
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent mesoporous nanorods as photocatalytic enzyme-like peroxidase surrogates†
M. Carmen
Ortega-Liebana
 ab,
Jose L.
Hueso
*ab,
Rodrigo
Fernandez-Pacheco
c,
Silvia
Irusta
ab and
Jesus
Santamaria
*ab
ab,
Jose L.
Hueso
*ab,
Rodrigo
Fernandez-Pacheco
c,
Silvia
Irusta
ab and
Jesus
Santamaria
*ab
aInstitute of Nanoscience of Aragon (INA), Department of Chemical Engineering and Environmental Technology, University of Zaragoza, 50018 Zaragoza, Spain. E-mail: jhueso@unizar.es; jesus.santamaria@unizar.es
bNerworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
cAdvanced Microscopy Laboratory (LMA), Institute of Nanoscience of Aragon (INA), University of Zaragoza, 50018 Zaragoza, Spain
First published on 24th August 2018
Abstract
Herein we report on a novel inorganic peroxidase-mimicking nanocatalyst activated under blue LED photoirradiation. A novel flash-pyrolysis method has been developed for the generation of strong blue photoluminescence (PL) centers attributed to silicon and carbon-based sites within a mesoporous SBA-15 silica nanorod platform. The type of centers and their PL response can be controlled by varying the flash thermal treatment conditions. By tailoring the operating conditions the system can be driven towards the preferential generation of carbon-based luminescent centers, with or without the simultaneous generation of silicon-based centers. The properties and the nature of these luminescent centers within the mesoporous nanorods have been thoroughly corroborated by a battery of characterization techniques including fluorescence spectroscopy, X-ray photoelectron spectroscopy (XPS) and electron energy loss spectroscopy (EELS) at the local level of the structures combined with scanning transmission electron microscopy (STEM) imaging. In addition, these luminescent mesoporous nanorods have been successfully tested as robust photocatalysts able to display peroxidase-like activity and indirect glucose sensing in a wider range of pH conditions compared to the natural enzyme, especially when carbogenic dots and oxygen-deficient silica centers are simultaneously present in the structure.
Introduction
Natural enzymes are widely used biocatalysts that combine high substrate specificity with excellent chemo-, regio-, and stereoselectivity under mild reaction conditions. They have lately attracted much interest in medical, biological and industry fields. However, natural enzymes suffer from inherent shortcomings such as high cost of preparation and purification, low stability due to denaturation, lack of sensitivity or catalytic activity due to environmental conditions and limited possibilities for recovery and reutilization. These drawbacks somehow restrain their practical applications. In order to overcome these problems, alternative candidates to mimic the working principles of natural enzymes are being currently explored. These alternative surrogates have been coined as artificial enzymes or enzyme-mimicking materials and are considered as promising, highly stable and affordable candidates to natural enzymes in a wide range of applications.1,2 As a consequence, the last decade has witnessed an increasing research effort devoted to the design and development of efficient artificial mimicking enzymes.3,4 Inorganic nanomaterials provide an appealing alternative due to their attractive properties, including stability, catalytic response and biocompatibility.Among enzymes peroxidases have received special attention due to their key role in multiple metabolic activities and their potential use as biomarkers in multiple diseases. Furthermore, peroxidases include a large family of isoenzymes found in almost all living organisms. These enzymes typically catalyze biological reactions where peroxides are reduced (e.g. hydrogen peroxide, H2O2), while a redox substrate acting as an electron donor is simultaneously oxidized. It should be noted that the nature of the electron donor is very much dependent on the structure of the enzyme. Through this catalytic process, peroxidases can scavenge H2O2, a naturally occurring byproduct of oxygen metabolism in human body, resulting in the formation of water and oxygen. In this way, peroxidases play an important role as highly efficient antioxidants to combat complications engendered by reactive oxygen species (ROS).5 In addition, peroxidases play a vital role in combination with the glucose oxidase (GOx) to determine glucose concentrations through cascade catalytic reactions.6 A variety of nanomaterials including magnetic nanoparticles (NPs),7–10 metallic NPs,11–13 carbon nanomaterials,14,15 silicon16,17 or their combinations18,19 have been recently tested as artificial inorganic enzymes mimicking the role of their natural peroxidase counterparts in environmental and biomedical fields. Nevertheless, the search for novel, innovative, affordable and stable enzymatic surrogates continues to be the focus of intense research activity. Particularly desirable are novel enzyme-mimicking nanomaterials with photo-enhanced response and capable of remote activation using light, a subject of great interest.20
Herein we present a novel inorganic nanoplatform based on luminescent mesoporous silica nanorods (hereafter LMS) with enhanced peroxidase-like activity, especially upon blue-light irradiation. This system does not require the presence of any metal or transition metal elements since it is based on the optical response of carbon and silica-based emitting centers. These centers have been generated in situ within the mesoporous network of SBA-15 nanorods. Remarkably, a direct carbonization of the surfactant molecules confined within the ordered mesoporous channels can be achieved after a few minutes of flash pyrolysis treatment under an inert gas stream. Alternatively, the flash-thermal treatment can be tuned to induce a carbothermic reduction of silica domains when static conditions (no carrier gas supply) are used. A very fast heating to the pyrolysis temperature was necessary to accomplish pyrolysis before the carbon precursors had time leave the silica pores. This fast heating could be achieved by immersion in a fluidized bed reactor at 500 °C.
This method contrasts with previous reports on the literature that generate luminescent mesoporous materials via the ex situ attachment of photoactive species such as quantum dots or organic dyes. These species are typically non-environmentally friendly, require complex functionalization steps and are often prone to photobleaching. Other impressive reports on mesoporous structures with embedded quantum-confined silicon centers typically require high thermal treatments and/or subsequent etching with strong acids.21–25 Furthermore, He et al. reported on the generation of luminescent oxygen deficient centers by calcination treatment of silica substrates.26,27 In this work, a different set of optically active centers based on carbon nanodots, Si–C(O) domains and/or oxygen-deficient silica centers have been generated within mesoporous silica nanorods by subjecting our silica nanostructures, with their pores still containing the structure-directing agent (SDA) to a fluidized-bed flash heating process. Species identification has been confirmed by a panoply of techniques: fluorescence spectroscopy, XPS, high-resolution (S)TEM imaging and EELS.
Moreover, we have shown the ability of this novel platform (made of abundant, environmentally friendly silicon and carbon based materials) to behave as a metal-free enzyme-like photocatalyst, offering a broad optical response in the visible range in combination with excellent thermal and chemical stability. Interestingly, the simultaneous presence of both carbon and silica-based emitting species produced nanostructures with the highest peroxidase-like activity upon LED irradiation. In contrast to previous studies on the peroxidase-mimic catalytic activity of freestanding carbon dots15 and silicon dots,16 the photo-activated LMSs structures demonstrated high enzyme-like activity over a broad pH range (5.0–7.0) even at neutral pH, which overcomes one of the major shortcomings of currently developed artificial enzymes (typically suffering from deactivation when operating at near neutral pH conditions).28 The high stability of the structures prepared in this work allowed these LMSs platforms to be successfully reused for multiple cycles. It is also worth mentioning that the peroxidase-mimicking role of the LMS photocatalysts was further evaluated in a cascade reaction in combination with other enzyme such as glucose oxidase (GOx). The combination of both systems enabled a rapid and robust quantification of glucose using a colorimetric assay. This new type of metal-free artificial enzyme exhibited high sensitive and selective response toward glucose detection only activated under LED blue irradiation.
