
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast detection and structural identification of carbocations on zeolites by dynamic nuclear polarization enhanced solid-state NMR†
Dong
Xiao‡
 abc,
Shutao
Xu‡
d,
Nick J.
Brownbill
c,
Subhradip
Paul
e,
Li-Hua
Chen
f,
Shane
Pawsey
g,
Fabien
Aussenac
h,
Bao-Lian
Su
fi,
Xiuwen
Han
a,
Xinhe
Bao
a,
Zhongmin
Liu
ad and
Frédéric
Blanc
*cj
abc,
Shutao
Xu‡
d,
Nick J.
Brownbill
c,
Subhradip
Paul
e,
Li-Hua
Chen
f,
Shane
Pawsey
g,
Fabien
Aussenac
h,
Bao-Lian
Su
fi,
Xiuwen
Han
a,
Xinhe
Bao
a,
Zhongmin
Liu
ad and
Frédéric
Blanc
*cj
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: frederic.blanc@liverpool.ac.uk
dNational Engineering Laboratory for Methanol to Olefins, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
eDNP MAS NMR Facility, Sir Peter Mansfield Imaging Centre, University of Nottingham, Nottingham NG7 2RD, UK
fState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luoshi Road, 430070, Wuhan, China
gBruker BioSpin Corporation, 15 Fortune Drive, Billerica, Massachusetts 01821, USA
hBruker BioSpin, 34 rue de I'Industrie BP 10002, 67166 Wissembourg Cedex, France
iCMI (Laboratory of Inorganic Materials Chemistry), University of Namur, 61 rue de Bruxelles, B-5000 Namur, Belgium
jStephenson Institute for Renewable Energy, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
First published on 2nd October 2018
Abstract
Acidic zeolites are porous aluminosilicates used in a wide range of industrial processes such as adsorption and catalysis. The formation of carbocation intermediates plays a key role in reactivity, selectivity and deactivation in heterogeneous catalytic processes. However, the observation and determination of carbocations remain a significant challenge in heterogeneous catalysis due to the lack of selective techniques of sufficient sensitivity to detect their low concentrations. Here, we combine 13C isotopic enrichment and efficient dynamic nuclear polarization magic angle spinning nuclear magnetic resonance spectroscopy to detect carbocations in zeolites. We use two dimensional 13C–13C through-bond correlations to establish their structures and 29Si–13C through-space experiments to quantitatively probe the interaction between multiple surface sites of the zeolites and the confined hydrocarbon pool species. We show that a range of various membered ring carbocations are intermediates in the methanol to hydrocarbons reaction catalysed by different microstructural β-zeolites and highlight that different reaction routes for the formation of both targeted hydrocarbon products and coke exist. These species have strong van der Waals interaction with the zeolite framework demonstrating that their accumulation in the channels of the zeolites leads to deactivation. These results enable understanding of deactivation pathways and open up opportunities for the design of catalysts with improved performances.
Introduction
Carbocations are important intermediates in many homogeneous1,2 and heterogeneous reactions,3–6 especially those catalysed by solid acids (e.g. acidic zeolites),7–9 and are formed from the corresponding hydrocarbons through protonation by the acidic protons of the Brønsted acid sites. They take part in a range of industrial processes such as cracking, isomerization, alkylation, etc., which account for the conversion of hydrocarbons to a range of products.8 For example, cyclic carbocations are proposed as important intermediates involved in the hydrocarbon pool mechanism for the conversion of methanol to hydrocarbons (MTH).9–17 Despite the significant roles of carbocations in heterogeneous reactions, their identifications in solid catalysts are not straightforward as they are reactive, transient, difficult to capture and exist in generally low concentrations,3,15,18–20 and therefore their spectroscopic characterization is very challenging.15,19–21Solid-state NMR is useful in detecting reactive carbocations on solid catalysts as shown in some limited cases on zeolites where their capture is achieved by quenching the reaction with liquid N2 (ref. 15 and 22) or stabilizing the intermediates with a base (e.g. ammonia).20 However, further development in extending the use of solid-state NMR to the study of carbocations is currently hindered by both the challenge associated with capturing enough highly reactive carbocations formed on solids (vide supra) and the intrinsically low sensitivity of NMR, especially when low natural abundance nuclei (e.g. 1.1% for 13C) are targeted. Although 13C isotopically enriched reagents are generally used to overcome this inherently poor sensitivity,15,19,22 the small amount of carbocations that can be captured in successful cases (typically 0.01 mmol g−1 in the MTH activated β-zeolite19) usually only permits the acquisition of one dimensional (1D) NMR signals, limiting the application of more informative multidimensional NMR experiments to obtain both the structures of these carbocations and their interaction with the solid catalysts. The structures of adsorbed carbocations are typically derived from such 1D 13C NMR spectra combined with gas chromatography-mass spectrometry (GC-MS) and density functional theory (DFT) calculations15,16 and therefore prior assumption of the existing structures is required.
We recently identified the carbocations formed in 13C enriched MTH activated ZSM-5 and investigated their host–guest interaction by obtaining limited structural constraints.23 However, the experimental times needed to acquire the multidimensional and multinuclear NMR data were prohibitively long (>5 days), even in this favourable case where the carbocation concentration is relatively high (>0.02 mmol g−1). This significantly hinders the systematic use of these powerful approaches on a wider range of solid acids with a lower amount of carbocation intermediates and addressing this challenge necessitates further dramatic boost in NMR sensitivity beyond 13C labelling.21
An emerging method with potential to delivering this increase in NMR sensitivity is dynamic nuclear polarization (DNP) which can enhance the NMR signals by multiple orders of magnitude by transferring the large polarization of electrons to nearby nuclei via microwave (μw) induced electron–nuclear transitions, thereby leading to a very significant reduction in experimental time.24–30 Insoluble samples for typical DNP experiments are impregnated with a solution of stable radicals as the source of electrons with the solvent providing the matrix for 1H polarization transfer. Although initial work focused on water-soluble radicals29,30 which are chemically incompatible with the investigated carbocations, water free radicals and matrices are now known31,32 and have enabled the structural characterization of a broad range of materials with DNP.30 In particular in heterogeneous catalysis, this approach is starting to provide detailed access to catalytic sites on the surface or in the pores of selected catalysts,28,33–40 including surface-enhanced NMR on mesoporous silica,28 organometallics on silica,33,34 SnVI-active sites in the Sn-β zeolite35–37 and Brønsted acid sites of aluminosilicates.38 However, DNP investigation on reactive carbocation intermediates confined in microporous zeolites is yet to be demonstrated.
