
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile folding of insulin variants bearing a prosthetic C-peptide prepared by α-ketoacid-hydroxylamine (KAHA) ligation†
Gábor N.
Boross
a,
Satomi
Shimura
a,
Melissa
Besenius
b,
Norbert
Tennagels
b,
Kai
Rossen‡
b,
Michael
Wagner
b and
Jeffrey W.
Bode
 *a
*a
aLaboratorium für Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland. E-mail: bode@org.chem.ethz.ch; Web: http://www.bode.ethz.ch/
bSanofi-Aventis Deutschland GmbH Industriepark Hoechst, 65926 Frankfurt am Main, Germany Web: http://www.sanofi.com
First published on 11th September 2018
Abstract
The chemical synthesis of insulin is an enduring challenge due to the hydrophobic peptide chains and construction of the correct intermolecular disulfide pattern. We report a new approach to the chemical synthesis of insulin using a short, traceless, prosthetic C-peptide that facilitates the formation of the correct disulfide pattern during folding and its removal by basic treatment. The linear precursor is assembled by an ester forming α-ketoacid-hydroxylamine (KAHA) ligation that provides access to the linear insulin precursors in good yield from two readily prepared segments. This convergent and flexible route provides access to various human, mouse, and guinea pig insulins containing a single homoserine mutation that shows no detrimental effect on the biological activities.
Introduction
Insulin is a peptide hormone of critical importance for the regulation of human metabolism and malfunction of its production or regulation is the underlying cause of diabetes mellitus. Administration of insulin is one of the pillars for diabetes treatment and several different insulins are administered to address basic medical needs.1 High purity therapeutic insulin is currently produced by a combination of recombinant expression and chemical/enzymatic modifications of the expressed proprotein. While this has largely solved the supply problem, the reliance on biotechnological production limits the development of new insulin variants that could be safer and more convenient.2Early chemical syntheses of insulin by chain combination methods are low yielding.2,3 Stepwise formation of disulfide bonds can bypass the problem of incorrect disulfide pairing but these methods suffer from a long synthetic route and a low overall yield.4–6 To improve synthetic access, Kent has reported a method for the synthesis of insulin Lispro variants, employing oxime-forming ligation for straightforward access to the folding precursors.7 Separately, Kent has successfully utilized a zero length C-peptide (ester bond between the GluA4 and ThrB30 sidechains) for folding of linear insulins prepared by native chemical ligation.8,9 The DiMarchi group has reported elegant methods to access single chain insulins as folding precursors. Oxime-forming ligation combined with a traceless C-peptide surrogate or enzyme-cleavable single-chain precursors resulted in a variety of correctly folded insulins.10,11
Inspired by these pioneering studies, here we report a convergent method to access insulin and its analogues with a traceless, base-labile prosthetic C-peptide that shepherds folding into the correct disulfide pattern (Scheme 1). The modestly sized linear insulin precursor is assembled from two fragments by the α-ketoacid-hydroxylamine (KAHA) ligation.12 The use of the chemoselective coupling of peptide α-ketoacids and (S)-5-oxaproline (Opr) proceeds under acidic conditions that readily solubilize the notoriously hydrophobic insulin chains and forms an ester linked linear insulin that offers improved solubilisation and handling. Tandem folding, linker cleavage, and O → N shift that products ThrB27Hse insulin variants that exhibit identical biological activity to medicinal forms.