Results and discussion
Flash-thermal assisted generation of luminescent mesoporous rod-shaped nanocatalysts
The ordered mesoporous silica (MS) nanorods were synthesized using Pluronic P123 as SDA and Tetraethyl orthosilicate (TEOS) as silicon source following a hydrothermal approach reported elsewhere.29 In order to induce the generation of light-emitting centers within the silica nanorods, these were placed in a thin wall 8 mm diameter quartz tube, which was immersed in a sand fluidized-bed reactor at 500 °C. The samples were flash-heated to the bed temperature in less than 40 s (Scheme 1 and Table S1†). This flash heating protocol induced luminescent sites within the mesoporous rods while keeping their ordered structure (Fig. S1 and Table S2†). Furthermore, the selection of different reaction ambients derived in a different photoluminescence response of the resulting materials (Scheme 1 and Fig. 1).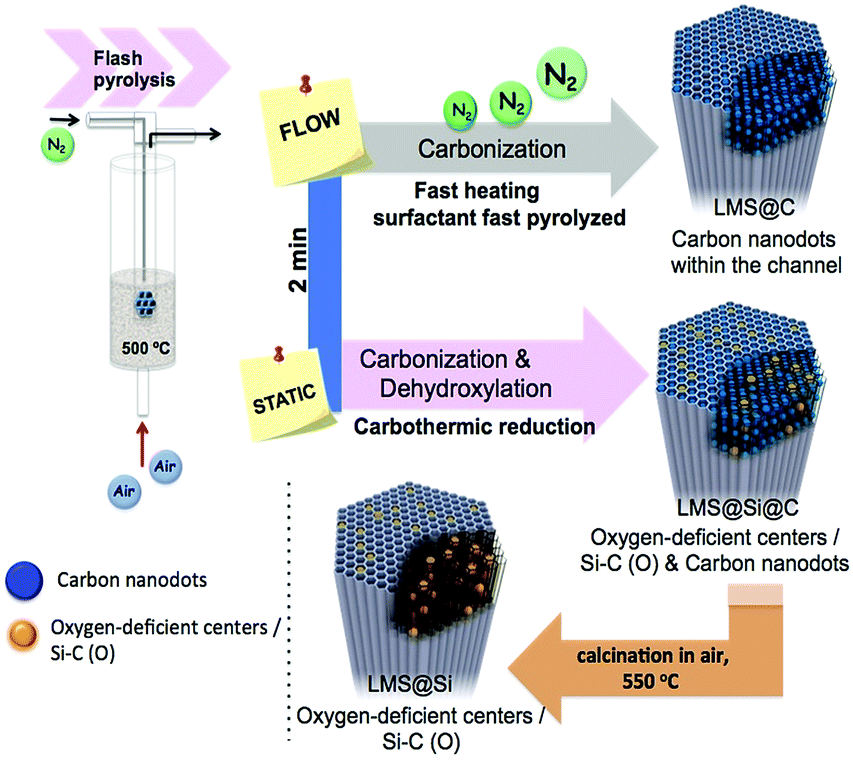 | ||
| Scheme 1 A flash heating of SDA-containing silica nanorods induced the generation of different light-emitting centers within its structure. The conditions could be tailored to obtain only carbon dots in the silica structure (LMS@C) or carbon dots plus silica-based emitting species (LMS@Si@C). A third set of samples containing only luminescent silicon-based species (LMS@Si) obtained by a slow oxidation post-treatment of the LMS@Si@C sample in a conventional furnace. Different reaction atmospheres (flowing N2 or static atmosphere) were used to tailor the preferential formation of the different luminescent sites during the flash-thermal treatments. | ||
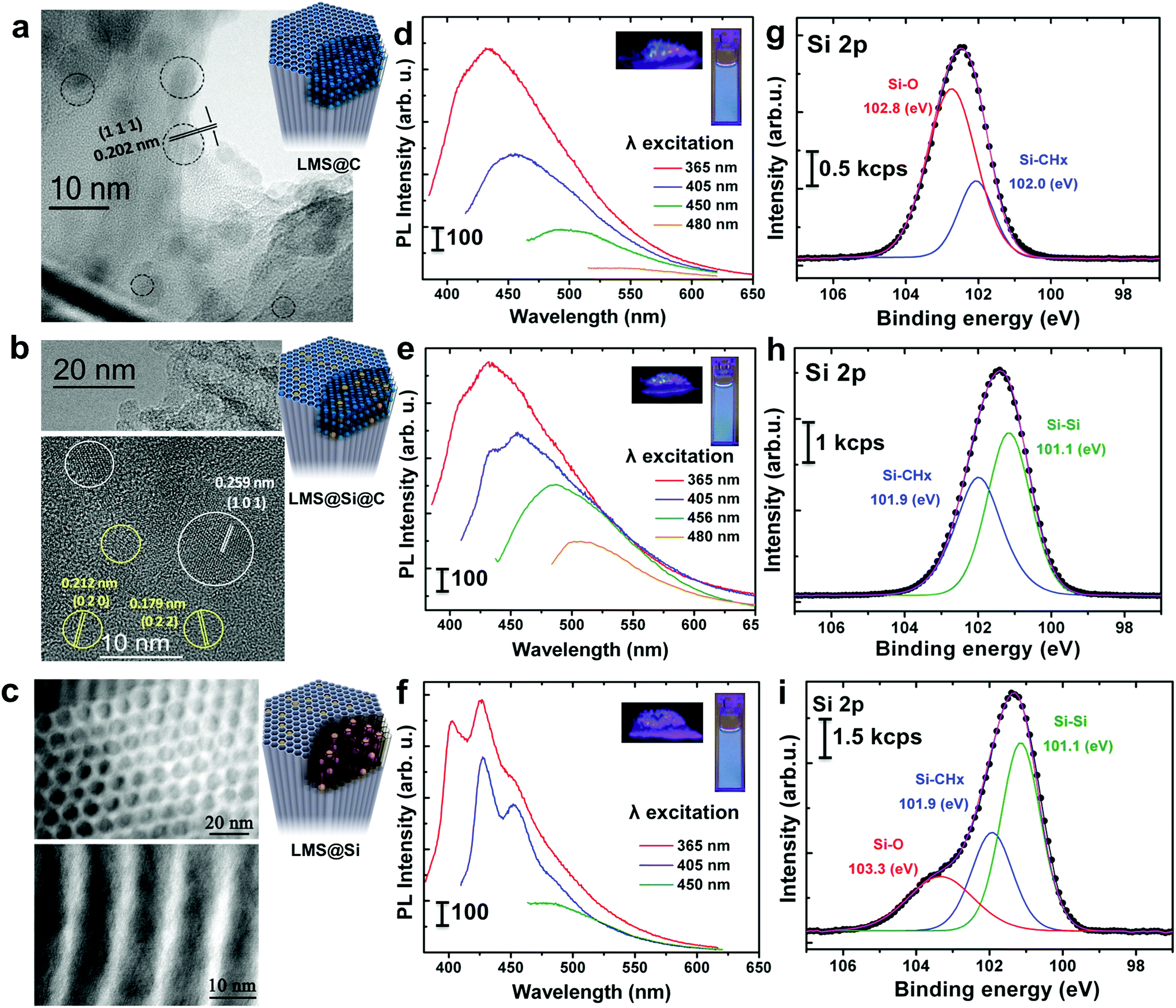 | ||
| Fig. 1 Characterization of the luminescent silica nanorods generated under different conditions (top row: flowing N2, middle: static conditions, bottom: static conditions plus post-synthesis calcination): (a–c) high resolution TEM images of small crystalline dots identified within the mesostructure of the LMSs networks attributable to graphitic-like carbon dots (black or yellow circles) or alternatively to silicon carbide domains (white circles); (d–f) photoluminescence spectra corresponding to the LMSs at different excitation wavelengths from 365 to 480 nm insets: digital photographs corresponding to the powdered LMSs and their aqueous suspension under irradiation with a UV lamp (λexc = 365 nm); (g–i) fitted X-ray photoemission contributions in the Si 2p region acknowledging different oxidation states for the LMS@C, LMS@Si@C and LMS@Si samples, respectively. | ||
Thus, flushing the samples with nitrogen during pyrolysis led to the preferential generation of carbogenic dots (sample hereafter referred as LMS@C) due to the in situ carbonization of the SDA encapsulated within the mesoporous channels. HR-TEM analysis further confirmed the presence of graphitic-like lattice distances (Fig. 1a and S2†). Interestingly, when a static atmosphere with no continuous flowing conditions was set, the simultaneous co-generation of a second type of emitting species took place. HR-TEM images and indexing confirmed the co-existence of graphitic dots and additional lattice fringes matching with silicon carbide (see Fig. 1b, S2 and Table S1†). The dynamics (static vs. flow conditions) in the system were even more important than the atmosphere used. Under static conditions (even when air was no removed from the reactor), the P123 surfactant molecules not only generated carbon dots, but also acted as carbonaceous reducing agents (i.e. carbothermic reduction) of the surrounding O3Si–O–SiO3 network that was in close contact (Scheme 1). The partial reduction of the silica units generated defect-luminescent features in the structure stemming from oxygen-deficient silica centers26 and/or alternatively carbon-doped silica domains30,31 (sample referred as LMS@Si@C). As a result, a different PL response could be identified (Fig. 1e) in comparison with the first LMS@C sample (Fig. 1d).
In order to clearly identify the emission tentatively attributed to defect-silica and/or carbon-doped silica sites, an additional calcination step was carried out using a slower (5 °C min−1) temperature ramp in order to remove all carbonaceous species (carbon dots and residues from the pyrolysis of the SDA still present in the pores) from samples LMS@C and LMS@Si@C (Scheme 1). In this way, the contribution of carbon dots could be ruled out. Interestingly, only the LMS@Si@C samples (i.e. those initially prepared under static conditions) were able to maintain the luminescence after the calcination step (Fig. 1f and Table S1†). After the removal of carbon dots, the specific contribution of partially reduced silica emission centers or Si–C–O domains could be clearly discerned, with characteristic emission peaks clearly distinguishable from the carbon dots typical luminescent features.30–34 This sample containing only silicon-related emitting species was labeled as LMS@Si for subsequent use and evaluation (Scheme 1). To corroborate that the generation of luminescent centers was due to the use of the flash pyrolysis method, slow conventional calcination treatments were carried out for comparison under identical reaction atmospheres and temperatures (Table S1†). None of the control experiments under conventional heating conditions rendered any luminescent nanostructures (Table S1†).
HR-TEM images also identified the presence of encased crystalline nanodomains (Fig. 1a and b). Previous researchers have also identified luminescent Si centers by using a variety of calcination procedures on Si-containing materials.34,35 Our previous research had already shown that it is possible to use a nanoporous structure to load organic precursors that, upon rapid pyrolysis, yield carbon dots confined within the pore channels.36 In this work, instead of loading an external C-containing species, we have used as precursor the SDA already present in the pores, since it is used during the synthesis of the silica nanostructure.