Here, we explore the use of DNP NMR to detect carbocations confined in porous zeolite catalysts during the MTH reaction.16,19 We show that combining 13C isotope enrichment and DNP enables the detection of low levels of carbocations (0.002–0.01 mmol g−1) in two types of β-zeolites within minutes. The considerable sensitivity increase obtained allows carbon connectivities to be obtained with 13C–13C through-bond experiments41 (see Fig. S1 in the ESI†) yielding the molecular structures of a series of carbocations. The spatial proximities between the surface sites of the zeolites and the confined carbon species are quantitatively probed via13C–29Si through-space NMR experiments.42 The identification of these carbocations reveals possible reaction routes for the formation of olefins and coke species in the MTH reaction while the quantification of host–guest interaction indicates dominant interaction contributing to the adsorption of hydrocarbon pool species in zeolites. In addition, while DNP application has been shown to be limited for microporous zeolites,35,37 here we suggest a potential way to optimize DNP efficiency on microporous zeolites, that is introducing hierarchical pores with different sizes ranging from micro (<2 nm), meso (2–50 nm) to macro pores (>50 nm).43
Results and discussion
DNP efficiencies on zeolites
Two different β-zeolites with different microstructures (microporous β-zeolite (M-β) and micro–meso–macroporous β-zeolite (MMM-β)44) were used in this study. The DNP efficiencies on these zeolites were initially explored on the pristine zeolites with organic templates. While moderate enhancements were only observed for M-β (εC CP = 14, εSi CP = 14), much larger enhancements of 54 on 13C (cross-polarization) CP and 72 on 29Si CP signals were obtained on MMM-β (Fig. 1, S2 and S3†), clearly showing the positive effects of introducing hierarchical porosities into zeolites. A more representative parameter to evaluate the DNP efficiency as compared with standard NMR at room temperature is the overall DNP gain Σ† (see calculation details in the ESI†). This takes into account the increase of sensitivity from thermal Boltzmann distribution going from 298 K to 110 K (temperature at which the 9.4 T DNP with μw on data were recorded), signal attenuation due to the paramagnetic relaxation effects (bleaching) from the exogenous stable biradicals and cross-effect induced depolarization under magic angle spinning (MAS) conditions.45–53Σ†C CP values of 25 and 53 for M-β and MMM-β and Σ†Si CP values of 34 and 97 for M-β and MMM-β, respectively, were obtained at 9.4 T (Tables S1 and S2 and Fig. 1, S2 and S3†). These large DNP gains Σ† are clearly reflected in Fig. S2 and S3† by the much larger signal-to-noise ratios obtained from the μw on DNP 29Si spectra (recorded in seconds) vs. the room temperature 29Si MAS NMR spectra (acquired in tens of minutes).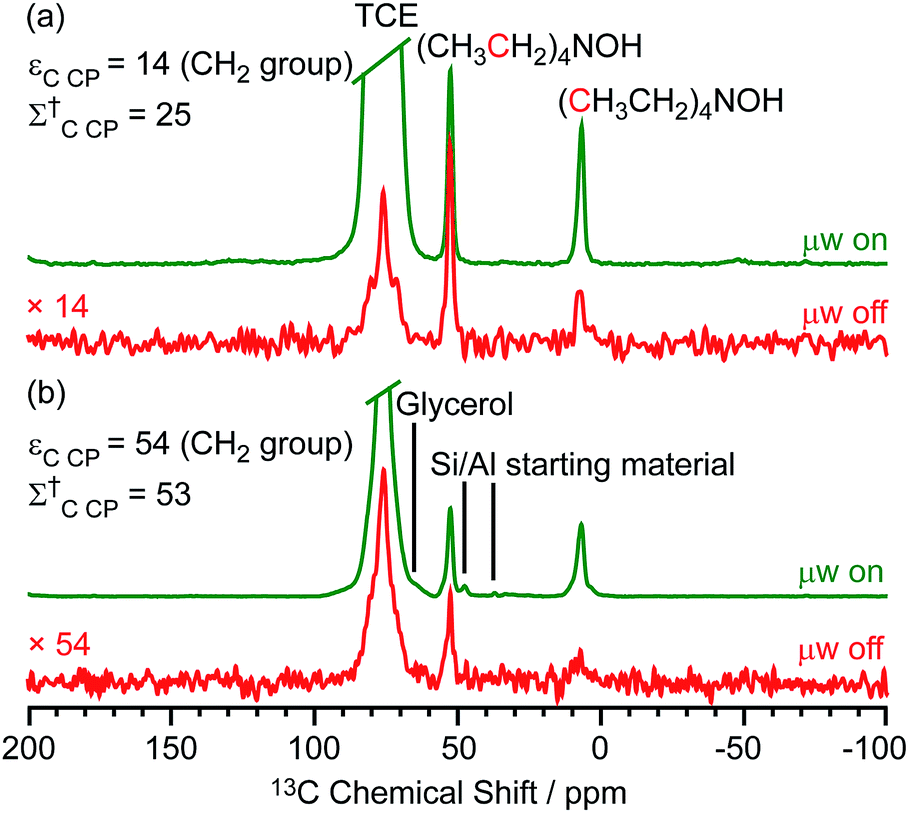 | ||
| Fig. 1 13C CP MAS DNP spectra of templated (a) M-β and (b) MMM-β with (μw on) (green) and without (μw off) microwave irradiation (red) at 9.4 T. TCE stands for 1,1,2,2-tetrachloroethane (solvent of impregnation). | ||
The effects of paramagnetic centres on the NMR signals can be evaluated by the contribution factor θ and the increase of the NMR signal full width at half maximum (FWHM). The contribution factor θ corrects for the loss of signals by paramagnetic bleaching such that signals from nuclei close to the paramagnetic centres are removed beyond detection limits and by depolarization induced by the cross-effect under MAS conditions.48–52 A high contribution factor of 0.7 was obtained on the 29Si NMR signals for M-β (see Table S1†) while θ decreased to 0.5 for MMM-β (Table S2†), suggesting less overall signal loss in M-β than in MMM-β. The paramagnetic effect will also cause additional line-broadening of the NMR signals if the radicals are in proximity to the observed nuclei due to the faster nuclear transverse relaxations ( ) induced by the radicals.45,54,55 Fig. S4 and S5† compare the 13C CP MAS NMR spectra of both M-β and MMM-β zeolites, respectively, in the presence and absence of radicals and no obvious change in FWHM is observed (as monitored by the CH2 resonance of the (CH3CH2)4NOH template). However, significant line broadening is observed when the zeolite is frozen in TCE at low temperature with the FWHM of the CH2 increasing from 100 Hz at room temperature to 210 Hz at 110 K (for M-β) and from 70 to 210 Hz (for MMM-β). This large line-broadening is attributed to the molecules being trapped in a variety of conformations by the low temperature and frozen solvent, leading to large inhomogeneous broadening (as generally observed in proteins).47,54,56 In this work, this broadening does not prevent the 13C resonances of the organic templates from being fully resolved.
