 eV), which is delocalized from the naphthalene core to the substituent. In contrast, the LUMO level is subjected to a smaller elevation from −1.003 to −0.630 eV.Such elevations in the frontier orbital energies agree well with one's chemical intuition: attachment of an EDG would make it easier for a conjugated molecule to lose an electron, thus lowering its ionization potential (IP); on the other hand, it should become more difficult to introduce an extra electron, which corresponds to a decreased electron affinity (EA). Although Koopmans' theorem does not hold rigorously within the DFT framework, a smaller IP and a decreased EA are still expected to accompany a raise in the HOMO and LUMO energies, respectively.
The ALMO analysis procedure, as outlined above, provides a quantitative justification for such highly intuitive results through detailed information about HOMO and LUMO at the intermediate stages between FRAG and FULL. From Fig. 4, it is clear that permanent electrostatics/Pauli repulsion (which drive the change between the FRAG and FRZ states) and polarization (which is responsible for the change from FRZ to POL) have only a marginal effect on the HOMO and LUMO energy levels, and thus on the gap in between. The most dramatic change to HOMO occurs with oo-mixing, where the pseudocanonical HOMO receives a substantial contribution from the dimethylamino group. This is consistent with the expectation that the dimethylamino group can serve as a π-donor. As a matter of fact, the HOMO obtained at the OOM stage already appears qualitatively very similar to the fully converged one. On the other hand, with a HOMO energy increase from −5.845 eV (POL) to −4.343 eV (OOM), oo-mixing clearly over-corrects the HOMO energy by around 50%, which then goes back down to its converged value (−4.833 eV) through ov-mixing and subsequent SCF steps.
Meanwhile, the LUMO receives a relatively smaller contribution from the dimethylamino group during the VVM, OVM, and the full SCF steps. The LUMO energy increases slightly from −1.116 eV (POL) to −0.966 eV upon the projection as shown in eqn (11) (not plotted in Fig. 4), and is then lowered to −1.004 eV due to the vv-mixing. The further relaxations in the OVM and full SCF steps raise the LUMO by roughly 0.4 eV, yielding its final energy −0.630 eV.
To fully understand the interactions between fragment orbitals, one can project HOMO and LUMO of the entire molecule (at the OOM + VVM, OVM, or FULL stage) onto polarized fragment orbitals (absolutely localized) using eqn (14). Recall that, in the basic MO theory, an out-of-phase (i.e. with a zero-value dividing surface between two constituents) superposition of fragment orbitals produces an orbital with a higher energy than its components (a good example is the 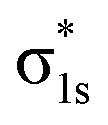 orbital of the H2 molecule). Within the current example as shown in Fig. 5, the dimethylamino HOMO forms an out-of-phase mixing with the naphthalene occupied orbitals (HOMO, HOMO−1 and HOMO−3), giving rise to an elevated energy for the full-system HOMO. Putting in the language of PMO theory (see, for example, Chapter 3.2 in ref. 29), the final HOMO includes three distinct components: a scaled-down contribution from naphthalene's HOMO, a first-order contribution from the dimethylamino HOMO, and a second-order contribution from naphthalene's HOMO−1 and HOMO−3. Fig. 5 also indicates that the dimethylamino HOMO has a minor out-of-phase mixing with naphthalene's LUMO, raising its energy from −1.116 to −0.630 eV.
orbital of the H2 molecule). Within the current example as shown in Fig. 5, the dimethylamino HOMO forms an out-of-phase mixing with the naphthalene occupied orbitals (HOMO, HOMO−1 and HOMO−3), giving rise to an elevated energy for the full-system HOMO. Putting in the language of PMO theory (see, for example, Chapter 3.2 in ref. 29), the final HOMO includes three distinct components: a scaled-down contribution from naphthalene's HOMO, a first-order contribution from the dimethylamino HOMO, and a second-order contribution from naphthalene's HOMO−1 and HOMO−3. Fig. 5 also indicates that the dimethylamino HOMO has a minor out-of-phase mixing with naphthalene's LUMO, raising its energy from −1.116 to −0.630 eV.