The SDA has the advantage of being already confined in the pores and to have a much stronger interaction with the silica walls (unlike adsorbed organic molecules, it cannot be simply desorbed: removing it from the structure requires calcination at temperatures in excess of 500 °C). As a result, the P123 micelles encapsulated within the mesoporous channels of LMSs are carbonized in situ, yielding confined carbon dots and generating oxygen-deficient silica centers, while preserving the silica mesostructure. On the other hand, the chemical structure of the SDA may also play a role in the process, since organic groups are known to preferentially induce dehydrogenation events in surrounding/neighboring Si–O networks. This induces the formation of multiple oxygen vacancies, surface defects and the appearance of oxygen deficient centers as previously postulated in previous works.26,30,31,33,34
Spectroscopic analysis of the silicon and carbon-based emitting centers present in the rod-shaped luminescent mesoporous nanocatalysts
The optical properties of the different LMSs were evaluated by fluorescence spectroscopy. The as-prepared rods exhibited no luminescent properties (Table S1†). However, after the flash-thermal treatment, remarkable PL properties of LMSs were observed. As displayed in Fig. 1d and e, as the excitation wavelength increased from 365 to 480 nm, the emission peak gradually red shifted from 434 to 516 nm, which suggests that the photo-luminescence of the LMS@C and LMS@Si@C are excitation wavelength dependent. Previous reports have attributed this optical behavior and blue emission to the presence of carbogenic dots subjected to quantum confinement effects.37,38 In order to corroborate this hypothesis, different control experiments were carried out. Both the LMS@C and LMS@Si@C and the non-heated nanorods were digested with NaOH to etch the mesoporous structure (Fig. S3†). After a purification process (centrifugation and dialysis), a yellow clear suspension was obtained. This suspension also exhibited PL response, that is, a strong luminescence remained after etching the silica supports (Fig. S3†). Strong photoluminescence was also observed when the aqueous suspension was irradiated under a UV lamp. Finally, TEM analysis confirmed the presence of the nanoparticles still present after the basic etching treatment (Fig. S3†) in the flash-thermally activated LMSs samples. These results and the absence of optical response in the non-treated nanorods (Fig. S3†) further confirmed the in situ generation of carbon nanodots within the mesochannels, as a consequence of the fast pyrolysis of the co-directing surfactant molecules present within the mesochannels. Previous theoretical and experimental reports have suggested a variety of potential mechanisms behind the PL response of carbon dots such as the presence of surface state defects,39 molecule-like state,40 sp2-carbon networks41 and quantum size effects,42etc. Mechanistically, the photoluminescence from@C may be attributed to the presence of surface energy traps that become emissive upon stabilization as a result of the surface passivation.43On the other hand, the emission spectra of the LMS@Si (Fig. 1f) exhibited a different structure tentatively associated to the formation of oxygen-deficient silica centers. Here, the formation process is attributed to the partial reduction of surface species favored by the strong interaction of the P123 surfactant molecules with the surrounding silica units that upon pyrolysis in addition to generating carbon dots acted as carbonaceous reducing agents of the O3Si–O–SiO3 network. An analogous control experiment digesting the LMS@Si sample with NaOH did not render any PL response, thereby confirming the origin of the emission on silica-related centers which are no longer available after the chemical etching treatment (see Fig. S4†). In summary, the blue photoluminescence observed can be ascribed to oxygen-deficient silica centers with Si–Si bonds.
The identification of the actual centers responsible for luminescence remains under discussion. Previous reports on blue photoluminescence of treated silica have been attributed to the surface passivation of small silicon nanocrystalline domains;32,44 or the presence of Si–Si–O3 species on the silicon NPs networks.33,45 More recent reports with similar temperature treatments showed that the PL response is highly dependent on the heating environment. Uchino et al. demonstrated that the dehydroxylation reaction under air resulted in the formation of a defect pair consisting of dioxasilirane (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(O2)) and silylene (Si:).46
Si(O2)) and silylene (Si:).46
Additional authors also correlated the appearance of PL properties to the appearance of oxygen defects and vacancies under air heating conditions.33,35 Alternatively, different authors have associated the origin of the different emission peaks to the presence of Si–C–O domains.30,31,47–49
To further explore the flash-thermal treatment effects, the different luminescent nanorods were also analyzed by X-ray photoelectron spectroscopy (XPS). High-resolution scanning of the Si 2p core level spectra is shown in Fig. 1g–i. The Si 2p region corresponding to the LMS@C sample was fitted into two main components (Fig. 1g), the one at higher binding energy (102.8 eV) associated to Si–O bonds and the peak at 102.0 eV attributed to Si–CHx bonds.47 This latter component would confirm the presence of carbon-enriched silica species on the material surface. Previous reports50 have also formed carbonaceous deposits inside mesoporous silica by calcination at a high temperature (900 °C for 3 h) under nitrogen protection. However in this case no luminescent carbon dots were observed, instead, significant carbonaceous deposits helped to maintain the silica structure in place. In a different work carbon dots were generated by calcination of carbon-containing precursors inside narrower silica pores, but a complex procedure was used involving extraction of the SDA under saturated solution of HCl, followed by functionalization of the silica previous to calcination (at 400 °C for 2 h). No luminescent Si centers were observed.51 The LMS@Si@C sample also showed two main contributions in the Si 2p spectrum at 101.9 and 101.1 eV, respectively (see Fig. 1h). Besides the Si–CHx bonds, the contribution at lower binding energy would indicate the presence of silicon atoms forming Si–Si bonds34 and confirming Si reduction as a result of partial dehydroxylation events in the silica network (Scheme 1).31,33 This is in agreement with our previous assumption that the P123 surfactant molecules act as reducing agent generating oxygen-deficient silica centers. After slow conventional calcination treatments, the Si 2p contribution of LMS@Si evolved into three components at 101.1, 101.9 and 103.3 eV indicating that some re-oxidation occurred, as shown in Fig. 1i (see also Table S3†). The analysis of C 1s and O 1s signals confirmed the presence of Si–CHx, C–O and C–OH surface groups (Table S3†).47,52,53
In order to analyze the chemical structure of the different materials at the nanoscale level, electron energy loss spectra were acquired in a probe-corrected microscope in scanning transmission (STEM) mode. The sample was scanned with a very thin probe (around 0.1 nm in diameter) so that High Angle Annular Dark Field (HAADF) images and EELS spectra were collected simultaneously for each point. The analysis of the low-loss region of the EEL spectrum is probably the most direct way to identify different silicon-based species.54–56 Different material phases were identified by measuring the plasmon energy of the obtained materials and comparing those to external ref. 57 (see Fig. 2). In the case of LMS@C, the presence of a plasmon peak at 22.2 eV matched well with the presence of silicon carbide (Si–C) and carbon structures (Fig. 2a). These assignations agree with the XPS analysis previous described (vide supra).
 | ||
| Fig. 2 STEM images (left) and localized EELS low-loss spectra (right) at specific positions of the different luminescent nanorods: (a) LMS@C; (b) LMS@Si@C; (c) LMS@Si samples. EEL spectra are compared with references of C, Si, SiC and SiO2, respectively. | ||
For the other structures, the overlapping of several peaks in a very narrow region increased the difficulty to clearly distinguish one single material. Therefore, a combination of several phases is the most probable scenario. Thus, for the LMS@Si@C sample, there is a broadening of the plasmon signals. This can be explained as a sum of the plasmon peak of SiO2 at 23.5 eV and a broad shoulder most probably attributable to the presence of Si in a reduced state at 16.8 eV.58 This is clearly visible throughout the analysis of multiple points of a single nanorod, either in the outer or inner areas (Fig. 2b). Regarding the LMS@Si structure, no presence of carbon or SiC contribution was observed. Instead, a combination of more reduced Si signal was mainly detected in the inner areas whereas the outer fraction of the nanorod was more compatible with the presence of more oxidized phases with the plasmon peak of SiO2 (Fig. 2c).
This latter outcome reinforces the hypothesis of a dehydroxylation pathway to render SiO2 and Si–Si domains.33,46 In conclusion, all the spectroscopic techniques further suggest that the origin of the induced photoluminescent properties in the rod-shaped mesoporous supports can be tentatively ascribed to the combined generation of carbogenic centers, silica domains in a reduced oxidation state (i.e. generation of oxygen vacancies) and/or the appearance of Si–C(O) centers. Indeed, only flash pyrolysis provides the effect sought, while conventional treatments at the same temperature did not show generation of luminescent centers. The very high speed of heating achieved by fluidized-bed aided flash heating is the key to understand this differential behavior. Under flash heating conditions, the material reaches the desired temperature almost instantly. This means that the organic molecules trapped inside the silica pores are quickly pyrolyzed and the decomposition products reach a high temperature before they have time to diffuse out of the pores, a process retarded also by the rod-like shape of the mesoporous support. It should be noted that these organic molecules present a close interaction with the surrounding silica units, since during synthesis they as structure-directing agents around which the silica pores are formed. During that brief heating period at 500 °C the structure directing agents are rapidly pyrolyzed, forming carbon-based nanodomains and giving carbon-based pyrolysis products. At high temperatures both species can act as reducers, affecting the neighboring Si–O centers present in the amorphous silica network of the rods. In contrast, under conventional heating conditions, we start from room temperature, with an increase of 10 °C min−1 until reaching 500 °C. This means that there is ample time for the gaseous species to diffuse out of the pores before they reach temperatures where they could act as in situ reducers. As a consequence, this treatment in spite of its longer duration (in total 2.8 h), did not induce reduction in the nanostructure (see Table S1†).