) induced by the radicals.45,54,55 Fig. S4 and S5† compare the 13C CP MAS NMR spectra of both M-β and MMM-β zeolites, respectively, in the presence and absence of radicals and no obvious change in FWHM is observed (as monitored by the CH2 resonance of the (CH3CH2)4NOH template). However, significant line broadening is observed when the zeolite is frozen in TCE at low temperature with the FWHM of the CH2 increasing from 100 Hz at room temperature to 210 Hz at 110 K (for M-β) and from 70 to 210 Hz (for MMM-β). This large line-broadening is attributed to the molecules being trapped in a variety of conformations by the low temperature and frozen solvent, leading to large inhomogeneous broadening (as generally observed in proteins).47,54,56 In this work, this broadening does not prevent the 13C resonances of the organic templates from being fully resolved.
The larger DNP enhancements in MMM-β vs. M-β can be attributed to the facilitated diffusion of both the large TEKPol radical (dTEKPol ≈ 2 nm, with d referring to the length of molecule in a DFT optimized structure34) and 1,1,2,2-tetrachloroethane (TCE) solvent which is improved by the existence of uniform mesopores and macropores in MMM-β (mesopore and macropore sizes of 2.5–4.0 and 100–300 nm,44 respectively). Since the spin polarization transfer relies on the 1H solvent spin diffusion, more efficient polarization transfer in MMM-β is expected, which translates to larger enhancements. Additionally, a larger amount of radical solution is needed to wet the MMM-β zeolite which leads to higher electron spin concentration as confirmed by electron paramagnetic resonance (EPR) spin counting experiments (Table S3†).
Fast detection of carbocations with DNP
The activated M-β was prepared by reacting M-β with 13CH3OH (see ESI† for experimental details). Both 13CH3OH and 13C2H4 were initially used to activate MMM-β; however the 13C2H4 activated MMM-β shows much stronger carbocation signals (Fig. S6†) and was therefore used in this work (unless otherwise specified). The chemical compatibility between the reactive cations, TCE and TEKPol solutions was investigated on these activated zeolites prior to DNP experiments. The M-β and MMM-β activated zeolites were impregnated with TCE and the 13C CP MAS spectra of both zeolites before and after impregnation with TCE are compared in Fig. S7.† The spectra show that, for both activated zeolites, the typical signals of carbocations (from 150 to 250 ppm), which structures identified in this work are shown in Fig. 2, and of the aromatics (120 to 150 ppm) remain unchanged after impregnation with TCE, indicating that the carbocations are well stabilised by confinement in pores. Note also that EPR spin counting experiments on the activated zeolites impregnated with the TCE/TEKPol biradical solution quantify the electron spin concentrations added (Table S3†) and illustrate the chemical compatibility of TCE/TEKPol with the carbocations and other carbon species formed in the activated zeolites. It is likely that the carbocations are mainly confined within the micropores of both β-zeolites (pore size < 1 nm), hence excluding the possibility of these cations reacting with bulky TEKPol (whose size is larger than the pore size of the micropores of the β-zeolite). It was demonstrated previously that immobilizing the reactive surface species inside a mesoporous support like MCM-41 (pore size of 2.5–3.0 nm) separates them from TEKPol and eliminates possible reactions between them while the polarization is still relayed by 1H spin diffusion of the solvent.34 This phenomenon is also responsible for the transfer of DNP polarization in the micropores of the Sn-β zeolite.35–37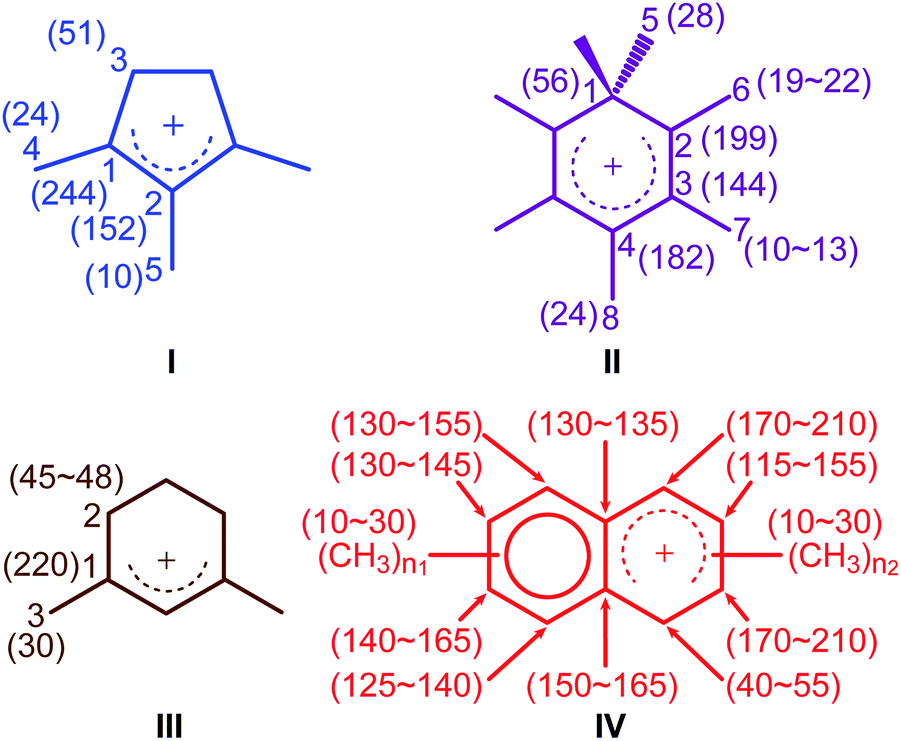 | ||
| Fig. 2 Carbocations identified in activated M-β and MMM-β coded with the same colors as their corresponding assignments and correlations in Fig. 3 and 4. Values in the parenthesis are 13C chemical shifts. n1 and n2 are the number of methyl groups with 3 ≤ (n1 + n2) ≤ 7. | ||