When a dimethylamino group is attached to 6-(propionyl)naphthalene to produce the prodan molecule (see Fig. 3), its effect turns out to be very similar. As shown in Fig. S3 and S4† in the ESI,† this EDG again substantially raises the HOMO energy of the whole molecule by mixing its HOMO with the occupied orbitals on the 6-(propionyl)naphthalene moiety.
3.3 Effect of the propionyl group
As shown in Fig. 6, the electron-withdrawing propionyl group reduces the HOMO–LUMO gap through a totally different mechanism. For all states in the ALMO analysis, the HOMO energy stays nearly constant, even after the HOMO receives a minor contribution from the oxygen atom of the propionyl group in the OOM + VVM state.Intuitively, one expects an EWG to cause a conjugated molecule to more readily accept an electron, which corresponds to a lowered LUMO energy. Indeed, upon the introduction of the propionyl group, the LUMO energy of the naphthalene ring is reduced from −0.985 eV (FRAG) to −1.672 eV (FULL). The most dramatic change to the LUMO occurs at the VVM stage, where it delocalizes onto the propionyl group, leading to a decrease of its energy from −1.168 eV (POL) to −1.735 eV (VVM). The LUMO energy then goes back up slightly to its fully converged value (−1.672 eV) through the OVM and full SCF steps. As in the case of the dimethylamino group, permanent electrostatics/Pauli repulsion and electric polarization have minimal effects on the HOMO–LUMO gap.
The orbital interaction diagram in Fig. 7 indicates that the propionyl LUMO is mixed with the LUMO and LUMO + 1 of the naphthalene core to produce the LUMO of 6-(propionyl)naphthalene, which acquires a lower energy than all its components thanks to an in-phase vv-mixing (i.e. without a zero-value dividing surface between two constituents). Such a constructive (orbital-energy-lowering) mixing of fragment orbitals resembles many other cases in MO theory, such as the σg orbital of the H2 molecule.
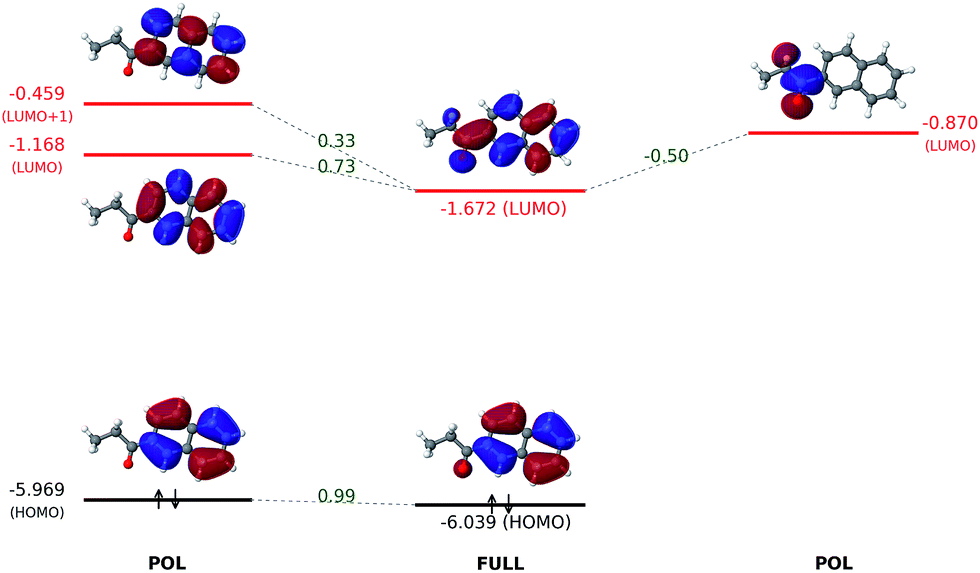 | ||
| Fig. 7 Interaction between polarized naphthalene and propionyl orbitals to produce the fully-converged molecular orbitals. All orbital energies are in eV. Numerical values in green refer to projection coefficients calculated based on eqn (15). Other superpositions of fragment unoccupied orbitals are not shown because they have higher energies. | ||
The propionyl group plays a very similar role when it is attached to 2-(dimethyl)-naphthalene (which also generates the prodan molecule). As shown in Fig. S5 and S6,† an in-phase vv-mixing of the low-lying virtual orbitals on 2-(dimethyl)-naphthalene and the propionyl group leads to a substantial reduction in the LUMO energy of the entire system.