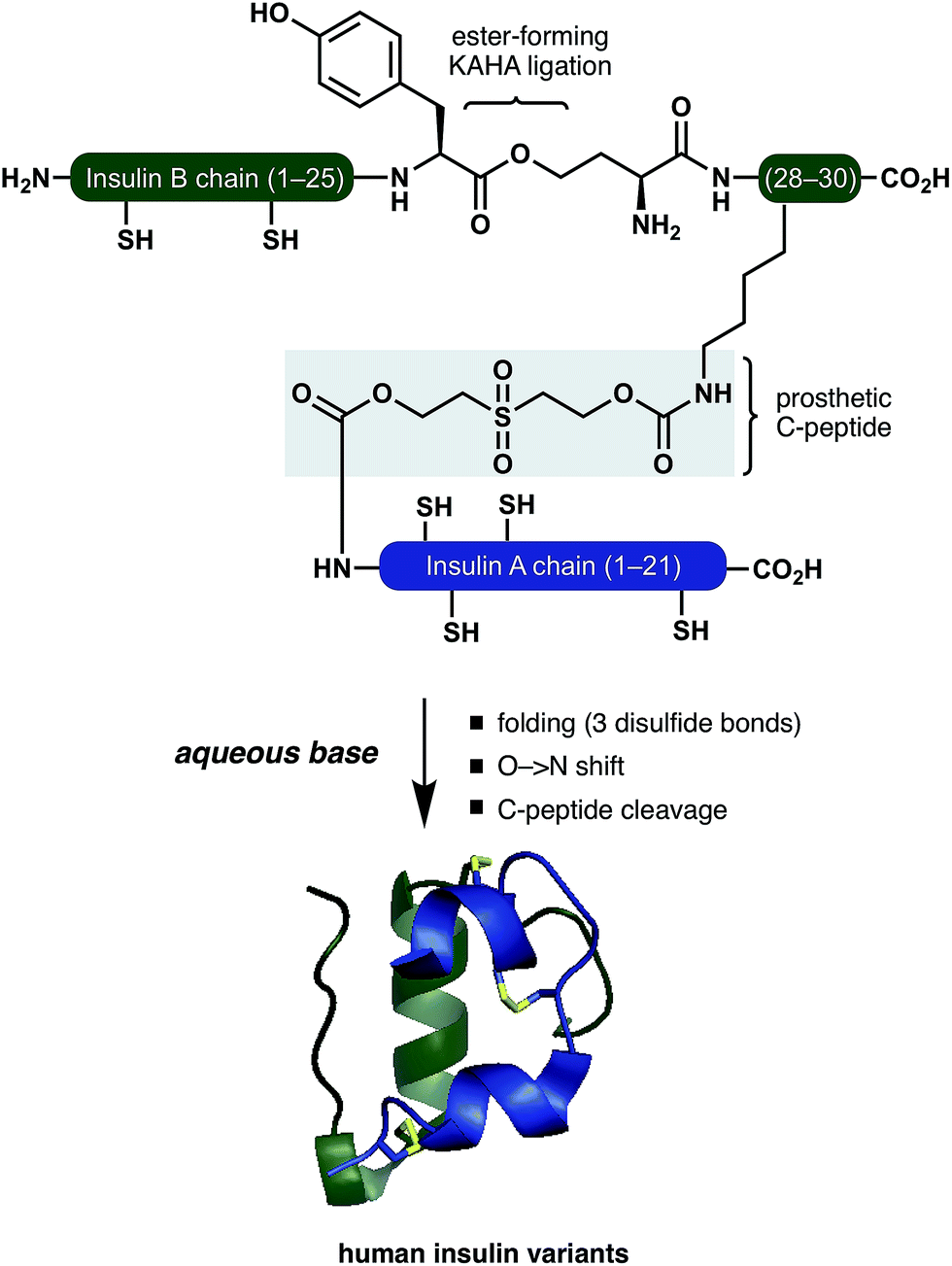 | ||
| Scheme 1 Folding of insulin variants directed by a prosthetic C-peptide. | ||
The basis for our prosthetic C-peptide is the work of Brandenburg et. al., who showed that an intermolecularly crosslinked (Nα GlyA1, Nε LysB29) native insulin induces the formation of the correct disulfide bonds upon reduction and subsequent refolding.13 Obermeier et. al. have designed a cleavable chemical linker that can mimic the function of the C-peptide of proinsulin and efficiently fold the reduced single-chain insulin into its native structure.14 Although a very promising as a strategy for folding synthetic insulin, this linker was employed only on isolated bovine insulin and – to the best of our knowledge – has not been used for the chemical synthesis insulin.
Results and discussion
We devised a convergent strategy for the chemical synthesis of insulin and its analogues based on Brandenburg's findings. The key intermediate in our approach is a linear reduced miniproinsulin in which the C-peptide is replaced by a short chemical linker inspired by the work of Obermeier.In our preliminary studies the necessary linear miniproinsulin could not be prepared directly by Fmoc SPPS (See ESI† page S62). We dissected our target into two shorter peptide segments that could be assembled by KAHA ligation. From the ligation product, folding, ester-to-amide rearrangement, and linker cleavage could all occur under basic conditions to access a variety of insulins.