The DNP-enhanced 13C CP MAS NMR spectra of the activated zeolites are shown in green in Fig. 3 and reveal 13C signal enhancements εC CP of 10 and 40 at 9.4 T for M-β and MMM-β, respectively. Under microwave irradiation at 9.4 T, the carbocation signals (150 to 250 ppm) can be clearly observed within minutes (experimental times of ≈7 minutes for M-β and ≈2 minutes for MMM-β) while, importantly, no carbocation signals emerge without microwave irradiation (red spectra). Other signals at around 0 to 50 ppm can be assigned to alkanes or alkyl groups from both aromatics and carbocations, while the two additional peaks at 50 and 60 ppm in activated M-β (Fig. 3a) correspond to methanol and dimethyl ether, respectively.19 The strong peak arising from the TCE solvent is located at about 75 ppm and does not interfere with the NMR signals of the adsorbed species (black spectra). The 13C signal enhancements above translate to overall DNP gains Σ†C CP of 30 and 161 for activated M-β (Table S4†) and MMM-β (Table S5†) zeolites, respectively. These gains correspond to a very significant reduction of experimental time compared to the standard experiments at room temperature, showing substantial DNP efficiency. Note the much better DNP efficiency on activated MMM-β than on activated M-β which validates the use of hierarchical pores. DNP experiments at 14.1 T were also recorded on both activated zeolites. While the 13C signal enhancements and DNP gains at 14.1 T are smaller than those at 9.4 T (Fig. 3), as expected from the inverse field-dependence of the DNP enhancement with cross-effect DNP,29,30 the carbocations can still be observed but no obvious increase in resolution is observed presumably due to strong inhomogeneous broadening and increase in chemical shift dispersion.
 | ||
| Fig. 3 13C CP MAS DNP spectra at 9.4 T, 110 K and 14.1 T, 125 K and room temperature experiments at 9.4 T of (a) activated M-β and (b) activated MMM-β. The assignments of the different carbocations (except methylnaphthalenium ions IV for clarity) are given with the same colors as their structures in Fig. 2. The experimental times for the spectra of activated M-β are ≈7 minutes at 9.4 T DNP, ≈9 minutes at 14.1 T DNP and ≈52 minutes at room temperature at 9.4 T without DNP while for activated MMM-β they are ≈2 minutes at 9.4 T DNP, ≈6 minutes at 14.1 T DNP and ≈1036 minutes at room temperature at 9.4 T without DNP. Σ† refers to the overall DNP gain and is calculated by comparing the DNP data to room temperature 9.4 T NMR spectra (see ESI†). All spectra were recorded at a MAS rate of 12.5 kHz with asterisks (*) denoting spinning sidebands. | ||
Large contribution factors (θ values ranging between 0.7 and 0.8) were obtained for the 13C signals of activated M-β and MMM-β (Tables S4 and S5†). These results indicate less signal loss by paramagnetic bleaching and depolarization in the two activated zeolites. The FWHM of the CH3OH signal in activated M-β was measured and used to monitor the changes of the 13C lineshapes in the presence of TEKPol and TCE and at low temperatures. Fig. S8† shows that adding TEKPol only increases the linewidth slightly by about 8% (from 455 to 490 Hz). However, a significantly larger line broadening is observed when activated M-β is frozen at low temperatures with the FWHM of CH3OH increasing by 25% (from 365 Hz at room temperature to 455 Hz at 110 K), resulting in a slight loss of resolution (e.g. the double peaks at 145 ppm and at 21 ppm are not resolved anymore, Fig. S8†). A similar observation is made in activated MMM-β (see Fig. S9†) with a clear loss of resolution of the signals arising from the alkyl groups. The results indicate inhomogeneous broadening due to molecules being trapped in a variety of conformations as the main contribution to the line-broadening, as on the zeolites with templates.47,54,56
Structure identification of the carbocations