4 Conclusions
An ALMO-based analysis procedure was introduced to divide the substituent effect on the frontier orbitals of conjugated molecules into several components: permanent electrostatics/Pauli exclusion, polarization, oo-mixing, vv-mixing, ov-mixing, etc. For the prodan example, it was found that pseudo-canonicalization (oo-mixing and vv-mixing) plays the most important role, where the dimethylamino group raises the HOMO energy through oo-mixing while the propionyl group lowers the LUMO energy through vv-mixing. Therefore, our analysis provides a quantitative theoretical foundation for the chemical intuition that EDGs raise HOMO while EWGs lower LUMO.While interactions through permanent electrostatics/Pauli repulsion and polarization between the chromophore and substituent orbitals do not significantly affect the HOMO–LUMO gap of the prodan molecule, they are expected to play a more pronounced role with more polar substituent groups. For example, a trifluoromethyl group is able to shift the HOMO and LUMO energies more significantly at the FRZ stage (see Fig. S7 in the ESI†).
The procedure outlined in our work is applicable to many other conjugated systems to understand how different substituent groups affect their frontier orbitals. On the other hand, in the study of substituent effects on absorption/emission wavelengths, its use will be mostly limited to systems where the excited state of interest is dominated by the HOMO–LUMO transition. In more complicated scenarios where multiple electronic configurations and/or states are involved, more advanced analysis tools need to be developed.
Overall, through a thorough investigation of all these effects on frontier orbitals coming out of an ALMO-based analysis, one will be able to acquire valuable quantitative insights into substituent effects on the electronic transition wavelengths of many dye molecules and then use the knowledge to guide the design of new chromophore/fluorophore analogs. In the near future, the fragment orbital preparation procedure established in this work will also be combined with a recently reported extension of ALMO-EDA to intermolecular interactions involving excited-state molecules,88 which can thus facilitate a direct probe of substituent effects on excitation energies and other excited-state properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
YS acknowledges financial support from the Department of Energy Office of Science (DE-SC0011297) and from the University of Oklahoma startup fund. YM and MHG were supported by Grant CHE-1665315 from the U.S. National Science Foundation (NSF). YS also thanks Yirong Mo, Jiali Gao, Yajun Liu, Wenjian Liu, Yingkai Zhang and Vardhan Satalkar for helpful discussions. Computational resources and services used in this work were provided by the OU Supercomputing Center for Education and Research.Notes and references
- R. Ma, P. Guo, H. Cui, X. Zhang, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. A, 2009, 113, 10119–10124 CrossRef PubMed.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef PubMed.
- R. G. Jinadasa, Y. Fang, S. Kumar, A. J. Osinski, X. Jiang, C. J. Ziegler, K. M. Kadish and H. Wang, J. Org. Chem., 2015, 80, 12076–12087 CrossRef PubMed.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- G. Canard, M. Ponce-Vargas, D. Jacquemin, B. Le Guennic, A. Felouat, M. Rivoal, E. Zaborova, A. D'Aléo and F. Fages, RSC Adv., 2017, 7, 10132–10142 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef.
- S. R. Gadre and C. H. Suresh, J. Org. Chem., 1997, 62, 2625–2627 CrossRef PubMed.
- V. E. Williams and R. P. Lemieux, J. Am. Chem. Soc., 1998, 120, 11311–11315 CrossRef.
- G. A. DiLabio, D. A. Pratt and J. S. Wright, J. Org. Chem., 2000, 65, 2195–2203 CrossRef PubMed.
- K. C. Gross, P. G. Seybold, Z. Peralta-Inga, J. S. Murray and P. Politzer, J. Org. Chem., 2001, 66, 6919–6925 CrossRef PubMed.
- K. C. Gross and P. G. Seybold, Int. J. Quantum Chem., 2001, 85, 569–579 CrossRef.