Luminescent mesoporous rods as enzyme-like peroxidase-like photocatalysts
Inspired by previous works on the use of freestanding carbon and silicon nanoparticles as peroxidase-mimicking artificial enzymes,4,15–17,59,60 our luminescent nanorods were tested as a novel metal-free inorganic peroxidase surrogate with high surface area and visible light response. In order to assess the peroxidase-like activity of the LMSs, the 3,3′,5,5′-tetramethylbenzidine (TMB)–H2O2 assay was used as model reaction. The catalytic oxidation of the peroxidase substrate TMB was monitored in the presence of H2O2 under light excitation. The maximum absorbance of the blue generated TMB oxidation product occurs at 652 nm and is typically ascribed to the charge-transfer complexes of diamine and diimine derived from the one-electron oxidation of TMB (see Fig. 3c and d).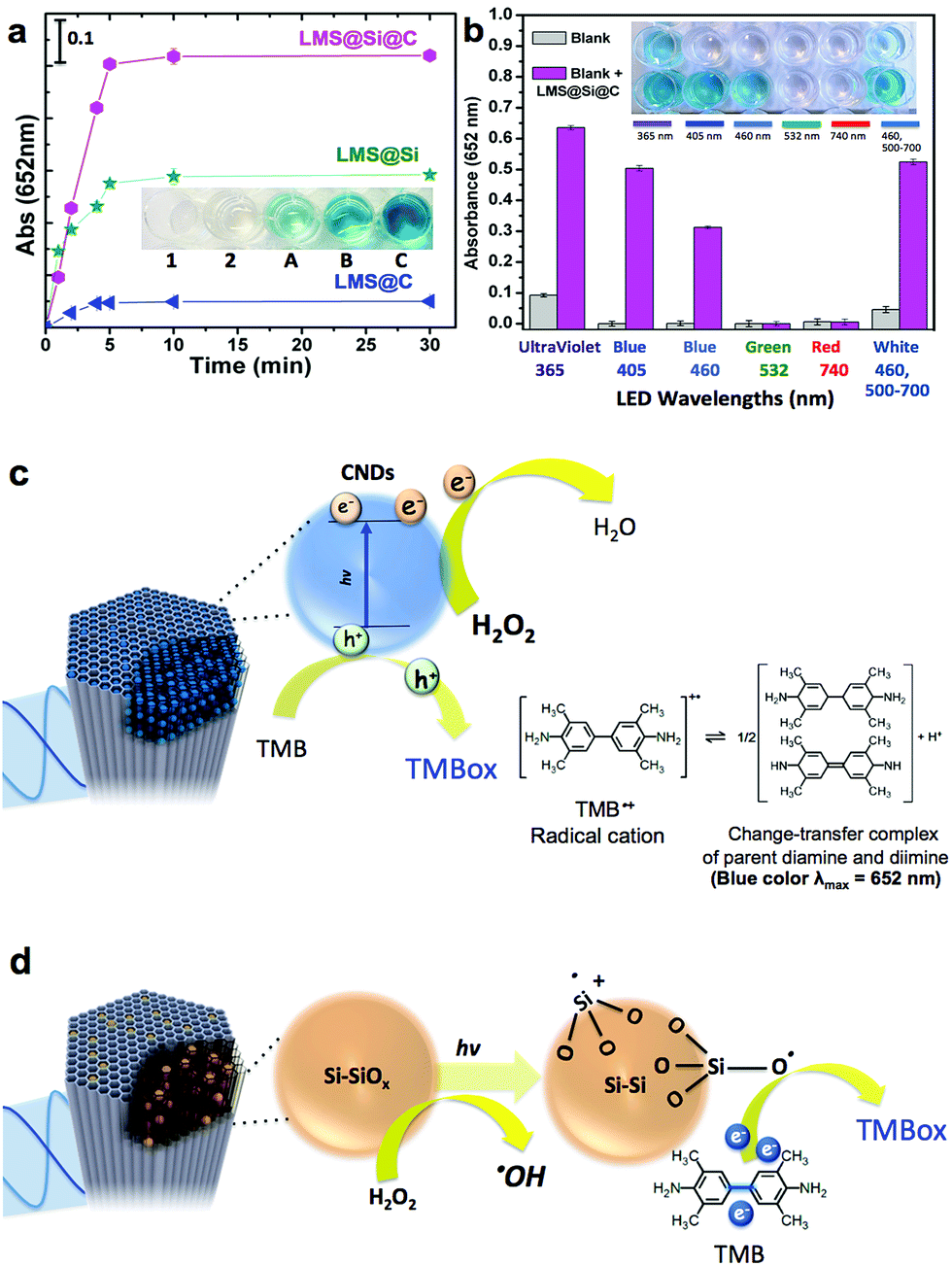 | ||
| Fig. 3 Evaluation of the peroxidase-like activity of the different LMSs. (a) Time-dependent absorbance changes TMB at 652 nm for the different luminescent mesoporous nanorods inset: digital image of wells containing reaction mixtures after 10 min or reaction with no light (1) or under irradiation with blue LED (2, A, B and C): (1) TMB + H2O2; (2) TMB + H2O2; (A) LMS@C; (B) LMS@Si and (C) LMS@Si@C; (b) Evaluation of the influence of irradiating the LMS@Si@C catalyst with different LED wavelengths. Experimental conditions: [catalyst] = 4 μg mL−1; [TMB] = 0.16 mM; [H2O2] = 10 mM; pH = 7.4 (0.2 M NaAc buffer); total volume = 2 mL; irradiation time = 5 min (inset: digital image of wells containing reaction mixtures after 5 min under irradiation with UV LED, blue LED-405, blue LED-460, green LED-532, red LED-740 and white LED, respectively). The error bars represent the standard deviation of three measurements; (c) proposed mechanism in the TMB–H2O2 involving the carbon dots embedded within the mesoporous template; (d) proposed peroxidase-like photoactivation mechanism involving Si–O active sites involving the generation of Si–O intermediates with affinity to withdraw electrons from TMB substrates. | ||
In previous studies, it was suggested that the wavelength of the LED used to trigger the oxidation of TMB should match the optimal excitation wavelength of the sensitizer.60 We initially selected the blue-LED at 405 nm for irradiation to maximize the performance of our photocatalysts based on the maximum absorption/emission spectra (see Fig. 2 and experimental setup in Fig. S9†). Fig. 3a (see also Fig. S5†), compares the enzyme-like activity of LMS@C, LMS@Si and LMS@Si@C. The LMSs showed no evidences of catalytic activity towards TMB oxidation in the absence of light after 30 min of reaction (data not shown). However, a slight catalytic activity for TMB oxidation under aerobic condition could be detected for expanded periods of time as expected from the charge transfer role of carbon and silicon nanodomains.15,16 On the other hand, LMS@C, LMS@Si and LMS@Si@C showed increasing activity. In particular, the LMS@Si@C sample exhibited the highest peroxidase-mimicking response (Fig. 3a and S5†), with an apparent synergistic effect that suggests the beneficial role of combining both type of emitting centers. Furthermore, no significant oxidation of TMB occurred in the control experiments (see Fig. S6† and inset in Fig. 3a). It is especially remarkable that no response was detected in the absence of H2O2, thereby confirming the selective peroxidase-like behaviour of our catalysts (see Fig. S7†). This further confirms that the carbogenic centers encased within our mesoporous supports do not resemble other graphene-like materials with selective photosensitizing capabilities to oxidize TMB in the absence of H2O2.59 Interestingly, the irradiation of the catalysts (in the presence of TMB–H2O2) with other LED wavelengths (Fig. 3b and also Fig. S8†) rendered a similar catalytic trend as the observed for the former experiments carried out at 405 nm. In contrast, green (532 nm) or red-emitting (740 nm) LEDs rendered a very limited or negligible response. This seems to confirm that the minimum threshold energy for photoactivation of our catalysts is given by wavelengths at 460 nm.
Taking into account the reaction mechanism reported in previous works15,61 we attribute the peroxidase-mimicking contribution of the carbogenic dots embedded within the mesochannels to their charge-transfer dual capacity to simultaneously accept electrons from the lonely pair available in the amine group of TMB and donate electrons from their conduction band to H2O2 that is reduced to water (Fig. 3c). The presence of light to induce electron–hole separation further accelerates the complete cycle TMB–H2O2 oxidation–reduction. In contrast, additional active sites that have rendered a major light-driven peroxidase activity (see Fig. 3a) must be associated to the preferential presence of O3Si–SiO3, Si–O, Si–OH dangling bonds including multiple surface and reactive defects (induced by the rapid pyrolysis conditions). According to previous studies62–64 H2O2 preferentially adsorbs onto Si/SiOx sites and reacts to form Si–O electrophilic intermediates that withdraw electron from TMB and promote its oxidation16,62 (see Fig. 3d). It seems also quite plausible that H2O2 alternatively or simultaneously reacts after a hemolytic cleavage of strained three-membered Si–O rings63 that ends up with the production of ˙OH radicals. This ˙OH radical generation was confirmed by using disodium terephthalate (NaTA) as a selective probe that especifically reacts with ˙OH radical groups to form 2-hydroxyl disodium terephthalate, a fluorescent compound emitting at 425 nm (Fig. S10a† and schematic reaction in Fig. S10b†). These ˙OH could further react with additional H2O2 molecules thereby accelerating the whole peroxidase-mimicking response.
It is also well established that the enzymatic activity can be strongly dependent on the reaction conditions. Therefore, we studied the influence on the observed activity of LMS@Si@C samples of the concentration of nanomaterials, pH and temperature. The pH was varied from 2 to 9 and the temperature from 20 to 65 °C. As shown in Fig. 4a, the catalytic activity increased in the pH range of 5–6, indicating that the oxidation reaction of TMB occurred easily under weakly acidic conditions. Likewise, the optimal performance was estimated at pH 5 for concentrations of 4 μg mL−1 of nanohybrid under irradiation at 405 nm (see Fig. S11†). Thus, unlike natural enzymes such as HRP that operate in a narrow interval of pH,60 our hybrid materials could perform and remain active in a wide range of pH (Fig. 4a) with an optimal performance around pH 5.
