
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design of a molecular triple photoswitch for wavelength-selective control†
Chong
Yang
a,
Chavdar
Slavov
 b,
Hermann A.
Wegner
c,
Josef
Wachtveitl
b and
Andreas
Dreuw
*a
b,
Hermann A.
Wegner
c,
Josef
Wachtveitl
b and
Andreas
Dreuw
*a
aInterdisciplinary Center for Scientific Computing, Heidelberg University, Im Neuenheimer Feld 205A, 69120 Heidelberg, Germany. E-mail: dreuw@uni-heidelberg.de
bInstitute for Physical and Theoretical Chemistry, Goethe University, Max-von-Laue Str. 7, 60438 Frankfurt am Main, Germany
cInstitute of Organic Chemistry, Justus Liebig University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
First published on 21st September 2018
Abstract
A small single molecule with multiple photoswitchable subunits, selectively and independently controllable by light of different wavelengths, is highly attractive for applications in multi-responsive materials and biological sciences. Herein, triple photoswitches are presented consisting of three independent azobenzene (AB) subunits that share a common central phenyl ring: the meta-trisazobenzenes (MTA). It is the unique meta-connectivity pattern leading to decoupling of all azo-subunits although they do overlap spatially. Based on this pattern, we design a triple MTA photoswitch, as proof-of-principle, with three different, electronically independent AB branches on the computer, which can be individually photo-excited to trigger ultra-fast E → Z isomerization at the selected AB branch.
1. Introduction
Azobenzene (AB) and its derivatives are by far the most popular molecular photoswitches with a wide range of application in the fields of photobiology, photochemistry and functional organic materials.1–7 Their attractive light-responsive properties are based on the ultra-fast photoisomerization between two long-lived E- and Z-configurations.1–13 In addition, their photochromic and isomerization properties and, in particular, their activation wavelength can be modulated on demand by suitable chemical modifications, e.g. by introduction of substituents to the benzene rings. Owing to their adjustable photochemical properties, AB-based compounds provide a versatile basis for numerous applications in data storage devices,3 optoelectronic devices,4 and molecular switches.5Recently, multi-photochromic compounds containing more than one switching unit that can possibly be selectively and independently controlled by external stimuli are attracting much interest.14–17 A single molecule integrating multiple different AB subunits may exhibit such a multi-functional nature, and may be applicable in supramolecular stimuli-responsive systems, organic liquid crystal photonics, functional nanomaterials, and multi-responsive biological systems.2–5
The photochromic behavior and the photoisomerization of AB and its derivatives have been intensively investigated so far,8–13 but only a few have recently demonstrated dual functionality. A general multi-addressable behavior of bisazobenzenes with two AB subunits was demonstrated, however, the individual AB subunits were not individually, i.e. selectively addressable.18–23 The switching behavior and the photochromic characteristics of four para-bisazobenzene derivatives connected by aryl–aryl linkers between two AB moieties were studied in an effort to optimize the photoswitching efficiency of linear multi-azobenzenes.19 Furthermore, other multi-photochromic compounds containing multiple switching subunits such as cyclo-trisazobenzene,24–26tris-[4-(phenylazo)-phenyl]-amine27,28 on a surface, and other multi-photochromic compounds29–36 have been addressed to explore new multi-photochromic effects and light-induced functionalities as well. More recently, orthogonal and reversible control of two distinct types of photoswitches in a multifunctional molecular system has been reported, giving rise to impressive dual functionality.36,37 However, the design of multi-state multi-photochromic compounds with multiple functionalities still poses a great challenge, because, in general, wavelength selective control of individual components of a multi-photochromic compound requires selective addressability and independent functionalities of the single photochromic subunits.
The ultrafast dynamics and photochromic properties of ortho-, meta- and para-bisazobenzenes have been investigated systematically, and their properties and cooperative behavior have been found to strongly depend on their connectivity pattern.21 The two AB branches are uncoupled in meta-bisazobenzenes and the absorption spectrum is a superposition of the spectra of two individual AB subunits. In contrast, a substantial red-shift of the ππ* absorption band occurs in para-bisazobenzenes and is attributed to strong π-conjugation and the resulting electronic coupling of the AB branches, ortho-bisazobenzenes reveal substantial excitonic coupling.20 In particular, the uncoupled meta-connectivity seems to allow for an independent photochemical behavior of the individual AB branches, indicating that the concept of meta-connectivity can be generally useful for the design of multi-photochromic compounds with different functionalities.