- B. Galabov, S. Ilieva and H. F. Schaefer, J. Org. Chem., 2006, 71, 6382–6387 CrossRef PubMed.
- T. Hirano, H. Nagai, T. Matsuhashi, Y. Hasumi, S. Iwano, K. Ito, S. Maki, H. Niwa and V. R. Viviani, Photochem. Photobiol. Sci., 2012, 11, 1281–1284 RSC.
- M. Kakiuchi, S. Ito, M. Yamaji, V. R. Viviani, S. Maki and T. Hirano, Photochem. Photobiol., 2017, 93, 486–494 CrossRef PubMed.
- W. J. Hehre, C. F. Pau, R. W. Taft, A. D. Headley and R. D. Topsom, J. Am. Chem. Soc., 1986, 108, 1711–1712 CrossRef.
- T. M. Krygowski, K. Ejsmont, B. T. Stepień, M. K. Cyrański, J. Poater and M. Solà, J. Org. Chem., 2004, 69, 6634–6640 CrossRef PubMed.
- J. B. Barbour and J. M. Karty, J. Phys. Org. Chem., 2005, 18, 210–216 CrossRef.
- I. Fernandez and G. Frenking, J. Org. Chem., 2006, 71, 2251–2256 CrossRef PubMed.
- W. P. OziminÌski and J. C. Dobrowolski, J. Phys. Org. Chem., 2009, 22, 769–778 CrossRef.
- T. M. Krygowski and N. Sadlej-Sosnowska, Struct. Chem., 2011, 22, 17–22 CrossRef.
- T. Siodla, W. P. Oziminski, M. Hoffmann, H. Koroniak and T. M. Krygowski, J. Org. Chem., 2014, 79, 7321–7331 CrossRef PubMed.
- T. M. Krygowski and W. P. Oziminski, J. Mol. Model., 2014, 20, 1–8 CrossRef PubMed.
- H. Szatylowicz, T. Siodla, O. A. Stasyuk and T. M. Krygowski, Phys. Chem. Chem. Phys., 2016, 18, 11711–11721 RSC.
- P. J. Baldry, J. Chem. Soc., Perkin Trans. 2, 1979, 951–953 RSC.
- C. Cao, G. Chen and Z. Yin, J. Phys. Org. Chem., 2008, 21, 808–815 CrossRef.
- N. Sadlej-Sosnowska and M. Kijak, Struct. Chem., 2012, 23, 359–365 CrossRef.
- J. C. Dobrowolski, P. F. J. Lipinski and G. Karpinska, J. Phys. Chem. A, 2018, 122, 4609–4621 CrossRef PubMed.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, John Wiley & Sons Inc, Hoboken, New Jersey, 2nd edn, 2013 Search PubMed.
- A. Imamura, Mol. Phys., 1968, 15, 225–238 CrossRef.
- L. Salem, J. Am. Chem. Soc., 1968, 90, 543–552 CrossRef.
- L. Libit and R. Hoffmann, J. Am. Chem. Soc., 1974, 96, 1370–1383 CrossRef.
- M.-H. Whangbo, B. Schlegel and S. Wolfe, J. Am. Chem. Soc., 1977, 99, 1296–1304 CrossRef.
- M. Dewar, The PMO Theory of Organic Chemistry, Springer, 2012 Search PubMed.
- J. B. Foresman, M. Head-Gordon, J. A. Pople and M. J. Frisch, J. Phys. Chem., 1992, 96, 135–149 CrossRef.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef.
- A. I. Krylov, Annu. Rev. Phys. Chem., 2008, 59, 433 CrossRef PubMed.
- J. Schirmer, Phys. Rev. A, 1982, 26, 2395 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef.
- K. Burke, J. Werschnik and E. K. U. Gross, J. Chem. Phys., 2005, 123, 062206 CrossRef PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef PubMed.
- M. E. Casida and M. Huix-Rotllant, Annu. Rev. Phys. Chem., 2012, 63, 287–323 CrossRef PubMed.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef PubMed.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef.
- K. Morokuma, Acc. Chem. Res., 1977, 10, 294–300 CrossRef.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef PubMed.