Synthesis of prosthetic C-peptides
Our approach to the chemical synthesis of various insulins required the preparation of base-cleavable sulfone-linkers bearing branched residues at the B30 site (Scheme 2). Based on Brandenburg's approach to crosslink Nα GlyA1 with Nε LysB29. Therefore we devised a common synthesis for the required building blocks, bearing a free carboxylic acid group on the C terminal of the GlyA21 and an Fmoc protected amino group on the N terminal of LysB29. The α carboxylic acid group of the Lys can be connected to an acid labile protecting group, leading to desB30 insulin variants, or to appropriately protected other amino acid residues, leading to a variety of insulin analogs. | ||
| Scheme 2 (a) Synthesis of building blocks with incorporated cleavable linkers. N-methylmorpholine (NMM) (b) synthesis of peptide segment 6 and incorporation of linker building block 5a by standard Fmoc-SPPS. | ||
Insulin glargine M2
Insulin glargine (Lantus®) – a marketed long acting insulin analog – has two additional Arg residues (ArgB31, ArgB32) that are accounting for a shift in the isoelectric point resulting the precipitation of the peptide at the injection site, which leads to prolonged therapeutic activity. Glargine has a further mutation on the A chain (AsnA21Gly).15 It is known that the absence of the ThrB30 residue has no major influence on the activity.16 In humans and animals, glargine undergoes rapid and significant metabolism, leading to early formation of the major metabolite M1 and M2 (AsnA21Gly, desThrB30), which have in vitro metabolic and mitogenic profiles comparable with human insulin.17In order to access the key synthetic intermediate of M2 insulin (ThrB27Hse) 9 (Scheme 2), we placed the ligation site on the B chain between TyrB26 and ThrB27. We chose this site because this disconnection resulted in two similarly sized peptide segments and our previous studies have shown that replacement of Ser/Thr residue with homoserine (Hse, T§) do not affect the folding or biological activity of synthetic proteins compared to wild type references.18–20
We prepared both peptide segments 6 and 7 by standard automated Fmoc solid phase peptide synthesis (SPPS). The segments were synthesized on ChemMatrix® resin with a loading of 0.2 mmol g−1 to avoid the aggregation of the growing peptide chains on resin. Due to the notoriously poor solubility of the insulin A chain and the single-chain insulin derivatives, we initiated the (S)-5-oxaproline segment (6) with a base-cleavable hexaarginine solubilizing tag (Arg-tag). The Arg-tag was attached to the C-terminus of the A chain via a base labile ester bond21 that can be concomitantly removed with the base-labile prosthetic C-peptide.
In our preliminary studies we prepared the peptide segments with unprotected Cys residues but observed premature formation and scrambling of disulfides during purification and ligation, regardless the presence of various reducing agents. To circumvent this we elected to keep the cysteine residues protected during purification of the segments and KAHA ligation. After evaluating several possibilities, we selected Acm protection for all of the Cys residues and prepared the requisite peptides without deviation from our optimized conditions.
The KAHA ligation of the two segments worked well in aqueous DMSO in the presence of oxalic acid. It was nearly complete after 18 hours without the formation of decomposition products. The ligated depsipeptide was separated by preparative reversed phase (RP) HPLC in 41% isolated yield. We retained the ester bond until the end of the synthesis to benefit from an expected increase in solubility from the additional unprotected primary amine in the linear insulin.22
The six Acm groups of ligated peptide 8 were removed by AgOAc under acidic conditions,23 which were ideal for solubilizing the linear, unfolded insulin 9. Although the starting material was completely converted after one hour, the isolated yield was modest, most likely due to the poor recovery from the preparative RP-HPLC.
With the reduced linear insulin 9 in hand, we proceeded to establish folding conditions using the prosthetic linker to direct the formation of the correct disulfide bond pattern. Unfolded, linear insulin variants are known to be poorly soluble in aqueous buffers and in the complete absence of denaturing agents and buffering salts, are very prone to form aggregates and precipitate.24
Based on folding conditions reported by Kent8 and DiMarchi,11 we decided to perform the folding in two steps. First, a strongly denaturing buffer containing 0.3 M Tris with 6 M guanidine hydrochloride (Gn·HCl) at lower pH (6.6) was used to solubilize the peptide (0.5 mg ml−1) at room temperature. Cysteine hydrochloride was added, as reducing agent to allow the formation of the thermodynamically most stable native disulfide pattern.