For individual addressability of the AB subunits, i.e. for individual electronic excitation by light, the absorption spectra of the AB subunits should be energetically well separated. This can in principle be achieved by tuning the main absorption bands through incorporation of appropriate electron-donating or -withdrawing functional groups. Optimally, each AB subunit can then be addressed individually by light of different wavelengths leading to the photo-isomerization of the selected AB subunit.
In this work we present multi-photochromic compounds integrating three azobenzene subunits within a single molecule: 1,3,5-tris[(E)-phenylazo]benzenes, or here referred to as meta-trisazobenzenes (MTAs). The basic structure of MTA consists of three AB subunits connected via a central shared phenyl ring (Fig. 1).38 Here, its photochemistry is investigated and all AB branches will be demonstrated below to be electronically decoupled, and unsubstituted MTA to behave like three essentially independent AB molecules. Furthermore, we explored a large pool of possible substituents at the at the terminal phenyl rings. This variation is shown to shift the spectra of the AB subunits making them in principle individually addressable by light with different wavelength. Relaxed scans along the E → Z isomerization coordinate of the individually excited AB branches of MTA reveal their capability of individual photoswitching. In other words, each branch of substituted MTA can be switched individually by light with the appropriate excitation wavelength.
 | ||
| Fig. 1 Molecular structure of substituted MTA photoswitches, in which different numbers of appropriate substituents are introduced at the terminal phenyl groups. | ||
2. Computational details
In previous theoretical studies, density functional theory (DFT) and time-dependent DFT (TDDFT)39–41 have been successfully proven to reproduce and accurately predict excited states of azobenzene-based compounds in good agreement with experimental results.8–10,12,13,18,19,21,22 To investigate optical properties of MTA and its substituted derivatives, their vertical absorption spectra were calculated in the gas phase and in ethanol (ε = 24.5) using TDDFT in combination with the BHHLYP42 functional and the 6-31G*43 basis set with and without conductor-like polarizable continuum model (C-PCM)44–46 for solvation. In previous works, this functional and basis set combination has been extensively tested for azobenzene and bisazobenzenes against ab initio ADC(2) results and experimental data.21,26 For consistency, all ground-state structure and excited-state geometry optimizations have been performed at BHHLYP/6-31G* level. In addition, the Tamm–Dancoff approximation to TDDFT and a state-tracking algorithm were employed for the excited state optimizations. All calculations were carried out with the Q-Chem 5.0 and the Orca 3.0 packages.47,483. Unsubstituted meta-trisazobenzene
As first step, the photophysical properties of unsubstituted MTA (Fig. 1) have been investigated. For this reason, the absorption spectra of the E- and Z-isomers for the parent AB and the (E,E,E)-, (E,E,Z)-, (E,Z,Z)-, (Z,Z,Z)-isomers of MTA were calculated using TDDFT with the BHHLYP functional and the 6-31G* basis set in the gas phase and in ethanol, respectively (Fig. 2). The absorption spectra of all isomers calculated in the gas phase and in ethanol resemble each other in shape. Owing to solvent effects, all absorption maxima of the main ππ* absorption band are significantly red-shifted in ethanol compared to those in the gas phase. The electronic coupling of the AB subunits ias well as all main spectral features are, however, not affected by the solvent. | ||
| Fig. 2 Comparison of the computed absorption spectra of isolated AB and MTA isomers at the optimized ground-state geometries in the gas phase (left) and in ethanol (right) at the TDDFT/BHHLYP/6-31G* level. The spectra are convoluted with a Lorentzian function with full-width-at-half-maximum of 10 nm. | ||
The absorption spectrum of (E,E,E)-MTA shows a strong ππ* transition at ∼295 nm with a tiny red-shift compared to single E-AB, and the intensity of this band is roughly three times as large as in E-AB, revealing the uncoupled nature of the three AB branches. Upon excitation into this band, all three azo branches possess equal probability to be switched due to the overlapping absorption bands. Overall, the photoswitching efficiency of all three AB branches is individually conserved, and thereby the total efficiency of MTA photoswitching of one AB subunit is increased by a factor of three compared to AB alone. This conclusion is completely consistent with previous theoretical work on meta-bisazobenzenes.20,21
However, switching one AB subunit of MTA reduces the sharpness and height of the main ππ* absorption band of (E,E,Z)-MTA compared to (E,E,E)-MTA. This can be explained by the fact that the intrinsic three-fold symmetry is broken upon E → Z switching of one AB branch, which leads to coupling between the branches. The same effect is observed when two AB branches are E → Z isomerized and the effect on the absorption spectrum is less clearly visible. The (E,Z,Z)-MTA and (E,E,Z)-MTA isomers are also affected by steric repulsions of the Z-AB subunits. Cisnetti and co-worker reported a similar photochromic behavior in the case of meta-bisazobenzene.22 The introduced coupling effects are overall small and will thus not exclude a possible separate control of each AB branch. However, the individual quantum yields of switching the three AB branches may depend on the switching order.