To identify the molecular structures of the confined carbon species, a 2D 13C–13C refocused INADEQUATE (Incredible Natural Abundance DoublE QUAntum Transfer Experiment),41 based on scalar J couplings and providing 13C–13C through-bond correlations, was performed. Spectra are shown in Fig. 4: three single ring carbocations can be identified which are trimethylcyclopentenyl cation I, heptamethylbenzenium cation II and dimethylcyclohexenyl cation III, confirming the previously postulated structures.16,19,57,58 More explicitly, the structure of I is obtained through the following correlations: C1(I) (244 ppm)–C2(I) (152 ppm), C1(I) (244 ppm)–C3(I) (51 ppm), C1(I) (244 ppm)–C4(I) (24 ppm) and C2(I) (152 ppm)–C5(I) (10 ppm) (see Fig. S10 and S11† for the derivation of II and III from these data). Note that cation I was previously identified as an intermediate over ZSM-5 (ref. 16 and 23) and SAPO-34 (ref. 58), but has only been postulated over β zeolites,19 with the data presented here therefore confirming its presence. These five- and six-membered ring cations suggest that both the paring and side-chain catalytic cycles may exist in the β zeolite for the conversion of methanol to hydrocarbons, and the experimentally structural identification of these cations here provides more directly spectroscopic support for the previously proposed mechanisms.15,16,19,59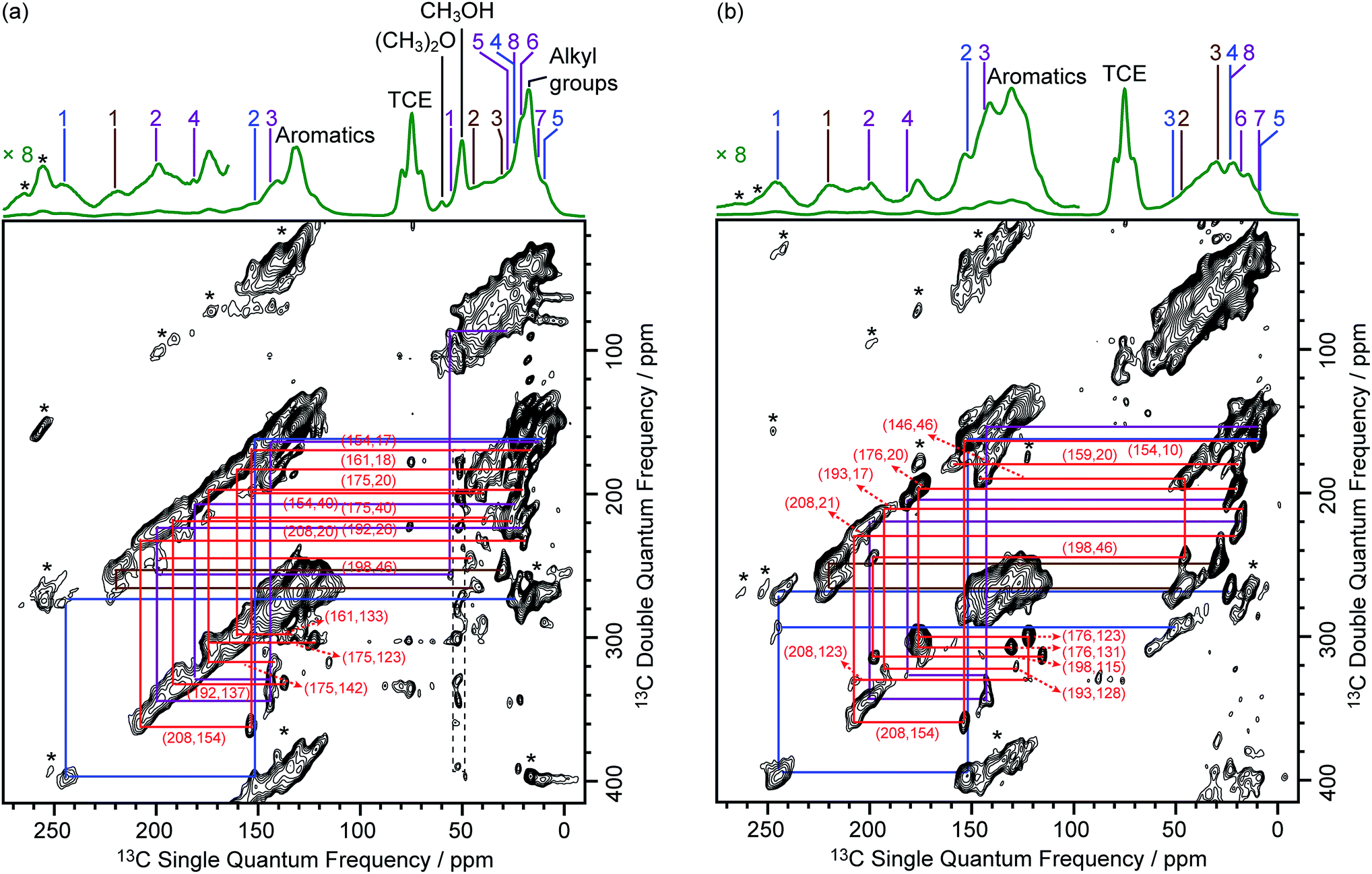 | ||
| Fig. 4 DNP enhanced 2D 13C–13C refocused INADEQUATE spectra of (a) activated M-β and (b) activated MMM-β. Data were recorded at B0 = 9.4 T and a MAS frequency of vr = 12.5 kHz. Experimental times for (a) and (b) are ≈20 and 14 hours, respectively. The correlations and spectral assignments are coded with the same colors as their corresponding carbocations in Fig. 2. Signals in the black dashed box in (a) correspond to t1 noise. Correlations corresponding to naphthalenium ions (IV) are shown with the 13C chemical shifts in the single quantum dimension of the two correlated carbon atoms given in parenthesis. Enlarged figures are shown in Fig. S10 and S11.† Asterisks (*) denote spinning sidebands. | ||
Neutral methylnaphthalenes have been identified by GC-MS as part of the hydrocarbon pool species in the β zeolite;19,59,60 however observation of their active cationic counterparts (methylnaphthalenium ions) and determination of those cations' structures have only been elusive so far even when 13C solid-state NMR was previously deployed.15,16,19 The extra sensitivity obtained with DNP permits the detection of additional 13C correlations (shown in red) involving lower field signals at 208 and 176 ppm to be also resolved (Fig. 4, S10 and S11†). These characteristic peaks have been observed in previous liquid-state 13C NMR studies of methylnaphthalenium ions bearing up to four methyl substituents and are obtained by protonation of methylnaphthalenes by magic acids.61,62
The DNP enhanced 2D 13C–13C NMR correlation experiments provide direct support for identifying the structure of these methylnaphthalenium ions. Characteristic correlations in the single quantum (horizontal) dimension at, for example, 208–154 ppm and 176–131 ppm are assigned to ring carbons while those at 208–21 and 176–20 ppm correspond to bonds between ring carbons and methyl substituents, and enable methylnaphthalenium ions with structure IV to be proposed (Fig. 2). The range of connectivities shown in Fig. 4 demonstrates that more than one methylnaphthalenium ion is present and highlights the actual complexity of these species in activated zeolites (Fig. 4).
These results confirmed previous computational63 and UV-vis spectroscopy64 studies that postulated the presence of methylnaphthalenium ions and methylnaphthalenes. This is also in agreement with previous GC-MS data which suggest that methylnaphthalenes with 3 to 7 methyl groups exist in the β zeolite.59,60 These species can act as both active hydrocarbon pool species to convert methanol into targeted hydrocarbons,60,63–65 and as coke precursors leading to zeolite deactivation.63,64
Correlations arising from neutral carbon species such as aromatics and alkanes19 are also shown in Fig. S12 and S13† from which structures such as methylnaphthalenes,60,66 hexamethylmethylenecyclohexadiene,59,67 hexamethylbenzene,66,68etc. could be possibly derived.