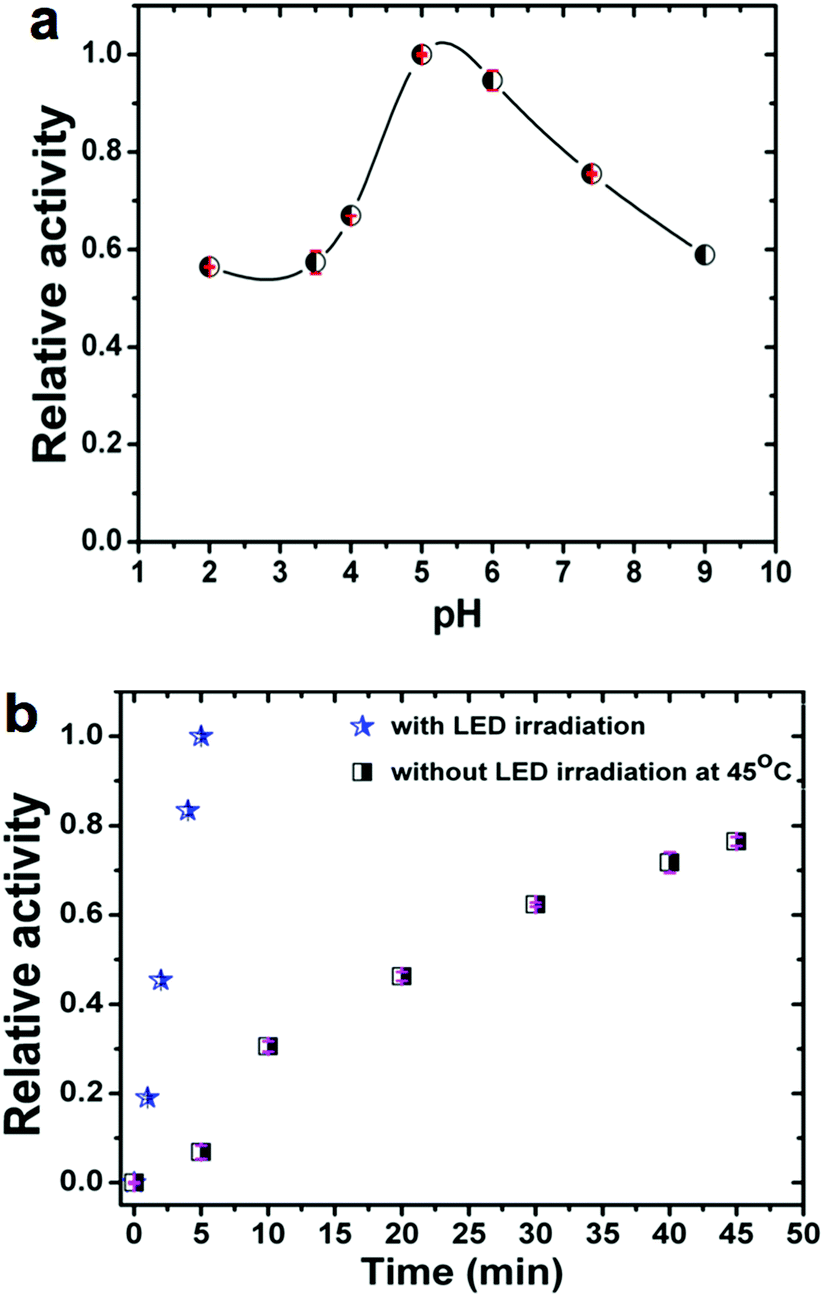 | ||
| Fig. 4 Evaluation of the overall peroxidase-mimicking performance of the LMS@Si@C sample against different reaction parameters: (a) influence of pH (experiments at 19–20 °C under irradiation with a blue LED at 405 nm for 5 min); and (b) comparison of the evolution of relative activity with time. Conversion under LED irradiation compared to thermally-driven reaction under optimal conditions (45 °C). The experiments were carried out using 4 μg mL−1 of catalyst in 2 mL of 0.2 M NaAc buffer with 0.16 mM TMB as substrate. The H2O2 concentration was 10 mM at pH 7.4 unless otherwise stated. The relative activity in each graph is referred to the maximum reaction rate observed for the experiments represented. The error bars represent the standard deviation of three measurements. | ||
In addition, to compare with other studies reported where there is no photo-enzymatic reaction, we evaluated the activity of the hybrid as a function of temperature in the range of 22–65 °C without irradiation. Fig. S12† shows the temperature dependent response curves. The optimal temperature was approximately 45–50 °C (as determined from the conversion after 45 min reaction time). It can be seen that the activity of the nanohybrid was much higher when working at room temperature under LED irradiation compared to thermal activation: 5 minutes of irradiation at room temperature produced a conversion well above that obtained at any working temperature without illumination (see Fig. 4b). It is also worth mentioning that the photo-catalyst also exhibited a high stability after several LED irradiation cycles (see Fig. S13†). The very small decrease observed per cycle is attributed to the loss of catalyst in the handling between samples.
In order to quantify the peroxidase-like activity of the LMS@Si@C a kinetic study of the oxidation of TMB by reduction of H2O2 was carried out. The kinetic parameters were obtained by performing a series of experiments changing the concentrations of one substrate and keeping constant the concentration of the other. To acquire the kinetics parameters, the data were fitted to the Michaelis–Menten equation and typical double-reciprocal Lineweaver–Burk plots were displayed (Fig. 5). The parallel trends observed at different substrate concentrations were identified as a clear signal of a so-called ping–pong mechanism,15,16 where a transitory catalyst complex is formed during the catalytic reaction. This indicates that, the LMS@Si@C, like horseradish peroxidase (HRP), binds and reacts with the first substrate (either TMB or H2O2), then releases the first product before reacting with the second substrate. As observed from the kinetic parameters, the active sites of LMS@Si@C created reagent intermediates for TMB oxidation.
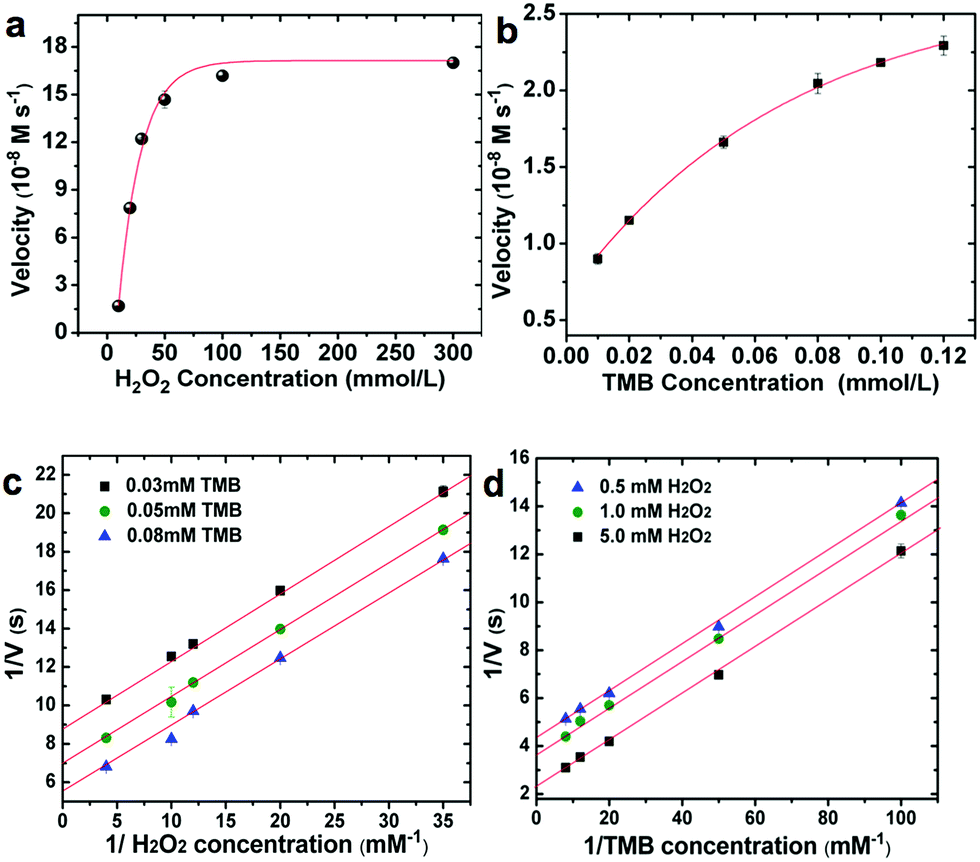 | ||
| Fig. 5 Steady-state kinetic assays using double-reciprocal Lineweaver–Burk plots and determination of the main kinetic constants during the blue-LED induced oxidation of TMB with H2O2 using LMS@Si@C as enzyme-like peroxidase photocatalyst. The velocity (v) of the reaction was measured using 4 μg mL−1 LMS@Si@C in 2 mL of 0.2 M NaAc buffer at pH 6 and photosensitization. The error bars represent the standard error derived from three repeated measurements. Experimental conditions in (a) [TMB] = 0.8 mM and [H2O2] was systematically varied; (b) [H2O2] = 50 mM and [TMB] was systematically varied; (c and d) double reciprocal plots of activity of NPs with the concentration of one substrate (H2O2 or TMB) fixed and the other varied. | ||
The combination of @Si@C species facilitated the formation of such intermediates, leading to enhanced catalytic activity. This ping–pong mechanism is also observed in analogous HRP-based systems. Important enzyme catalytic parameters such as the Michaelis–Menten constant (Km) and the maximum initial velocity (Vmax) were obtained. Km is recognized as an indicator of the affinity of an enzyme to its substrate. A strong affinity of the nanoenzyme-substrate is reflected by a smaller Km and vice versa.9 The apparent Km and Vmax values of LMS@Si@C and other enzyme-like nanomaterials have been summarized for comparison in Table S4.† The low Km values for the LMS@Si@C catalyst indicate high affinity towards both TMB and H2O2. It is also worth mentioning that Km values relative to both TMB and H2O2 were remarkably lower than in previously reported artificial enzymes with analogous compositions and/or photocatalytic response (Table S4†). This may be due to the fact that photoactive centers are abundant and homogeneously distributed on the high surface area of the mesoporous nanorods.19 Hence, the lower magnitude of Km values for H2O2 also suggests that a lower concentration of H2O2 was required to successfully complete the TMB oxidation in comparison with many other enzyme-like peroxidase systems.19,65 The apparent Km and the maximum reaction rate value of photo-activated LMS@Si@C with TMB as substrate was 0.0525 mM and 1.488 nM s−1, respectively. Compared with that of HRP (Km = 0.155 mM),15 the Km value of the photo-activated LMS@Si@C with TMB as substrate was much lower, indicating that LMS@Si@C possess a higher affinity towards the TMB substrate than the natural counterpart (HRP). In addition, the LMS@Si@C with TMB as the substrate had much smaller Km value than other reported nanomaterials with peroxidase-like activities, such as freestanding Si-dots (Km = 1.502 mM),16 graphene oxide (GO)-AuNCs nanocomposites (Km = 0.16 mM)66 or Fe3O4@carbon (Km = 0.072 mM).66 Furthermore, it is worth mentioning that our photocatalyst exhibit negligible enzymatic activity in the absence of H2O2 (Fig. S7†) or light external stimuli (Fig. S6–S8†). While DNA-complexes have reported an analogous response,60 graphene oxide has reported an opposite behaviour under photoirradiation.59
Based on the positive peroxidase-mimicking response of the LMS@Si@C sample, a colorimetric method for glucose detection was additionally designed as alternative to HRP (see also Experimental section for further details). It consisted on the combination of a natural enzyme such as glucose oxidase (GOx) and the LMS@Si@C peroxidase-like catalyst to carry out a cascade catalytic process. This process enabled an indirect determination of the concentration of glucose (Fig. S14†). Glucose was indirectly determined by measuring the in situ generated H2O2 by-product stemming from the glucose oxidation reaction catalyzed by the glucose oxidase (GOx) natural enzyme. The color variation associated to the TMB oxidation catalyzed by the LMS@Si@C was H2O2 concentration-dependent. First we determined the detection limit of H2O2 and the absorbance measurements at 652 nm (TMB oxidation catalyzed by LMS@Si@C) was proportional to H2O2 concentration from 0.001–0.10 mM (Fig. S14†) exhibiting a linear correlation with a detection limit (DL) of 1.5 μM. The color variation was obvious by visual observation (inset in Fig. S14b†). Secondly, when combined with GOx, the linear range for glucose detection was set from 10 to 130 μM and the DL was 0.5 μM (Fig. S14c†). Likewise, colorimetric variations were clearly identified by visual observation (inset in Fig. S14c†). Generally, the serum glucose concentrations in healthy and diabetic individuals range from 3–8 mM and 9–40 mM, respectively.15 In the light of the linear range described above, the proposed method could be applied to detect glucose in diluted serum. In addition, to test the selectivity of the above proposed method, it was also tested in the presence of other saccharides such as fructose, lactose, sucrose and maltose. Fig. S14d† shows that the absorbance of these glucose analogs was negligible compared with that of glucose even at concentrations as high as 10 mM. This is due to the specificity of GOx to glucose. These results further indicate that the method can be potentially applied to determine the glucose content in real samples, which is important for biological and biomedical applications.
Conclusions