It appears thus reasonable to assume that independent Z → E isomerization of each AB branch can in principle be achieved if the individual AB branches can be individually excited. Overall, this motivated us to design MTA-based compounds integrating differently substituted AB subunits possibly acting as truly independent photoswitches.
4. Substituted MTA derivatives
In unsubstituted MTA all three AB branches are identical and electronically practically uncoupled, but due to its three-fold symmetry the AB branches are not individually addressable. To achieve the latter, MTA derivatives are needed in which the three-fold symmetry is broken and the absorption peaks of the individual AB branches are sufficiently spectrally separated. A straightforward strategy to achieve this is the introduction of different substituents at the terminal phenyl groups with different types of electron-donating and electron-withdrawing electronic effects. For instance, F, Cl, Br, Me, CN, NH2, OH, SH, OMe or NMe2 groups in the ortho-, meta- and para-positions of the terminal phenyl rings can easily be introduced in silico (Fig. 1). Following this procedure, a series of mono-substituted MTA, di-substituted MTA and multi-substituted MTA derivatives were designed to achieve a maximum separation of the main absorption peaks of the individual AB branches. At the same time, the individual AB subunits are required to remain electronically decoupled or at least only weakly coupled to be individually addressable and photoswitchable.The simulated absorption spectra of a series of substituted MTA derivatives are presented in Fig. S1–S5 in the ESI.† From the analysis of these absorption spectra, it is found that only a few MTA derivatives like, for instance 4-NH2-MTA, successfully yield a combination of a red-shifted ππ* absorption band for the substituted AB branch and an original ππ* absorption band for the remaining unsubstituted ones. Most absorption spectra of the tested substituted MTA derivatives (Fig. S1–S5†) exhibit the desired weak electronic coupling. Overall, a large number of substituted MTA derivatives were evaluated, however, we chose to present the results for CN and SH substituted MTAs as representative examples of the design principle as proof-of-principle, since a particularly large spectral separation of the individual ππ* absorption bands is achieved while the AB branches remain uncoupled as shown below.
The simulated absorption spectra of unsubstituted MTA, single branch substituted 4′-SH-MTA and 2′,4′,6′-tri-CN-MTA and double branch substituted (2′,4′,6′-tri-CN-4′′-SH)-MTA are presented in Fig. 3. The vertical excitation energies and corresponding intensities of MTA and its derivatives are summarized in Table 1. The absorption spectra of 4′-SH-MTA and 2′,4′,6′-tri-CN-MTA exhibit two main ππ* absorption bands in contrast to a single one of MTA (Fig. 3b). Apart from the original ππ* absorption band of the unsubstituted branches, both of them exhibit a red-shifted ππ* absorption band, which differs in intensity and displacement. The red-shifted ππ* absorption bands at ∼314 nm for 4′-SH-MTA and at ∼335 nm for 2′,4′,6′-tri-CN-MTA compared to the original ππ* absorption band (∼295 nm), allow in principle for selective excitation of these AB branches.
 | ||
| Fig. 3 Comparison of the simulated absorption spectra of unsubstituted MTA (a), single-branch substituted 4′-SH-MTA and 2′,4′,6′-tri-CN-MTA (b) and double branch-substituted 2′,4′,6′-tri-CN-4′-SH-MTA (c). TD-DFT/BHHLYP with the 6-31G* basis set has been used employed for vertical excitation energy calculations in the gas phase. The spectra are convoluted with Lorentzian function with full-width at half-maximum (FWHM) of 10 nm. | ||
| State | MTA | 4′-SH-MTA | 2′,4′,6′-tri-CN-MTA | 2′,4′,6′-tri-CN-4′′-SH-MTA |
|---|---|---|---|---|
| S1 | 2.92 (0.00) | 2.92 (0.00) | 2.63 (0.00) | 2.63 (0.00) |
| S2 | 2.93 (0.00) | 2.93 (0.00) | 2.92 (0.00) | 2.92 (0.00) |
| S3 | 2.93 (0.00) | 2.95 (0.00) | 2.93 (0.00) | 2.95 (0.00) |
| S4 | 4.15 (0.00) | 3.95 (1.47) | 3.52 (0.04) | 3.40 (0.04) |
| S5 | 4.19 (1.41) | 4.11 (0.04) | 3.70 (1.02) | 3.66 (1.02) |
| S6 | 4.19 (1.41) | 4.19 (1.35) | 4.21 (1.30) | 3.95 (1.06) |
| S7 | 4.40 (0.00) | 4.36 (0.12) | 4.29 (0.43) | 4.16 (0.70) |
| S8 | 4.93 (0.00) | 4.85 (0.03) | 4.29 (0.00) | 4.30 (0.00) |
To further extend this concept, 2′,4′,6′-tri-CN and 4′-SH substitution have been combined in the double-branch substituted (2′,4′,6′-tri-CN-4′′-SH)-MTA derivative, which possesses now three different AB branches. Its simulated absorption spectrum (Fig. 3 (c)) shows three distinct ππ* absorption bands which are spectrally well separated at 290, 315 and 335 nm, corresponding to the unsubstituted, the 4′′-SH and the 2′,4′,6′-tri-CN substituted AB branches. Comparison with the simulated absorption spectrum of MTA and 2′,4′,6′-tri-CN-MTA and 4′-SH-MTA reveals that the peaks are not energetically shifted, only the intensity of the unsubstituted AB branch is reduced to half and the corresponding peak of the substituted branch is added. The absorption spectrum of double-substituted (2′,4′,6′-tri-CN-4′′-SH)-MTA is thus the sum of the intensities of three ππ* absorption bands of three uncoupled AB branches.