It is noteworthy that fairly similar cyclic hydrocarbon pool species were identified in both 13CH3OH activated M-β and 13C2H4 activated MMM-β zeolites (Fig. 4). A reasonable route for the formation of the cyclic hydrocarbon species can be proposed here and starts with the initial C–C bond formation from C1 reactants such as CH3OH and its derivatives to produce ethylene first69 which can then produce cyclic hydrocarbon species via polymerization and cyclization (Fig. S14†).
In addition to the scalar coupling based INADEQUATE discussed above, a 2D 13C–13C Proton Driven Spin Diffusion (PDSD) Dipolar Assisted Rotational Resonance (DARR) MAS correlation experiment on activated MMM-β (Fig. S15†) was also obtained. In this experiment, cross peaks arise from spatial proximities of the species or chemical exchange.70–72 Using a short mixing time of 30 ms, intramolecular correlations are observed,47 and indeed, correlations from the directly bonded 13C nuclei from the same carbocations are identified in Fig. S15,† which is consistent with the INADEQUATE results. A very small number of correlations from non-bonded 13C nuclei can also be observed in the PDSD DARR spectrum (for example, C1(I) (244 ppm)–C5(I) (10 ppm)), reinforcing the INADEQUATE spectral interpretation and the identified carbocations (Fig. 2).
Investigation of the reaction process
Based on the structural determination of the carbon species and the largely increased sensitivity provided by DNP, we further investigated the reaction process by monitoring ex situ the evolution of the DNP enhanced 13C NMR spectra of activated M-β zeolites with variable 13CH3OH activation times (Fig. 5). The spectra show that even after a very short activation of only one minute, signals from aromatics and carbocations (e.g. resonances at 244 and 152 for I) are detected. The results demonstrate that the hydrocarbon pool species can be formed at a very early stage of the induction period of the MTH reaction. As activation times increased, the signals of aromatics and carbocations increase in intensity pointing out to the accumulation of these species. The spectrum of M-β activated for 60 minutes is fairly similar to the one after 20 minutes, suggesting that the components of the hydrocarbon pool species are steady over these reaction times. | ||
| Fig. 5 μw on 13C CP MAS DNP spectra of M-β activated for (a) one minute, (b) 20 minutes and (c) 60 minutes. Spectra were recorded at 9.4 T and at a MAS rate of 12.5 kHz. Asterisks (*) denote spinning sidebands. Additional spectra at different MAS rates of M-β activated for 20 minutes are shown in Fig. S16.† | ||
Investigation of host–guest interaction using 29Si{13C} REDOR (Rotational Echo DOuble Resonance)42
The DNP enhanced 29Si CP MAS spectra of activated M-β and activated MMM-β are shown in Fig. 6 and both show multiple resonances at −96, −103, −107, −111 and −116 ppm which are characteristic of the (SiO)2Si(OH)2 (Q2), (SiO)3SiOH (Q3), Si(OSi)3(OAl) (Si(1Al)), Si(OSi)4 (Q4) and the crystallographically inequivalent Si(OSi)4 (Q4′) sites, respectively, of which the Si(1Al) sites contribute to the Brønsted acid sites.73,74εSi CP DNP enhancement of 9 and overall DNP gain Σ†Si CP of 96 for M-β (Fig. 6a) and εSi CP of 45 and Σ†Si CP of 462 for MMM-β (Fig. 6c) were obtained. | ||
| Fig. 6 (a and c) 29Si CP MAS DNP spectra and (b and d) μw on experimental 29Si CP MAS DNP spectra with spectral deconvolution, overall simulated lineshape and difference between experimental and simulated spectra of activated M-β (a and b) and activated MMM-β (c and d). All spectra were recorded at 9.4 T and at a MAS rate of 8 kHz. Σ† refers to the overall DNP gain and is calculated by comparing the DNP data to room temperature 9.4 T NMR spectra (see ESI†). | ||