We have reported here the synthesis of a new type of peroxidase-like enzyme surrogate. Under suitable conditions, the flash pyrolysis of the structure directing agent confined in the mesopores of silica nanorods produced both carbon nanodots and oxygen-deficient centers within the silica structure that could be simultaneously activated under blue-emitting LED irradiation sources. A flash-thermal heating of the mesoporous rods (attained by immersion in a fluidized-bed reactor) was essential to achieve the formation of carbon dots and the carbothermic reduction of the silica before the products of SDA decomposition could diffuse out of the mesoporous structure: a slower conventional heating process to the same or even higher temperature did not produce the desired results. In addition, the type of atmosphere during the flash pyrolysis process can also be used to tune the type of emitting centers generated. The simultaneous activation of both of these luminescent centers gave rise to a cooperative effect that increased the catalytic activity of the hybrid. The resulting peroxidase-like catalyst is active in a wider range of pH than natural peroxidases and its activity can be switched on and off at will by controlling the sample illumination. It is also worth mentioning that the combination of these novel catalysts with GOx to establish a cascade reaction for the indirect and selective quantification of H2O2 and/or glucose paves the way for the development of novel colorimetric sensors based on inexpensive and readily abundant raw precursors such as carbon and silicon. Furthermore, these metal-free materials possess an ideal combination of robustness, mechanical stability, inertness and appealing optical properties that make them excellent candidates for deployment as low-cost, durable, sensing materials.Experimental
Material
Tetraethylorthosilicate (TEOS), ammonium fluoride (NH4F, 98%), heptane (99%), poly(ethylene glicol)-block-poly(propylene glycol)-block-poly(ethylene glycol) average Mn ∼ 5800 (Pluronic® P-123), hydrogen peroxide (H2O2, 30%), hydrochloric acid (HCl, 37%), 3,3,5,5-tetramethylbenzidine (TMB, 99%), terephtalic acid (TA, 89%), and sodium hydroxide (NaOH, > 97%) were obtained from Sigma-Aldrich. All chemicals were of analytical purity grade.Characterization techniques
The morphologies and particle size distributions were determined by Transmission Electron Microscopy (TEM) in a Tecnai T20 and F30 (Thermofisher scientific, formerly FEI), operated at 200–300 kV, respectively, and coupled with a CCD camera (Gatan). Scanning Transmission Electron Microscopy (STEM) was performed in a probe-corrected Titan (Thermofisher Scientific, formerly FEI) at 300 kV. High angle annular dark field images were obtained with a HAADF detector (Fischione). Electron energy loss spectra were obtained with a Tridiem energy filter (Gatan) working as a spectrometer. To prepare the samples, the nanoparticle suspensions were diluted with water prior to casting on a holey carbon TEM grid. Fluorescence emission spectra were collected on a LS55 Fluorescence Spectrometer (PerkinElmer) equipped with a xenon arc lamp as the light source and a quartz cell (10 × 10 mm). The nanoparticles surface was analyzed by X-ray photoelectron spectroscopy (XPS) with an Axis Ultra DLD (Kratos Tech.). A monochromatic Al Kα source (1486.6 eV) was employed with multiple runs at 12 kV, 10 mA and pass energy of 20 eV was used. The binding energies were calibrated to the internal C 1s (284.2 eV) standard. Analyses of the peaks were performed with CasaXPS software, using a weighted sum of Lorentzian and Gaussian component curves after Shirley background subtraction. The specific surface area and pore-volume distribution were measured by nitrogen adsorption at 77 K in a Micromeritics ASAP 2020; samples were out-gassed at 26.7 Pa and 623 K for 6 h before the measurement. The ordered mesoporous structure, phase identification and purity of the LMSs were studied by low angle X-ray diffraction. The patterns were recorded in a Philips X-Pert diffractometer equipped with a monochromatized Cu-Kα radiation (40 kV, 20 mA) over the range 0.6–10.0° with a step of 0.02° and an analysis time of 5 s. All measurements were performed at room temperature. Thermogravimetric analyses were conducted in an SDTA-851 Mettler Toledo equipment (using 50 mL min−1 of synthetic air, heating ramp of 5 °C min−1 up to 800 °C) and 40% weight losses corresponding to the organic matrix where identified in the as prepared silica nanorods. The samples subjected to flash pyrolysis treatment (LMS@C and LMS@Si@C) kept a 7–8% wt and samples subjected to slow calcination in air rendered values below 1.5% wt attributable to organic species.Synthesis of SBA-15 mesoporous silica nanorods
The ordered mesoporous silica nanorods were prepared according to previously reported procedures.29,67 In brief, 1.2 g of P123 and 0.014 g of NH4F were first dissolved at 20 °C in 40 mL of HCl (1.75 M) solution under stirring. Upon complete dissolution, 2.75 mL of TEOS and 8.5 mL heptane were added drop-wise and left under stirring for 8 min. The aged precursor mixture was hydrothermally heated at 100 °C for 24 hours in a Teflon-lined autoclave. The solid product was then filtered, washed three times with distilled water, and dried at 60 °C overnight.Flash-thermal treatment to induce the generation of carbon and silica-based emitting centers
Typically, 20 mg of the mesoporous nanorods were placed in a quartz capillary well (Øin = 8 mm) designed with inlet–outlet outfits (Øin = 6 mm) for inert gas feeding (flow rate 3 L h−1). The capillary tube was introduced inside a sand bed (8 cm bed height) used to induce the formation of a bubbling fluidized bed via air bubbling (flow feeding rate 15 L h−1). The fluidized bed was kept at 500 °C for 2 min with or without nitrogen flow and allowed to cool down to room temperature, producing LMS@C and LMS@Si@C, respectively. After the flash pyrolysis process, a yellowish-pale solid was obtained. The generation of the LMS@Si sample was carried out using the same protocol to form the LMS@Si@C followed by a subsequent calcination step in air with a heating ramp of 1 °C min−1 up to 550 °C and dwell time of 6 h. These reaction conditions were performed and reproduced with multiple batches and showed similar trends. Likewise, additional experiments with up to 100 mg rendered analogous response but a proper scale up would require the design and detailed analysis of multiple parameter settings not carried out in this work. The experiments have been performed by the platform of production of biomaterials and nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials & Nanomedicine (CIBER-BBN).Peroxidase enzymatic assays using TMB
The experiments were carried out at room temperature in MW24 cell culture plates and upon irradiation with a blue LED (λexc = 405 nm). The peroxidase-like activity of the as-prepared LMS nanostructures was tested via the catalytic oxidation of the peroxidase substrate TMB in the presence of H2O2 (see Fig. 3 and S5†). The reactions were carried out in 2.0 mL of a 0.2 M sodium acetate buffer solution, pH 7.4, containing 4 μg mL−1 of the catalysts in the presence of 10 mM H2O2 and 0.16 mM TMB as the substrates (8 μL of 40 mM TMB and 200 μL of 100 mM H2O2, stock solution, respectively). A photograph of the progress of the reaction solutions was taken under LED irradiation (see Fig. S5†). The blue solutions appearing along with the reaction proceeding were monitored in time scan mode at 652 nm using a spectrophotometer.To evaluate the photo-responsive properties at different wavelength, different LEDs irradiating at different wavelengths have been additionally tested in the colorimetric assay with the substrate TMB to assess the response of this novel material to other light wavelengths (see Fig. 3b and S8†). A maximum power supply of 2 W was used for all the LEDs. The distance between the wells containing the reacting suspension and the LED source was set at 15 mm (see Fig. S9†). The actual irradiance for each LED has been additionally included and is based on estimations provided by the manufacturer (LED ENGIN, LZ4 models): 365 nm (Ultraviolet-LED): 12 mW cm−2; 405 nm (Blue-LED): 68 mW cm−2; 460 nm (Blue-LED): 42.4 mW cm−2; 532 nm (Green-LED): 210 mW cm−2; 704 nm (Red-LED): 51.4 mW cm−2 and 460 + 500–700 nm (White-LED): 81 mW cm−2.