Once the peptide was completely dissolved, the buffer was rapidly diluted with deionized water to a final peptide concentration of 0.25 mg ml−1, the pH was set to 8.2 and the reaction was incubated at 4 °C. After 12 hours the solution was incubated for 4 hours at room temperature, in order to induce the O → N acyl shift. The folded peptide 10 was separated by preparative RP-HPLC. The purified folded peptide was treated with 0.1 M NaOH at 0 °C for 10 minutes in order to cleave C-peptide and the Arg-tag. After quenching the reaction with acetic acid the final peptide was isolated by preparative RP-HPLC. The denaturing, folding, O → N acyl shift, base mediated cleavage of the prosthetic C-peptide, Arg-tag removal and two RP-HPLC purification steps gave M2 insulin (ThrB27Hse) (11) in 10% overall yield from the unprotected linear precursor. The purity and identity of the isolated product was confirmed by analytical RP-HPLC and high resolution (HR) MS. The tertiary structure of the peptide was characterized by circular dichroism (CD) and compared with authentic recombinant insulin. The synthetic M2 insulin variant exhibits similar negative bands at 208 nm and 222 nm and a positive band at 193 nm as the reference compound; indicative of the presence of α-helices (Scheme 3).25
 | ||
| Scheme 3 (a) Sequences of synthetically prepared insulin variants. (b) Chemical synthesis of M2 insulin variant (11). (c) Course of KAHA ligation monitored by analytical RP HPLC. (d) Course of the peptide folding and O → N shift. Bottom HPLC trace of purified folding precursor (9). Middle HPLC trace after 12 hours folding and 4 hours basic incubation. Top HPLC trace of purified folded peptide (10). (e) HPLC of purified M2 insulin variant (11). (f) HR MS trace of purified M2 insulin variant (11). (g) CD spectra of 11. | ||
Mouse and guinea pig insulin
The sequence of insulin shows similarity across vertebrates. Mouse insulin differs only in four residues (GlnB3Lys, HisB9Pro, ThrB30Ser, GluA4Asp) from the human insulin and served as an excellent model to test the applicability of our approach to other insulin variants. We targeted the synthesis of a modified mouse insulin (ThrB27Hse, AsnA21Gly) with the same workflow (for details see the ESI†). We were pleased to see that without any change in our methods we could isolate mouse insulin with an overall isolated yield of 14% after denaturing, folding, O → N acyl shift, base mediated cleavage of the prosthetic C-peptide, Arg-tag and two RP-HPLC purification.The recombinant production of guinea pig insulin has long posed a challenged and a successful expression in yeast has only very recently been described.26 Guinea pig insulin is known to lack the ability to form hexamers, rending it of importance for studying the oligomer forming properties of different insulin variants. In guinea pig insulin the key structural motive of the three disulfide bonds is conserved, but the primary amino acid sequence differs from human insulin in 18 residues out of the 51.
We could successfully apply our workflow on the synthesis of a modified guinea pig insulin (ThrB27Hse, AsnA21Gly), demonstrating that our approach readily tolerates changes in the sequence of the targeted peptides and offers a reliable approach to distinct insulin variants.
Human insulin
Having succeeded with the synthesis of M2 and rodent insulins, we have turned our attention to the synthesis of human insulin variant. We choose side chain anchoring of the C-terminal Asn residue27 of the A-chain to our resin bound Arg-tag via a photo labile, ortho-nitrobenzyl based linker first reported by Brown et al.28 This is readily introduced by coupling of the side chain of Fmoc-Asp-OtBu. Upon photolysis the linker yields natural AsnA21 residue.We prepared the (S)-5-oxaproline segment 12 with the photolabile solubilizing tag (Scheme 4) by following our established protocol. The ThrB30 residue was introduced with an appropriately modified linker (5d, Scheme 1). The KAHA ligation preceded smoothly, and after 18 h 97 mg of ligated depsi peptide (13) was isolated by preparative RP-HPLC in 61% yield. The Acm protecting groups were removed as described above and the folding precursor was isolated in 46% yield. The folding, subsequent O → N acyl shift were performed and the protein purified as described for M2 insulin. During folding the peptide behaved similar to the base labile analogues. The correctly folded peptide was observed as the main peak on the analytical RP-HPLC and purified by preparative RP-HPLC. We did not observe premature cleavage of the photolabile Arg tag during any of the purification, handling, or coupling steps.