- P. R. Horn, E. J. Sundstrom, T. A. Baker and M. Head-Gordon, J. Chem. Phys., 2013, 138, 134119 CrossRef PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- Y. Mao, P. R. Horn and M. Head-Gordon, Phys. Chem. Chem. Phys., 2017, 19, 5944–5958 RSC.
- Y. Mo, J. Gao and S. D. Peyerimhoff, J. Chem. Phys., 2000, 112, 5530–5538 CrossRef.
- Y. Mo, L. Song and Y. Lin, J. Phys. Chem. A, 2007, 111, 8291–8301 CrossRef PubMed.
- Y. Mo, P. Bao and J. Gao, Phys. Chem. Chem. Phys., 2011, 13, 6760–6775 RSC.
- E. D. Glendening and A. Streitwieser, J. Chem. Phys., 1994, 100, 2900–2909 CrossRef.
- E. D. Glendening, J. Phys. Chem. A, 2005, 109, 11936–11940 CrossRef PubMed.
- Q. Wu, P. W. Ayers and Y. Zhang, J. Chem. Phys., 2009, 131, 164112 CrossRef PubMed.
- Q. Wu, J. Chem. Phys., 2014, 140, 244109 CrossRef PubMed.
- M. J. Phipps, T. Fox, C. S. Tautermann and C.-K. Skylaris, Chem. Soc. Rev., 2015, 44, 3177–3211 RSC.
- D. Cappel, S. Tullmann, A. Krapp and G. Frenking, Angew. Chem., Int. Ed., 2005, 44, 3617–3620 CrossRef PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef PubMed.
- M. von Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 Search PubMed.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- D. S. Levine, P. R. Horn, Y. Mao and M. Head-Gordon, J. Chem. Theory Comput., 2016, 12, 4812–4820 CrossRef PubMed.
- D. S. Levine and M. Head-Gordon, Proc. Natl. Acad. Sci., 2017, 114, 12649–12656 CrossRef PubMed.
- D. Fedorov and K. Kitaura, J. Comput. Chem., 2007, 28, 222–237 CrossRef PubMed.
- J. F. Gonthier and C. Corminboeuf, J. Chem. Phys., 2014, 140, 154107 CrossRef.
- R. M. Parrish, J. F. Gonthier, C. Corminbœuf and C. D. Sherrill, J. Chem. Phys., 2016, 143, 051103 CrossRef PubMed.
- P. Su, Z. Chen and W. Wu, Chem. Phys. Lett., 2015, 635, 250–256 CrossRef.
- P. Su, H. Chen and W. Wu, Sci. China: Chem., 2016, 59, 1025–1032 CrossRef.
- L. Goodman, V. Pophristic and F. Weinhold, Acc. Chem. Res., 1999, 32, 983–993 CrossRef.
- Y. Mo and J. Gao, Acc. Chem. Res., 2007, 40, 113–119 CrossRef PubMed.
- Y. Mo, Nat. Chem., 2010, 2, 666–671 CrossRef PubMed.
- Y. Mo, J. Phys. Chem. A, 2012, 116, 5240–5246 CrossRef PubMed.
- H. Zhang, X. Jiang, W. Wu and Y. Mo, Phys. Chem. Chem. Phys., 2016, 18, 11821–11828 RSC.
- J. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef.
- Y. Wang, Phys. Chem. Chem. Phys., 2018, 20, 13792–13809 RSC.
- M. Head-Gordon, P. E. Maslen and C. A. White, J. Chem. Phys., 1998, 108, 616 CrossRef.
- R. Z. Khaliullin, M. Head-Gordon and A. T. Bell, J. Chem. Phys., 2006, 124, 204105 CrossRef PubMed.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef PubMed.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2016, 110, 2579–2619 CrossRef PubMed.
- G. Weber and F. J. Farris, Biochemistry, 1979, 18, 3075–3078 CrossRef PubMed.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York, 1971 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. Hanson-Heine, P. H. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef.
- Q. Ge, Y. Mao and M. Head-Gordon, J. Chem. Phys., 2018, 148, 064105 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02990c |
| This journal is © The Royal Society of Chemistry 2018 |