Detection of hydroxyl radical using terephtalic acid
The peroxidase-mimicking activity of the LMS nanostructures was initially determined from their catalytic ability to decompose H2O2 into ˙OH radicals. A fluorimetric method to detect the interaction of these OH radicals with terephtalic acid (TA)20 was selected and optimized (see also Fig. S7†). We found it was important to select basic conditions (pH ∼ 7–9) or alternatively use disodium terephthalate (NaTA) to conduct the experiments in water due to the limited solubility of TA in water. NaTA is a non-fluorescent molecule that may stoichiometrically react with ˙OH to produce its fluorescent derivative (Fig. 3a). The fluorescence intensity of this derivative correlates with the level of ˙OH radical formation.68 The preparation of NaTA was carried out by neutralization of 0.8 g of TA with 50 mL of aqueous 0.05 N NaOH solution with constant stirring for 1 h. Then, 8.3 g of NaOH were added to reach 14% by weight concentration and favor the precipitation of the desired product. Under these conditions, the precipitate NaTA was filtered and dried in an oven at 110 °C for 12 hours. Finally, for detecting ˙OH, as-prepared LMS (0.004 mg mL−1) was redispersed in the probe NaTA solution (2 mL, 5 mM). After irradiation with a blue-emitting LED at 405 nm for, the required period of time (typically 5 minutes), the mixture suspension was centrifuged to remove the solid nanocatalysts. The fluorescence emission spectrum of the generated 2-hydroxy disodium terephtalate in the supernatant was subsequently monitored under the excitation wavelength of 315 nm.Determination of the kinetic parameters
All kinetic assays were carried out at room temperature in 2.0 mL MW24 cell culture plates and irradiation with a blue LED at 405 nm. Unless otherwise stated, a 0.2 M NaAc solution (pH 7.4) was used as the reaction buffer (total volume = 2 mL). The reaction kinetic measurements were carried out in time course mode by monitoring the absorbance variation of 652 nm. The initial experiments were performed with 4 μg mL−1 of LMS nanostructures in the presence of 50 mM H2O2 and using different concentrations of TMB as the substrate. Likewise, the same amount of catalyst in a 2 mL buffer reaction volume was employed in a series of experiments with 0.8 mM of TMB, and different concentrations of H2O2. Additional experiments varying TMB and H2O2 concentrations were tuned to 0.01, 0.02, 0.05, 0.08, 0.10, 0.12 mM and 10, 20, 30, 50, 100, 300 mM, respectively. After the addition of LMS@Si@C catalyst to the substrates (TMB and H2O2), the absorbance values (with especial attention to the maximum at 652 nm) were monitored as function of time in 1 minute intervals. These “absorbance vs. time” plots were then used to obtain the slope at the initial point (SlopeInitial) of each reaction by conducting the first derivation of each curve using OriginPro 8.0 software. The initial reaction velocity (ν) was calculated by SlopeInitial/(εTMB-652 nm × l), where εTMB-652 nm is the molar extinction coefficient of TMB at 652 nm, which equals 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.15 The plots of ν against substrate concentrations were fitted using nonlinear regression of the Michaelis–Menten equation.9 The apparent kinetic parameters were calculated based on the Michaelis–Menten equation using the Lineweaver–Burk plot: 1/ν = Km/Vmax(1/[S] + 1/Km), where Vmax represents the maximal reaction velocity, [S] is the concentration of substrate, and Km is the Michaelis constant. Parameters Km and Vmax were obtained from the double reciprocal plot (see Fig. 5).
000 M−1 cm−1.15 The plots of ν against substrate concentrations were fitted using nonlinear regression of the Michaelis–Menten equation.9 The apparent kinetic parameters were calculated based on the Michaelis–Menten equation using the Lineweaver–Burk plot: 1/ν = Km/Vmax(1/[S] + 1/Km), where Vmax represents the maximal reaction velocity, [S] is the concentration of substrate, and Km is the Michaelis constant. Parameters Km and Vmax were obtained from the double reciprocal plot (see Fig. 5).
Selective detection of glucose in a cascade reaction
For glucose detection, 20 mM glucose and 0.5 mg mL−1 GOx in 1 mL of PBS buffer (pH 7.4) were incubated at 37 °C for 1 h. Then, 0.1 mL of 16 mM TMB (final concentration: 0.8 mM), 0.2 mL of the LMS@Si@C stock solution (80 μg mL−1) (final concentration: 4 μg mL−1) and 0.7 mL of NaAc buffer (0.2 M, pH 7.4) were added to the above glucose reaction system (1 mL). Finally, the mixed solution was irradiated with a blue LED source at an excitation wavelength of 405 nm for 10 min to allow development of the blue color. The absorbance of the oxidation product was monitored at 652 nm. In the control experiments, 10 mM maltose, 10 mM fructose and 10 mM lactose were used instead of glucose.Detection limits (DL)
A typical colorimetric analysis was performed as follows: 0.1 mL of TMB (2.5 mM) (final conc., 0.16 mM), 0.2 mL of the LMS@Si@C stock solution (80 μg mL−1) (final conc., 4 μg mL−1) and 0.2 mL of H2O2 with several concentration ranges (to finally obtain concentrations: 0.001–0.5 mM) were added into 1.5 mL of NaAc buffer (2 M, pH 7.4). Then, the absorbance of the reaction solution at 652 nm of each sample was immediately measured. Colorimetric assays for glucose detection were performed as follows: (i) 0.5 mg mL−1 GOx in 1 mL and different concentrations of glucose (0.001–0.5 mM) in 1 mL of PBS buffer (pH 7.4) were incubated at 37 °C for 1 h to produce H2O2; (ii) 0.1 mL of 2.5 mM TMB, 0.2 mL of LMS@Si@C (final conc., 4 μg mL−1) and 0.7 mL of NaAc buffer (0.2 M, pH 7.4) were added to the above glucose reaction system (1 mL); and (iii) the mixed solution was irradiated LED with 405 nm for 10 min to allow development of the blue color. The absorbance of the oxidation product was monitored at 652 nm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the European Research Council for funding through an advanced grant research project (HECTOR grant number 267626; CADENCE grant number 742684) and a CIG-Marie Curie Reintegration Grant (NANOLIGHT REA grant number 294094). The TEM measurements were conducted at the Laboratorio de Microscopias Avanzadas, Instituto de Nanociencia de Aragon, Universidad de Zaragoza, Spain. The synthesis of materials has been performed by the Platform of Production of Biomaterials and Nanoparticles of the NANOBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials & Nanomedicine (CIBER-BBN). M. C. O. acknowledges the Spanish Government for an FPU predoctoral fellowship.Notes and references
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef PubMed.
- R. Breslow and L. E. Overman, J. Am. Chem. Soc., 1970, 92, 1075–1077 CrossRef PubMed.
- H. Wei and E. K. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- Y. B. Zhou, B. W. Liu, R. H. Yang and J. W. Liu, Bioconjugate Chem., 2017, 28, 2903–2909 CrossRef PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- Y. F. Zhang, S. Tsitkov and H. Hess, Nat. Commun., 2016, 7, 13982 CrossRef PubMed.
- H. Wei and E. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef PubMed.
- S. Y. Fu, S. Wang, X. D. Zhang, A. H. Qi, Z. R. Liu, X. Yu, C. F. Chen and L. L. Li, Colloids Surf., B, 2017, 154, 239–245 CrossRef PubMed.
- L. Z. Gao, J. Zhuang, L. Nie, J. B. Zhang, Y. Zhang, N. Gu, T. H. Wang, J. Feng, D. L. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef PubMed.
- L. Z. Gao, K. L. Fan and X. Y. Yan, Theranostics, 2017, 7, 3207–3227 CrossRef PubMed.
- J. B. Liu, X. N. Hu, S. Hou, T. Wen, W. Q. Liu, X. Zhu and X. C. Wu, Chem. Commun., 2011, 47, 10981–10983 RSC.
- Y. Jv, B. X. Li and R. Cao, Chem. Commun., 2010, 46, 8017–8019 RSC.
- Y. Ju and J. Kim, Chem. Commun., 2015, 51, 13752–13755 RSC.
- Y. J. Song, K. G. Qu, C. Zhao, J. S. Ren and X. G. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef PubMed.
- W. Shi, Q. Wang, Y. Long, Z. Cheng, S. Chen, H. Zheng and Y. Huang, Chem. Commun., 2011, 47, 6695–6697 RSC.