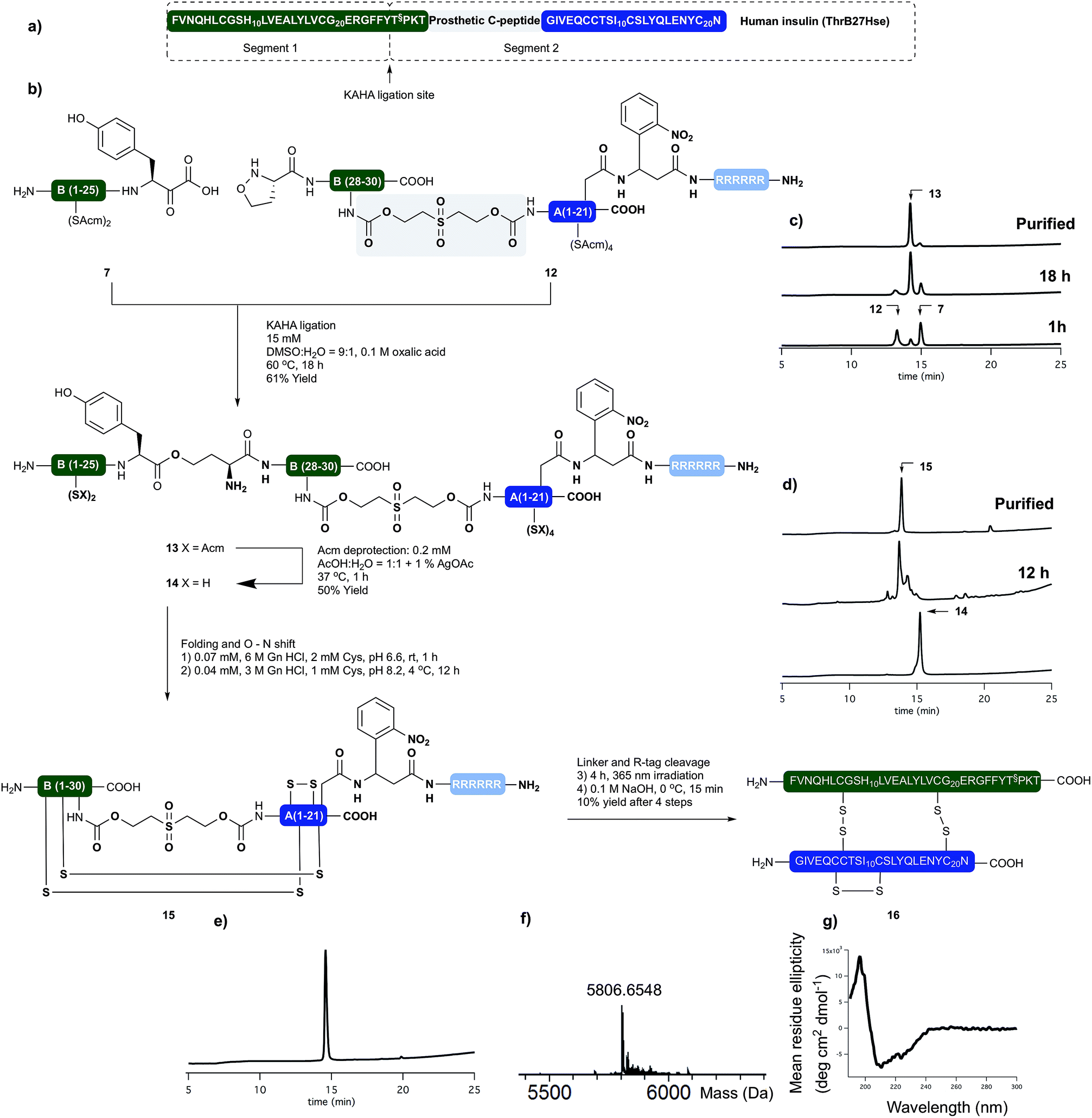 | ||
| Scheme 4 (a) Sequences of synthetically prepared human insulin variant. (b) Chemical synthesis of human insulin variant. (c) Course of KAHA ligation monitored by analytical RP-HPLC. (d) Course of the peptide folding and O → N shift. Bottom RP-HPLC trace of purified folding precursor (14). Middle RP-HPLC trace after 12 hours folding and 4 hours basic incubation. Top RP-HPLC trace of purified folded peptide (15). (e) RP-HPLC of purified human insulin variant (16). (f) HR MS trace of purified human insulin variant (16). (g) CD spectra of 16. | ||
The combined preparative RP-HPLC fractions containing pure folded insulin (15) were irradiated with a hand held UV lamp at 365 nm. The progress of the reaction was followed by MALDI-MS. After 4 hours the sample was lyophilized and treated with 0.1 M NaOH at 0 °C for 10 min in order to cleave the C-peptide. After neutralizing the mixture with acetic acid, the final product (16) was isolated by preparative RP-HPLC. The folding, O → N acyl shift, photorelease of the Arg tag and base mediated cleavage of the C-peptide and two HPLC purification steps gave human insulin (ThrB27Hse) in 10% yield.