In analogy to the single-branch substituted derivatives, the three different branches of 2′,4′,6′-tri-CN-4′′-SH-MTA can in principle be excited selectively, and since the coupling between the AB branches is obviously very small, it is suggestive that the AB branches can be photoswitched individually.
Computed detachment and attachment densities plots of the first ten excited states of (2′,4′,6′-tri-CN-4′′-SH)-MTA supports the individuality of the AB branches, because the excited states decisive for photoswitching are located at the individual branches (Fig. 4). The detachment density is that part of the density which is removed upon excitation and re-arranged as attachment density. An analysis of all plots reveals the 1st-3rd excited states to correspond to the typical nπ* states each localized at one AB branch. The 4th–7th excited states are characterized as ππ* states localized at three N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds of the AB branches. Due to the different substitution of the AB branches, we observe also charge transfer states, e.g. S4 in (2′,4′,6′-tri-CN-4′′-SH)-MTA, with little oscillator strengths which start to interfere with the localized ππ* states.
N double bonds of the AB branches. Due to the different substitution of the AB branches, we observe also charge transfer states, e.g. S4 in (2′,4′,6′-tri-CN-4′′-SH)-MTA, with little oscillator strengths which start to interfere with the localized ππ* states.
 | ||
| Fig. 4 Detachment (red) and attachment (blue) densities for the energetically lowest ten excited singlet states S1 to S10 of (2′,4′,6′-tri-CN-4′′-SH)-MTA at the BHHLYP/6-31G* level of theory. | ||
To investigate whether excitation into the individual optically bright ππ* states of the three AB branches can lead to individual photoswitching, relaxed scans of the potential energy surfaces along the individual switching coordinates have been computed. In other words, the excited state geometry of the corresponding ππ* state has been optimized starting at the ground-state equilibrium geometry, and then the E → Z isomerization has been studied along the CNNC dihedral angle coordinate. We started in the S5, S6 or S7 state for the isomerization of the 2′,4′,6′-tri-CN, the 4′′-SH and the unsubstituted AB branches, respectively. As a representative example, the isomerization of the unsubstituted AB branch starting in S7, which is the most complicated case, is described in detail here.
Fig. 5 displays the relaxation pathway of the locally excited unsubstituted AB branch of (E,E,E)-(2′,4′,6′-tri-CN-4′′-SH)-MTA starting in S7 and leading barrierless via E → Z isomerization to the corresponding (E,E,Z) conformer, in which the unsubstituted branch is now photoisomerized. Initially the geometrical structure of S7 state has been freely optimized (Fig. 5(b)), during which the S7 state crosses S6–S2 and decays non-radiatively into the second lowest excited state, which corresponds to the nπ* state of the unsubstituted AB branch. The excited state population does not decay into the other states located on the other branches, since the coupling between them is negligible, as revealed previously by the computed spectra. The structural distortions along this pathway are small while-(2′,4′,6′-tri-CN-4′′-SH)-MTA stays essentially planar. The CNNC dihedral angle of the unsubstituted branch varies only slightly (Fig. 5(a)) along this initial relaxation. Once excited-(2′,4′,6′-tri-CN-4′′-SH)-MTA molecules arrive in the nπ* state of the unsubstituted AB branch, barrierless E → Z isomerization of this branch via CNNC rotation is possible in analogy to the parent azobenzene molecule, as is revealed by the corresponding relaxed potential energy surface scan (Fig. 5c). Hence, the computed potential energy curves suggest that excitation into the S7 (ππ*) state at the unsubstituted AB branch will eventually lead to E → Z isomerization of that particular AB branch. Similar curves are found for excitation of the ππ* states located at the SH-substituted (S6) and the 2′,4′,6′-tri-CN substituted (S5) branches (ESI†). Hence, our calculations suggest that indeed individual photoswitching of the different AB branches is in principle possible, when the AB branches are individually excited.