- Q. Chen, M. Liu, J. Zhao, X. Peng, X. Chen, N. Mi, B. Yin, H. Li, Y. Zhang and S. Yao, Chem. Commun., 2014, 50, 6771–6774 RSC.
- H. W. Wang, W. W. Jiang, Y. W. Wang, X. L. Liu, J. L. Yao, L. Yuan, Z. Q. Wu, D. Li, B. Song and H. Chen, Langmuir, 2013, 29, 3–7 CrossRef PubMed.
- Y. Z. Wang, W. J. Qi and Y. J. Song, Chem. Commun., 2016, 52, 7994–7997 RSC.
- M. C. Kim, D. Lee, S. H. Jeong, S. Y. Lee and E. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 34317–34326 CrossRef PubMed.
- M. C. Ortega-Liebana, J. L. Hueso, R. Arenal and J. Santamaria, Nanoscale, 2017, 9, 1787–1792 RSC.
- Z. Y. Xie, E. J. Henderson, O. Dag, W. Wang, J. E. Lofgreen, C. Kubel, T. Scherer, P. M. Brodersen, Z. Z. Gu and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5094–5102 CrossRef PubMed.
- M. Guan, W. D. Wang, E. J. Henderson, O. Dag, C. Kubel, V. S. K. Chakravadhanula, J. Rinck, I. L. Moudrakovski, J. Thomson, J. McDowell, A. K. Powell, H. X. Zhang and G. A. Ozin, J. Am. Chem. Soc., 2012, 134, 8439–8446 CrossRef PubMed.
- J. H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef PubMed.
- L. Gu, D. J. Hall, Z. T. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326 CrossRef PubMed.
- M. J. Sailor and E. C. Wu, Adv. Funct. Mater., 2009, 19, 3195–3208 CrossRef.
- Q. J. He, J. L. Shi, X. Z. Cui, C. Y. Wei, L. X. Zhang, W. Wu, W. B. Bu, H. R. Chen and H. X. Wu, Chem. Commun., 2011, 47, 7947–7949 RSC.
- Q. J. He, C. H. Xu, J. Q. Luo, W. Wu and J. L. Shi, Chem. Commun., 2014, 50, 13944–13947 RSC.
- Z. J. Zhang, X. H. Zhang, B. W. Liu and J. W. Liu, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef PubMed.
- E. M. Johansson, M. A. Ballem, J. M. Cordoba and M. Oden, Langmuir, 2011, 27, 4994–4999 CrossRef PubMed.
- Y. Ishikawa, A. V. Vasin, J. Salonen, S. Muto, V. S. Lysenko, A. N. Nazarov, N. Shibata and V. P. Lehto, J. Appl. Phys., 2008, 104, 083522 CrossRef.
- S. Y. Seo, K. S. Cho and J. H. Shin, Appl. Phys. Lett., 2004, 84, 717–719 CrossRef.
- D. Z. Tan, Z. J. Ma, B. B. Xu, Y. Dai, G. H. Ma, M. He, Z. M. Jin and J. R. Qiu, Phys. Chem. Chem. Phys., 2011, 13, 20255–20261 RSC.
- C. M. Carbonaro, R. Corpino, P. C. Ricci, M. Salis and A. Anedda, J. Mater. Sci., 2013, 48, 4452–4458 CrossRef.
- V. S. Kortov, A. F. Zatsepin, S. V. Gorbunov and A. M. Murzakaev, Phys. Solid State, 2006, 48, 1273–1279 CrossRef.
- V. S. Kortov, A. F. Zatsepin, V. A. Pustovarov, A. A. Chudinov and D. Y. Biryukov, Radiat. Meas., 2007, 42, 891–893 CrossRef.
- M. C. Ortega-Liebana, N. X. Chung, R. Limpens, L. Gomez, J. L. Hueso, J. Santamaria and T. Gregorkiewicz, Carbon, 2017, 117, 437–446 CrossRef.
- S. J. Zhu, Y. B. Song, X. H. Zhao, J. R. Shao, J. H. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- M. L. Liu, L. Yang, R. S. Li, B. B. Chen, H. Liu and C. Z. Huang, Green Chem., 2017, 19, 3611–3617 RSC.
- L. Wang, S. J. Zhu, H. Y. Wang, Y. F. Wang, Y. W. Hao, J. H. Zhang, Q. D. Chen, Y. L. Zhang, W. Han, B. Yang and H. B. Sun, Adv. Opt. Mater., 2013, 1, 264–271 CrossRef.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef PubMed.
- S. Kim, S. W. Hwang, M. K. Kim, D. Y. Shin, D. H. Shin, C. O. Kim, S. B. Yang, J. H. Park, E. Hwang, S. H. Choi, G. Ko, S. Sim, C. Sone, H. J. Choi, S. Bae and B. H. Hong, ACS Nano, 2012, 6, 8203–8208 CrossRef PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. F. Wang, P. J. G. Luo, H. Yang, M. E. Kose, B. L. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef PubMed.
- J. D. Holmes, K. J. Ziegler, R. C. Doty, L. E. Pell, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2001, 123, 3743–3748 CrossRef PubMed.
- M. J. L. Portoles, F. R. Nieto, D. B. Soria, J. I. Amalvy, P. J. Peruzzo, D. O. Martire, M. Kotler, O. Holub and M. C. Gonzalez, J. Phys. Chem. C, 2009, 113, 13694–13702 CrossRef PubMed.
- A. Aboshi, N. Kurumoto, T. Yamada and T. Uchino, J. Phys. Chem. C, 2007, 111, 8483–8488 CrossRef.
- L. R. Zheng, B. B. Huang, J. Y. Wei, Y. Dai and M. H. Whangbo, J. Lumin., 2011, 131, 218–224 CrossRef.
- V. Nikas, S. Gallis, M. Huang, A. E. Kaloyeros, A. P. D. Nguyen, A. Stesmans and V. V. Afanas'ev, Appl. Phys. Lett., 2014, 104, 061906 CrossRef.
- M. Narisawa, T. Kawai, S. Watase, K. Matsukawa and A. Iwase, AIP Adv., 2014, 4, 017118 CrossRef.
- Q. He, M. Ma, C. Wei and J. Shi, Biomaterials, 2012, 33, 4392–4402 CrossRef PubMed.
- H. Chen, Z. Zhen, W. Tang, T. Todd, Y.-J. Chuang, L. Wang, Z. Pan and J. Xie, Theranostics, 2013, 3, 650–657 CrossRef PubMed.
- A. Malumbres, G. Martinez, J. L. Hueso, J. Gracia, R. Mallada, A. Ibarra and J. Santamaria, Nanoscale, 2015, 7, 8566–8573 RSC.
- G. S. Fu, X. Z. Wang, H. N. Feng, X. Yu, W. L. Dai, W. B. Lu and W. Yu, J. Alloys Compd., 2013, 579, 284–289 CrossRef.
- E. Pippel, J. Woltersdorf, H. O. Olafsson and E. O. Sveinbjornsson, J. Appl. Phys., 2005, 97, 034302 CrossRef.
- A. Eljarrat, L. Lopez-Conesa, J. Lopez-Vidrier, S. Hernandez, B. Garrido, C. Magen, F. Peiro and S. Estrade, Nanoscale, 2014, 6, 14971–14983 RSC.
- E. Pippel, O. Lichtenberger and J. Woltersdorf, J. Mater. Sci. Lett., 2000, 19, 2059–2060 CrossRef.
- P. Ewels, T. Sikora, V. Serin, C. P. Ewels and L. Lajaunie, Microsc. Microanal., 2016, 22, 717–724 CrossRef PubMed.
- P. E. Batson, Microsc., Microanal., Microstruct., 1991, 2, 395–402 CrossRef.
- X. F. Zhang, L. Deng, C. P. Huang, J. Y. Zhang, X. D. Hou, P. Wu and J. W. Liu, Chem.–Eur. J., 2018, 24, 2602–2608 CrossRef PubMed.
- X. F. Zhang, C. P. Huang, S. X. Xu, J. B. Chen, Y. Zeng, P. Wu and X. D. Hou, Chem. Commun., 2015, 51, 14465–14468 RSC.
- Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef PubMed.
- Z. H. Kang, C. H. A. Tsang, N. B. Wong, Z. D. Zhang and S. T. Lee, J. Am. Chem. Soc., 2007, 129, 12090–12091 CrossRef PubMed.
- H. Y. Zhang, D. R. Dunphy, X. M. Jiang, H. Meng, B. B. Sun, D. Tarn, M. Xue, X. Wang, S. J. Lin, Z. X. Ji, R. B. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef PubMed.
- T. Yamada, M. Nakajima, T. Suemoto and T. Uchino, J. Phys. Chem. C, 2007, 111, 12973–12979 CrossRef.
- L. N. Song, C. Huang, W. Zhang, M. Ma, Z. W. Chen, N. Gu and Y. Zhang, Colloids Surf., A, 2016, 506, 747–755 CrossRef.
- Y. Tao, Y. H. Lin, Z. Z. Huang, J. S. Ren and X. G. Qu, Adv. Mater., 2013, 25, 2594–2599 CrossRef PubMed.
- L. Uson, J. L. Hueso, V. Sebastian, R. Arenal, I. Florea, S. Irusta, M. Arruebo and J. Santamaria, Catal. Commun., 2017, 100, 93–97 CrossRef.
- J. Fu, Y. Shao, L. Wang and Y. Zhu, Nanoscale, 2015, 7, 7275–7283 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with additional experimental synthesis conditions, additional information regarding the spectroscopic characterization of the catalysts and additional details on the enzymatic assays carried out with the luminescent nanorods. See DOI: 10.1039/c8sc03112f |
| This journal is © The Royal Society of Chemistry 2018 |