These sensitivities permit the fast collection of 29Si detected 29Si{13C} REDOR experiments with high signal-to-noise (S/N) ratios which would otherwise be extremely time consuming. These experiments reintroduce the 29Si–13C dipolar couplings under MAS42 allowing the spatial proximities between the confined carbon species and surface sites of the zeolite to be quantitatively probed. At a recoupling time of 28.5 ms for M-β (Fig. 7a) and 30 ms for MMM-β (Fig. S17†), the 13C dephased 29Si detected signals S′ show significant reduction in intensities compared with the spin echo signal S0, demonstrating spatial proximities between 29Si and 13C nuclei. The high S/N ratios permit small differences in the evolutionary pattern of the REDOR fraction ΔS/S0 as a function of the recoupling times for individual Si sites to be distinguished. The REDOR curves for different Si sites are overlaid in Fig. 7b (for M-β) and S17† (for MMM-β) and demonstrate clear differences between these Si sites. Separate figures for each Si site can also be found in Fig. S18 and S19.†
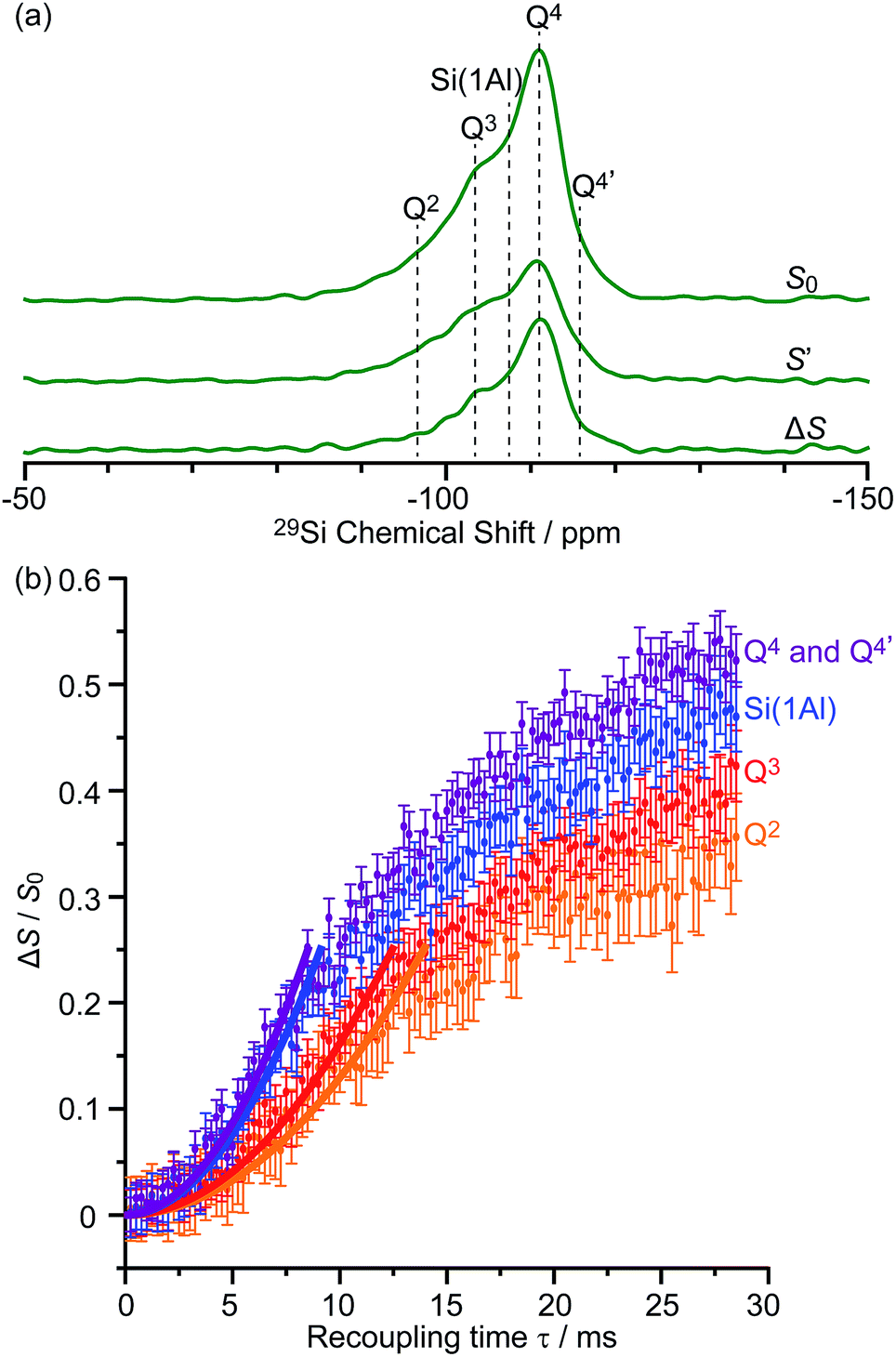 | ||
| Fig. 7 (a) DNP enhanced 29Si CP spin echo spectrum (S0) and 29Si{13C} REDOR spectrum (S′) with the reintroduction of dipolar couplings at a recoupling time of 28.5 ms. ΔS is the difference spectrum S0 − S′. Spectra were recorded at 9.4 T on activated M-β. (b) 29Si{13C} REDOR fraction ΔS/S0 as a function of the recoupling time up to 28.5 ms. The experimental time is ≈15 hours. The solid lines are the best-fit of the REDOR curves up to a ΔS/S0 of 0.25 using a first-order approximation and eqn (S4) in the ESI.†75 The 29Si–13C dipolar coupling values are given in Table 1. The vertical error bars correspond to the error analysis as given in Section 3 in the ESI.† The REDOR curves for each 29Si site are also shown separately in Fig. S18.† | ||
Considering the number of retained carbon species and their unknown geometries in the zeolites, a geometrically independent REDOR curve model which only requires data at short dipolar evolution times (ΔS/S0 up to 0.25)75 was used to fit the REDOR data (see ESI† for further details) and the results are summarized in Table 1. By further assuming a 29Si–13C single spin pair model, an estimation of the 29Si–13C dipolar coupling strengths and distances is also given.
| Zeolite | 29Si sites | ∑Di2/Hz2 | D /Hz | r /Å |
|---|---|---|---|---|
| a Assuming a simplified 29Si–13C single spin pair model. | ||||
| Activated M-β | Q2 | 1200 ± 400 | 35 ± 7 | 5.6 ± 0.4 |
| Q3 | 1500 ± 500 | 39 ± 7 | 5.4 ± 0.3 | |
| Si(1Al) | 2800 ± 800 | 53 ± 8 | 4.8 ± 0.3 | |
| Q4 + Q4′ | 3200 ± 800 | 57 ± 8 | 4.7 ± 0.3 | |
| Activated MMM-β | Q2 | 65 ± 25 | 8 ± 2 | 9.1 ± 0.7 |
| Q3 | 105 ± 40 | 10 ± 2 | 8.4 ± 0.7 | |
| Si(1Al) | 155 ± 40 | 12 ± 2 | 7.8 ± 0.4 | |
| Q4 + Q4′ | 235 ± 45 | 15 ± 2 | 7.3 ± 0.3 | |
The 29Si{13C} REDOR data (Table 1) show that both Q4 and Si(1Al) sites have the strongest interaction with the hydrocarbon pool species in activated M-β with ∑Di2 being 3200 ± 800 Hz2 and 2800 ± 800 Hz2, respectively. This major contribution to the 29Si{13C} REDOR curves is expected as neutral aromatic species are the main hydrocarbon pool species, as shown in the 13C CP spectra, and interact more strongly with the zeolite frameworks. The similar interaction between the hydrocarbons and both Q4 and Si(1Al) sites also suggests that the van der Waals interaction with the zeolite framework dominates the adsorption of these hydrocarbon pool species within the micropores (known as the confinement effects)76 and that, surprisingly, there is no evidence for preferential interaction with the Brønsted acid sites of the zeolites. The zeolite deactivation during the MTH process is therefore likely due to the accumulation of aromatics in the channels of the zeolite and their further growth to form polycyclic cokes blocking the reactants' accesses to the catalytic acid sites supporting previous computational studies.76,77 The quantitative information provided here also promotes understanding of the nature of the previously proposed supramolecular reaction centers78 and yields structural constraints (distance of around 4.8 Å) between the hydrocarbon pool species and zeolite frameworks. Table 1 also shows that the Q2 and Q3 sites have much weaker interaction with the hydrocarbon pool species suggesting that these silanol defects are mainly located at the external surface.