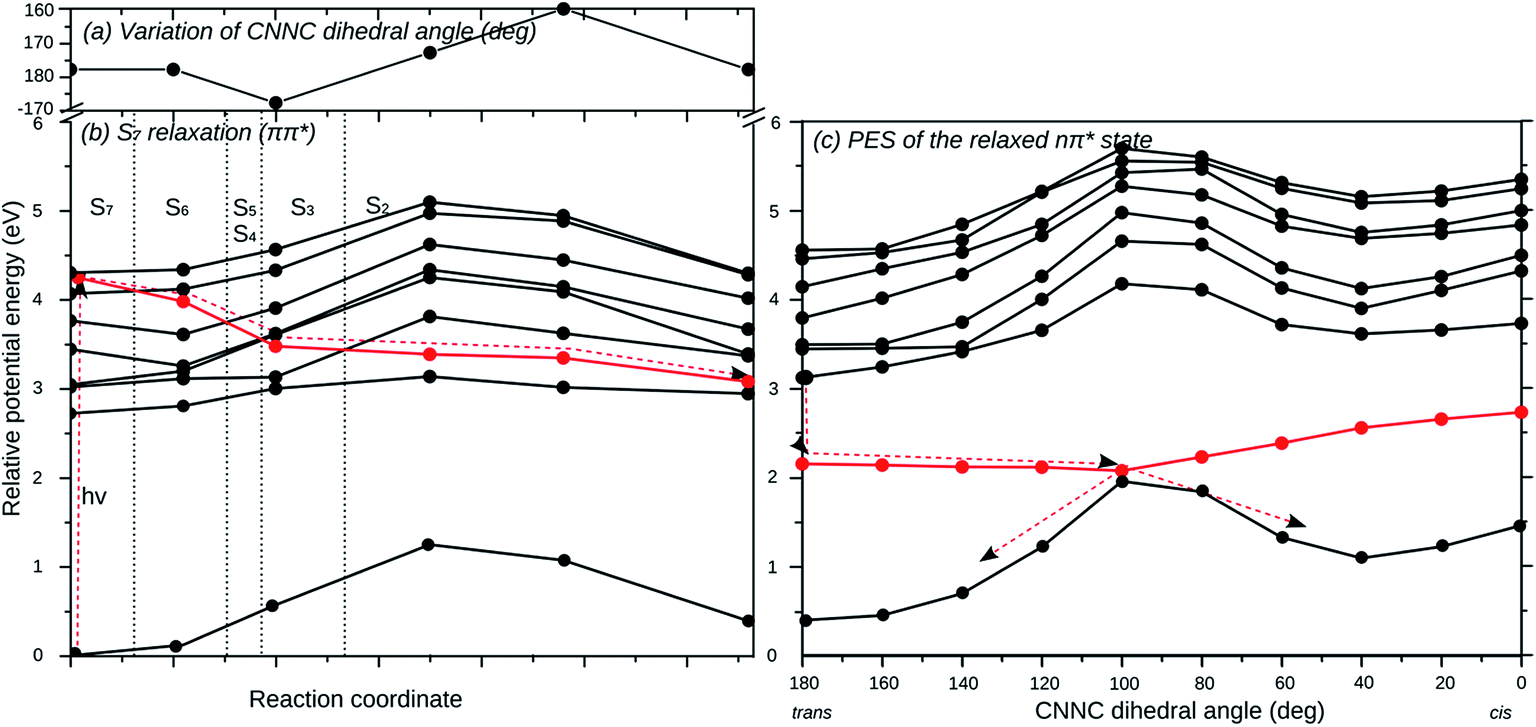 | ||
| Fig. 5 Relaxed scans of the potential energy surfaces along the isomerization pathway of the unsubstituted AB branch of (2′,4′,6′-tri-CN-4′′-SH)-MTA. (a) Variation of CNNC dihedral angle (deg) along the unconstrained relaxation of S7 starting at the ground state equilibrium geometry. (b) Potential energy curves along the unconstrained relaxation of S7, the ππ* excited state of the unsubstituted AB branch, to decay non-radiatively into S2, the nπ* state of the unsubstituted AB branch. (c) Potential energy surface scan of the relaxed nπ* excited state of the unsubstituted AB branch along the CNNC dihedral angle. The red line corresponds to the state, which is optimized, and the vertical dotted lines in (b) separate the regions where the optimized state is S7, S6, etc. down to S2. | ||
A remaining question to discuss is the quantum yield of photoswitching of the individual AB branches. In principle, every crossing encountered during the initial relaxation process corresponds to a potential loss channel, because the excited molecules could switch to another excited state. However, the states crossed are located at the other branches, i.e. spatially separated, and hence a non-radiative decay into these states would correspond to an energy transfer from one branch to another. The efficiency of energy transfer on the other hand depends on the coupling strength between the electronic states located at the different AB branches, and this is negligible, as we have demonstrated above. Therefore, energy transfer can be expected to be slow, at least much slower than the well-known ultra-fast E → Z isomerization of azobenzene, which occurs in about 200 fs. In summary, we expect the quantum yields of switching the individual AB branches in MTAs to be similar to the ones of the corresponding isolated ABs.