The purity and identity of the isolated human insulin was confirmed by analytical RP-HPLC and HRMS. In the CD spectra negative bands at 208 nm and 222 nm and a positive band at 193 nm were detected as indicative for the presence of α-helices and characteristic for authentic recombinant human insulin.
With the folded, human insulin (ThrB27Hse) in hand the disulfide bond pattern could be confirmed by endoproteinase Glu-C disulfide mapping (ESI† S55). The fragments obtained from digesting the synthetic insulin by Glu-C enzyme corresponded to the ones from commercial recombinant insulin.10
Biological activity measurements
The biological activity of synthetic insulin variants was assessed by an In-Cell Western human insulin receptor (IR) autophosphorylation assay.29 Recombinant human insulin (Sanofi) was used as reference and the EC50 values were determined as shown in Fig. 1. The synthetic human insulin variant 16 and the synthetic M2 insulin variant 11 showed similar values to the recombinant human insulin, whereas the synthetic mouse insulin variant 33 and synthetic guinea pig variant 39 showed lower activity on human insulin receptors. These data show that the insulins containing ThrB27Hse are equally active as the native references and could be considered for further development due to their improved synthetic access.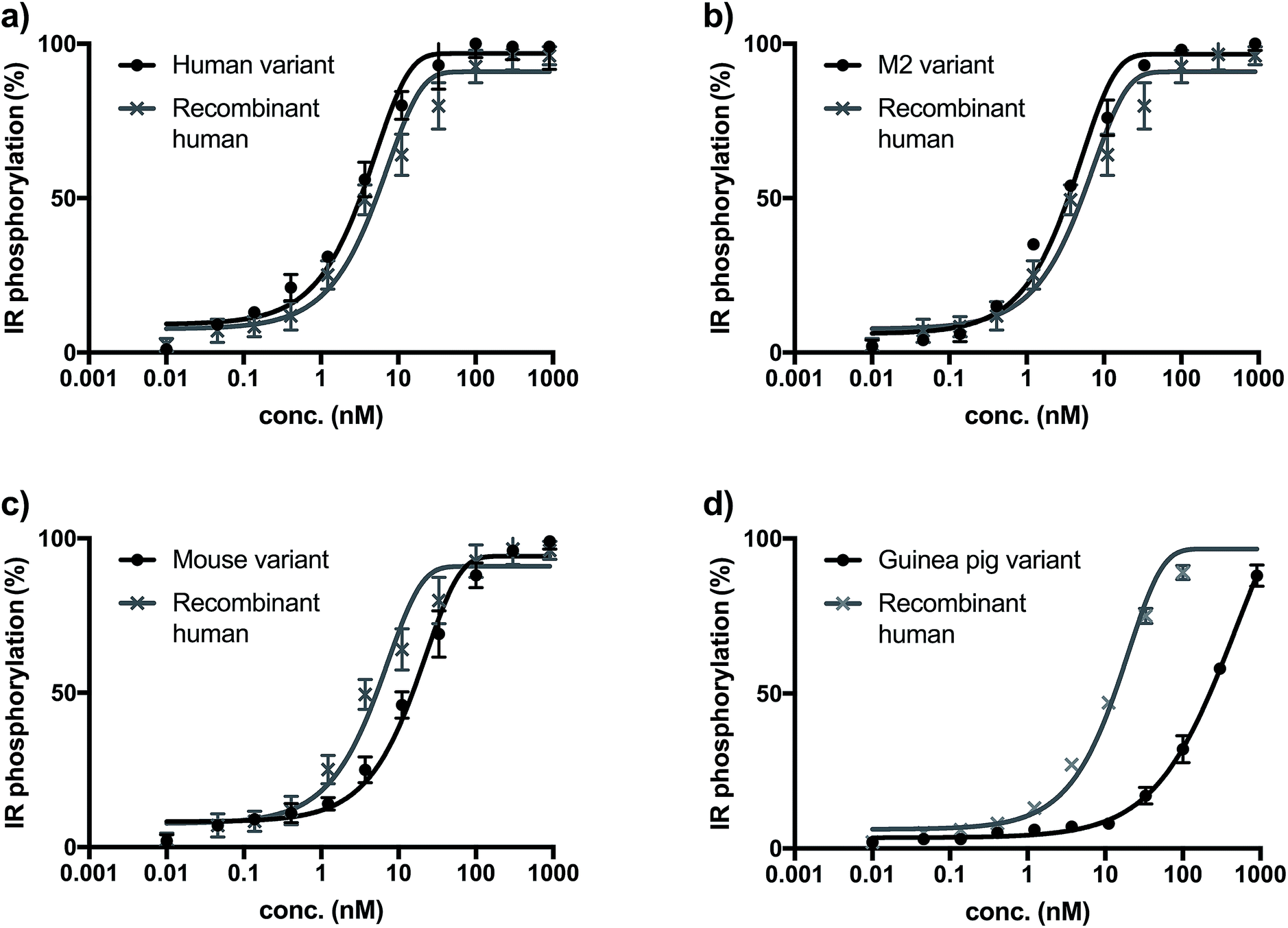 | ||
| Fig. 1 (a) Insulin receptor phosphorylation of recombinant insulin (gray, EC50 = 7.43 nM) synthetic human insulin (16) variant (black, EC50 = 3.01 nM). (b) Insulin receptor phosphorylation of recombinant insulin (gray, EC50 = 6.34 nM) synthetic M2 (11) variant (black, EC50 = 3.41 nM). (c) Insulin receptor phosphorylation of recombinant insulin (gray, EC50 = 7.43 nM) synthetic mouse insulin (33) variant (black, EC50 = 23.67 nM). (d) Insulin receptor phosphorylation of recombinant insulin (gray, EC50 = 12.36 nM) synthetic guinea pig insulin (39) variant (black, EC50 > 100 nM). | ||
Conclusions