5. Summary and outlook
In summary, we have successfully designed a multi-photochromic compound in silico based on a meta-connectivity principle applied to multi-azobenzenes. The smallest trisazobenzene consists of three azobenzene (AB) branches, which share the central phenyl ring. We have shown the low-lying excited electronic states of the three AB branches to be electronically decoupled despite of the large spatial overlap. It is highly astonishing that four phenyl rings are sufficient to build a photoswitch that exhibits the properties of three independent azobenzene molecules.(2,4,6-CN;4-SH)-meta-trisazobenzene served as representative example and proof-of-principle for a triple photoswitch, in which all AB-branches can be, in principle, selectively excited because the individual ππ* bands are energetically well separated. In addition, computation of the isomerization pathways of individually excited AB branches demonstrated selective excitation to lead to selective photoswitching.
We believe the meta-connectivity principle to be generally applicable for the construction of multi-photoswitchable systems, as it leads to maximally uncoupled individual subsystems retaining their individual photochemistry. A general limitation of course are the timescales of photoswitching compared to other non-radiative and radiative decay channels. In particular, excitation energy transfer from one subunit to another can significantly interfere. However, as long as the switching process is fast enough, the other deactivation pathways do not play a relevant role.
While in this work static quantum chemical calculations are sufficient to demonstrate the concept of meta-connectivity and the computational design of a first triple-photoswitch based on azobenzene, in general nuclear quantum dynamics are required to study the efficiency of photoswitching and to determine timescales and relevant loss channels. In the future we plan to synthesize and spectroscopically investigate the suggested meta-trisazobenzene derivatives in analogy to previous works.49–51 This work indeed paves the way for future development of selectively photoswitchable control systems being used in advanced materials and nano-electronic devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the China Scholarship Council for the financial support of Yang's CSC Ph.D fellowship and the HGS MathComp (GSC 220) and acknowledge funding by the DFG (WA 1850/4-2).References
- H. Rau, in Photochromism: Molecular and Systems, ed. H. Durr and H. Bouas-Lauran, Elsevier, Amsterdam, 1990, pp. 165–192 Search PubMed.
- D. Bléger, Z. Yu and S. Hecht, Toward optomechanics: Maximizing the photodeformation of individual molecules, Chem. Commun., 2011, 47, 12260–12266 RSC.
- B. H. M. Dhammika and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- W. Feng, W. Luo and Y. Feng, Photo-responsive carbon nanomaterials functionalized by azobenzene moieties: structures, properties and application, Nanoscale, 2012, 4, 6118–6134 RSC.
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- S. Spörlein, H. Carstens, H. Satzger, C. Renner, R. Behrendt, L. Moroder, P. Tavan, W. Zinth and J. Wachtveitl, Ultrafast spectroscopy reveals sub-nanosecond peptide conformational dynamics and validates molecular dynamics simulation, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7998–8002 CrossRef PubMed.
- J. Bredenbeck, J. Helbing, A. Sieg, T. Schrader, W. Zinth, J. Wachtveitl, C. Renner, R. Behrendt, L. Moroder and P. Hamm, Picosecond conformational transition and equilibrium of a cyclic peptide, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6452–6457 CrossRef CAS PubMed.
- S. Monti, G. Orlandi and P. Palmieri, Features of the photochemically active state surfaces of azobenzene, Chem. Phys., 1982, 71, 87–99 CrossRef CAS.
- T. Fujino, S. Y. Arzhantsev and T. Tahar, Femtosecond Time-Resolved Fluorescence Study of Photoisomerization of trans-Azobenzene, J. Phys. Chem. A, 2001, 105, 8123–8129 CrossRef CAS.
- T. Nägele, R. Hoche, W. Zinth and J. Wachtveitl, Femtosecond photoisomerization of cis-azobenzene, Chem. Phys. Lett., 1997, 252, 489–495 CrossRef.
- E. W. A. Diau, New Trans-to-Cis Photoisomerization Mechanism of Azobenzene on the S1(n,π*) Surface, J. Phys. Chem. A, 2004, 108, 950–956 CrossRef CAS.
- C. R. Crecca and A. E. Roitberg, Theoretical Study of the Isomerization Mechanism of Azobenzene and Disubstituted Azobenzene Derivatives, J. Phys. Chem. A, 2006, 110, 8188–8193 CrossRef CAS PubMed.
- E. Kazuma, M. Han, J. Jung, J. Oh, T. Seki and Y. Kim, Elucidation of Isomerization Pathways of a Single Azobenzene Derivative Using an STM, J. Phys. Chem. Lett., 2015, 6(21), 4239–4243 CrossRef CAS PubMed.
- J. Bahrenburg, C. M. Sievers, J. B. Schönborn, B. Hartke, F. Renth, F. Temps, C. Näther, D. Frank, F. D. Sönnichsen and J. Bahrenburg, Photochemical properties of multi-azobenzene compounds, Photochem. Photobiol. Sci., 2013, 12, 511–518 RSC.
- J. Vapaavuori, A. Goulet-Hanssens, I. T. S. Heikkinen, C. J. Barrett and A. Priimagi, Are Two Azo Groups Better than One? Investigating the Photoresponse of Polymer-Bisazobenzene Complexes, Chem. Mater., 2014, 26(17), 5089 CrossRef CAS.
- F. Zhao, L. Grubert, S. Hecht and D. Bleger, Orthogonal switching in four-state azobenzene mixed-dimers, Chem. Commun., 2017, 53, 3323–3326 RSC.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Multiphotochromic molecular systems, Chem. Soc. Rev., 2015, 44(11), 3719–3759 RSC.
- S. Bellotto, R. Reuter, C. Heinis and H. A. Wegner, Synthesis and Photochemical Properties of Oligo-ortho-azobenzenes, J. Org. Chem., 2011, 76(23), 9826–9834 CrossRef CAS PubMed.
- D. Bléger, J. Dokic, M. V. Peters, L. Grubert, P. Saalfrank and S. Hecht, Electronic Decoupling Approach to Quantitative Photoswitching in Linear Multiazobenzene Architectures, J. Phys. Chem. B, 2011, 115, 9930–9940 CrossRef PubMed.
- G. Floß and P. Saalfrank, The Photoinduced E → Z Isomerization of Bisazobenzenes: A Surface Hopping Molecular Dynamics Study, J. Phys. Chem. A, 2015, 119, 5026–5037 CrossRef PubMed.
- C. Slavov, C. Yang, L. Schweighauser, C. Boumrifak, A. Dreuw, H. A. Wegner and J. Wachtveitl, Connectivity matters -ultrafast isomerization dynamics of bisazobenzene photoswitches, Phys. Chem. Chem. Phys., 2016, 18, 14795–14804 RSC.
- F. Cisnetti, R. Ballardini, A. Credi, M. T. Gandolfi, S. Masiero, F. Negri, S. Pieraccini and G. P. Spada, Photochemical and Electronic Properties of Conjugated Bis(azo) Compounds: An Experimental and Computational Study, Chem.–Eur. J., 2004, 10, 2011–2021 CrossRef CAS PubMed.
- J. Robertus, S. F. Reker, T. C. Pijper, A. Deuzeman, W. R. Browne and B. L. Feringa, Kinetic analysis of the thermal isomerisation pathways in an asymmetric double azobenzene switch, Phys. Chem. Chem. Phys., 2012, 14, 4374–4382 RSC.
- R. Reuter and H. A. Wegner, Synthesis and isomerization studies of cyclotrisazobiphenyl, Chem.–Eur. J., 2011, 17, 2987–2995 CrossRef CAS PubMed.
- R. Reuter and H. A. Wegner, A Chiral Cyclotrisazobiphenyl: Synthesis and Photochemical Properties, Org. Lett., 2011, 13, 5908–5911 CrossRef CAS PubMed.
- C. Slavov, C. Yang, L. Schweighauser, H. A. Wegner, A. Dreuw and J. Wachtveitl, Ultrafast excited-state deactivation dynamics of cyclotriazobenzene-a novel type of UV-B absorber, ChemPhysChem, 2017, 18, 2137 CrossRef CAS PubMed.