We have established a convergent chemical route for the preparation of ThrB27Hse variants of different insulins. Our strategy employs a traceless, short, chemically cleavable linker (prosthetic C-peptide) for facile folding of linear precursors. The linear key intermediate can be accessed in good yield by KAHA ligation and the use of C-terminal solubilizing tag and the late stage Cys-deprotection improved the recovery from RP-HPLC purification steps and prevented premature formation of disulfide bonds. Additionally, the use of a prosthetic C-peptide enables the efficient, intramolecular folding of the linear insulins, allowing folding to occur under conditions that minimize the formation of aggregates or precipitation. The synthetic human and M2 insulin variants showed comparable biological activity to human insulin (insulin receptor autophosphorylation).This convergent approach tolerates numerous mutations on the insulin sequence and can be applied to other members of the insulin protein family. It allows the synthesis of insulin variants, such as guinea pig insulin, that are difficult to produce by recombinant methods. A fully synthetic approach will also facilitate the generation of new insulin analogues with improved therapeutic utility.
Conflicts of interest
“M. B., M. W. and N. T. are employees of Sanofi, the other authors declare no conflicts of interest.”Acknowledgements
We thank Sanofi for financial support. Dr Jacques Lagouardat and Dr Vincent Ferey (Sanofi, France) are gratefully acknowledged for their help in project coordination. We thank the MS service of the Laboratorium für Organische Chemie at ETH Zürich for analyses.Notes and references
- J. P. Mayer, F. Zhang and R. D. DiMarchi, Biopolymers, 2007, 88, 687–713 CrossRef PubMed.
- P. G. Katsoyannis, Diabetes, 1964, 13, 339–348 CrossRef PubMed.
- P. G. Katsoyannis, A. C. Traketellis, C. Zalut, S. Johnson, A. Tometsko, G. Schwartz and J. Ginos, Biochemistry, 1967, 9, 2658–2668 Search PubMed.
- F. Liu, E. Y. Luo, D. B. Flora and J. P. Mayer, Org. Lett., 2013, 15, 960–963 CrossRef PubMed.
- F. Liu, E. Y. Luo, D. B. Flora and A. R. Mezo, Angew. Chem., Int. Ed., 2014, 53, 3983–3987 CrossRef PubMed.
- M. M. Disotuar, M. E. Petersen, J. M. Nogueira, M. S. Kay and D. H. C. Chou, Org. Biomol. Chem., 2018 10.1039/c8ob01212a.
- Y. Sohma and S. B. H. Kent, J. Am. Chem. Soc., 2009, 131, 16313–16318 CrossRef PubMed.