- T. G. Gopakumar, T. Davran-Candan, J. Bahrenburg, R. J. Maurer, F. Temps, K. Reuter and R. Berndt, Broken Symmetry of an Adsorbed Molecular Switch Determined by Scanning Tunneling Spectroscopy, Angew. Chem., Int. Ed., 2013, 52, 11007–11010 CrossRef CAS PubMed.
- K. Scheil, T. G. Gopakumar, J. Bahrenburg, F. Temps, R. J. Maurer, K. Reuter and R. Berndt, Switching of an Azobenzene-Tripod Molecule on Ag(111), J. Phys. Chem. Lett., 2016, 7, 2080–2084 CrossRef CAS PubMed.
- E. Titov, G. Granucci, J. P. Götze, M. Persico and P. Saalfrank, Dynamics of Azobenzene Dimer Photoisomerization: Electronic and Steric Effects, J. Phys. Chem. Lett., 2016, 7, 3591–3596 CrossRef CAS PubMed.
- Y. Lim, S. Choi, K. B. Park and C. Cho, Synthesis of Novel 1,3,5-Tris(arylazo)benzenes via Pd-Catalyzed Couplings and Cu(I)-Mediated Direct Oxidations, J. Org. Chem., 2004, 69, 2603–2606 CrossRef CAS PubMed.
- M. Müri, K. C. Schuermann, L. De Cola and M. Mayor, Shape-Switchable Azo-Macrocycles, Eur. J. Org. Chem., 2009, 2562–2575 CrossRef.
- Á. Moneo, G. C. Justino, M. F. N. N. Carvalho, M. C. Oliveira, A. M. M. Antunes, D. Bléger, S. Hecht and J. P. Telo, Electronic Communication in Linear Oligo(azobenzene) Radical Anions, J. Phys. Chem. A, 2013, 117, 14056–14064 CrossRef PubMed.
- O. A. Blackburn, B. J. Coe and M. Helliwell, Tetrapalladium(II) Bisazobenzene and Azoazoxybenzene Complexes: Syntheses, Electronic Structures, and Optical Properties, Organometallics, 2011, 30, 4910–4923 CrossRef CAS.
- A. Fihey, A. Perriera, W. R. Browne and D. Jacquemin, Multiphotochromic molecular systems, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- F. Zhao, L. Grubert, S. Hecht and D. Bléger, Orthogonal switching in four-state azobenzene mixed-dimers, Chem. Commun., 2017, 53, 3323–3326 RSC.
- M. M. Lerch, M. J. Hansen, W. A. Velema, W. Szymanski and B. L. Feringa, Orthogonal photoswitching in a multifunctional molecular system, Nat. Commun., 2016, 7, 12054 CrossRef CAS PubMed.
- F. Zhao, L. Grubert, S. Hecht and D. Bleger, Orthogonal switching in four-state azobenzene mixed-dimers, Chem. Commun., 2017, 53, 3323–3326 RSC.
- Y.-W. Choi, Y.-K. Lim, S. U. Lee, C.-G. Cho, Y. Lee and D. Sohn, Thermal cis–trans isomerization of triazo-benzene, Curr. Appl. Phys., 2007, 7, 513–516 CrossRef.
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. E. Casida Recent Advances in Density Functional Methods, World Scientific, 1995 Search PubMed.
- A. Dreuw and M. Head-Gordon, Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J. Liu and W. Liang, Analytical approach for the excited-state Hessian in time-dependent density functional theory: formalism, implementation, and performance, J. Chem. Phys., 2011, 135, 184111 CrossRef PubMed.
- J. Liu and W. Liang, Analytical Hessian of electronic excited states in time-dependent density functional theory with Tamm-Dancoff approximation, J. Chem. Phys., 2011, 135, 014113 CrossRef PubMed.
- J. Liu and W. Liang, Analytical second derivatives of excited-state energy within the time-dependent density functional theory coupled with a conductor-like polarizable continuum model, J. Chem. Phys., 2013, 138, 024101 CrossRef PubMed.
- Y. Shao, et al., Advances in molecular quantum chemistry contained in the Q-Chem 4 program package, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- Y.-K. Lim, S. Choi, K. B. Park and C.-G. Cho, J. Org. Chem., 2004, 69, 2603–2606 CrossRef CAS PubMed.
- S. Bellotto, R. Reuter, C. Heinis and H. A. Wegner, J. Org. Chem., 2011, 76, 9826–9834 CrossRef CAS PubMed.
- R. Reuter and H. A. Wegner, Beilstein J. Org. Chem., 2012, 8, 877–883, DOI:10.3762/bjoc.8.99.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03379j |
| This journal is © The Royal Society of Chemistry 2018 |