- Y. Sohma, Q. Hua, J. Whittaker, M. A. Weiss and S. B. H. Kent, Angew. Chem., Int. Ed., 2010, 49, 5489–5493 CrossRef PubMed.
- M. Avital-Shmilovivi, K. Mandal, Z. P. Gates, N. B. Philips, M. A. Weiss and S. B. H. Kent, J. Am. Chem. Soc., 2013, 135, 3173–3185 CrossRef PubMed.
- K. Thalluri, B. Kou, V. Gelfanov, J. P. Mayer, F. Liu and R. P. DiMarchi, Org. Lett., 2017, 19, 706–709 CrossRef PubMed.
- A. N. Zaykov, J. P. Mayer, V. M. Gelfanov and R. D. DiMarchi, ACS Chem. Biol., 2014, 9, 683–691 CrossRef PubMed.
- A. O. Ogunkoya, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9693–9697 CrossRef PubMed.
- D. Brandenburg and A. Wollmer, Hoppe-Seyler's Z. Physiol. Chem., 1973, 354, 613–627 CrossRef PubMed.
- R. Obermeier and R. Geiger, Hoppe-Seyler's Z. Physiol. Chem., 1975, 356, 1631–1634 CrossRef PubMed.
- R. H. Rosskamp and G. Park, Diabetes Care, 1999, 22, 109–113 Search PubMed.
- J. G. Menting, Y. Yang, S. J. Chan, N. B. Phillips, B. J. Smith, J. Whittaker, N. P. Wickramasinghe, L. J. Whittaker, V. Pandyarajan, Z. Wan, S. P. Yadav, J. M. Caroll, N. Strokes, C. T. Roberts, F. Ismail-Beigi, W. Milewski, D. F. Steiner, V. S. Chauhan, C. W. Ward, M. A. Weiss and M. C. Lawrence, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3395–3404 CrossRef PubMed.
- G. U. Kuerzel, U. Shukla, H. E. Scholtz, S. G. Pretorius, D. H. Wessels, C. Venter, M. A. Potgieter, A. M. Lang, T. Koose and E. Bernhardt, Curr. Med. Res. Opin., 2003, 19, 34–40 CrossRef PubMed.
- C. He, S. S. Kulkarni, F. Thuaud and J. W. Bode, Angew. Chem., Int. Ed., 2015, 54, 12996–13001 CrossRef PubMed.
- T. G. Wucherpfennig, S. Müller, C. Wolfrum and J. W. Bode, Helv. Chim. Acta, 2016, 99, 897 CrossRef.
- F. Rohrbacher, A. Zwicky and J. W. Bode, Chem. Sci., 2017, 8, 4051–4055 RSC.
- M. A. Hossain, A. Belgi, F. Lin, S. Zhang, F. Shabanpoor, L. Chan, C. Belyea, H. Troung, A. R. Blair, S. Andrikopoulos, G. W. Tregear and J. D. Wade, Bioconjugate Chem., 2009, 20, 1390–1396 CrossRef PubMed.
- T. G. Wucherpfennig, F. Rohrbacher, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12244–12247 CrossRef PubMed.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef PubMed.
- J. Winter, H. Lilie and R. Rudolph, Anal. Biochem., 2002, 310, 148–155 CrossRef PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 10, 119–139 CrossRef PubMed.
- E. Engholm, T. H. Hansen, E. Johansson, H. M. Strauss, T. N. Vinther, K. J. Jensen, F. Hubálek and T. B. Kjeldsen, ChemBioChem, 2015, 16, 954–958 CrossRef PubMed.
- Y. Garcia-Ramos, M. Paradís-Bas, J. Tulla-Puch and F. Albericio, J. Pept. Sci., 2010, 16, 675–678 CrossRef PubMed.
- B. B. Brown, D. S. Wagner and H. M. Geysen, Mol. Diversity, 1995, 1, 4–12 CrossRef PubMed . Upon removal of the base-clavable solubilizing tag from C-terminal asparagine residue (in human insulin AsnA21) aspartimide formation was observed. Further explanation can be found in the ESI† (page S101).
- M. R. Sommerfeld, G. Müller, G. Tschank, G. Seipke, P. Habermann, R. Kurrle and N. Tennagels, PLoS One, 2010, 5, 9540 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data of all new compounds. See DOI: 10.1039/c8sc03738h |
| ‡ Current address: H. Lundbeck A/S Ottiliavej 9, 2500 Valby, Denmark, Web: http://www.lundbeck.com |
| This journal is © The Royal Society of Chemistry 2018 |