Comparison of the 29Si{13C} REDOR data for both activated zeolites (Table 1) revealed that the Si sites show much weaker interaction with the hydrocarbon pool species in MMM-β than in M-β. We ascribe this phenomenon to the presence of the mesopores in MMM-β which weaken the confinement effects and suggest that some carbon species should predominantly locate in the mesopores.
Note that the 29Si{13C} REDOR results do not exclude the possibility that carbocations have strong interaction with the Brønsted acid sites, likely via the formation of ion-pair complexes,79 since the 29Si{13C} REDOR experiments measure the overall dipolar coupling to 29Si from 13C spins of different molecules while not distinguishing the source of the contribution.
Conclusions
In summary, we demonstrate that a small concentration of carbocations confined in zeolites can be detected within minutes by DNP enhanced multinuclear NMR spectroscopy. The large DNP signal enhancements enable acquisition of two-dimensional 13C–13C NMR correlation experiments in hours which would otherwise take days or even weeks without DNP. These correlations permit the identification of a series of five- and six-membered ring carbocations serving as intermediates in the MTH reaction. In particular, methylnaphthalenium ions are identified and reinforce their importance as hydrocarbon pool species for the formation of targeted hydrocarbon products and coke precursors leading to zeolite deactivation. Additionally, the host–guest interaction between various silicon sites of zeolites and hydrocarbon pool species is quantitatively determined via DNP enhanced 29Si{13C} REDOR experiments, which indicate that van der Waals interaction with the zeolite frameworks dominates the adsorption of the majority hydrocarbon pool species, suggesting accumulation of these species in the channels and leading to zeolite deactivation. Finally, we show that introducing hierarchical pores into zeolites is a promising way to improve DNP efficiency on this type of materials. The implications of this strategy to tackle the understanding of hierarchically structured porous materials of considerable interest in catalysis, gas adsorption, sensing, etc.43,80 are potentially very large.Author contributions
D. X. and F. B. designed the project, conducted the DNP experiments, analysed the data and wrote the manuscript with inputs from all co-authors. S. X. prepared the activated zeolites, contributed to the DNP experiments and data analysis. N. B., S. P. and F. A. aided in carrying out the DNP experiments. F. A. performed the EPR spin counting experiments. L. C. and B. S. prepared the MMM-β zeolite. All authors discussed the results. D. X. and S. X. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from BP, University of Liverpool and Dalian Institute of Chemical Physics for a studentship for D. X. (75139), from the National Natural Science Foundation of China (91545104 and 21473182) and the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2014165) to S. X., and the Engineering and Physical Science Research Council (EPSRC) for Doctoral Training Studentships to N. J. B. and grants EP/M00869X/1 and EP/P511353/1 to F. B. and the University of Nottingham Advanced Molecular Materials RPA funding scheme to F. B. are acknowledged. D. X. thanks Ashlea R. Hughes (University of Liverpool) for recording some of the room temperature 13C and 29Si CP MAS NMR spectra and fruitful discussions. Access to the University of Nottingham DNP MAS NMR Facility used in this research to record the 14.1 T DNP data and funded by the EPSRC (EP/L022524/1) is gratefully acknowledged. The experimental data are provided as a supporting dataset from the University of Liverpool Data Catalogue portal at http://datacat.liverpool.ac.uk/556/.Notes and references
- G. A. Olah, Angew. Chem., Int. Ed., 1995, 34, 1393–1405 CrossRef CAS.
- R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905–6948 CrossRef CAS PubMed.
- V. B. Kazansky, Acc. Chem. Res., 1991, 24, 379–383 CrossRef.
- V. Adeeva, H.-Y. Liu, B.-Q. Xu and W. M. H. Sachtler, Top. Catal., 1998, 6, 61–76 CrossRef CAS.
- Y. V. Kissin, Catal. Rev.: Sci. Eng., 2001, 43, 85–146 CrossRef CAS.
- J. Weitkamp, ChemCatChem, 2012, 4, 292–306 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- I. Kiricsi, H. Förster, G. Tasi and J. B. Nagy, Chem. Rev., 1999, 99, 2085–2114 CrossRef CAS PubMed.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS.
- T. Xu and J. L. White, US Pat., 6734330B1, 2004.
- T. Xu and J. L. White, US Pat., 6743747B1, 2004.
- J. L. White, Catal. Sci. Technol., 2011, 1, 1630–1635 RSC.
- S. Xu, A. Zheng, Y. Wei, J. Chen, J. Li, Y. Chu, M. Zhang, Q. Wang, Y. Zhou, J. Wang, F. Deng and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 11564–11568 CrossRef CAS PubMed.
- C. Wang, Y. Chu, A. Zheng, J. Xu, Q. Wang, P. Gao, G. Qi, Y. Gong and F. Deng, Chem.–Eur. J., 2014, 20, 12432–12443 CrossRef CAS PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- J. F. Haw, J. B. Nicholas, T. Xu, L. W. Beck and D. B. Ferguson, Acc. Chem. Res., 1996, 29, 259–267 CrossRef CAS.
- M. Zhang, S. Xu, J. Li, Y. Wei, Y. Gong, Y. Chu, A. Zheng, J. Wang, W. Zhang, X. Wu, F. Deng and Z. Liu, J. Catal., 2016, 335, 47–57 CrossRef CAS.
- W. Dai, C. Wang, X. Yi, A. Zheng, L. Li, G. Wu, N. Guan, Z. Xie, M. Dyballa and M. Hunger, Angew. Chem., Int. Ed., 2015, 54, 8783–8786 CrossRef CAS PubMed.
- G. Buntkowsky and T. Gutmann, Angew. Chem., Int. Ed., 2015, 54, 9450–9451 CrossRef CAS PubMed.
- J. F. Haw, P. W. Goguen, T. Xu, T. W. Skloss, W. Song and Z. Wang, Angew. Chem., Int. Ed., 1998, 37, 948–949 CrossRef CAS PubMed.
- D. Xiao, S. Xu, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2017, 8, 8309–8314 RSC.
- A. W. Overhauser, Phys. Rev., 1953, 92, 411–415 CrossRef CAS.
- T. R. Carver and C. P. Slichter, Phys. Rev., 1953, 92, 212–213 CrossRef CAS.
- T. R. Carver and C. P. Slichter, Phys. Rev., 1956, 102, 975–980 CrossRef CAS.
- D. A. Hall, D. C. Maus, G. J. Gerfen, S. J. Inati, L. R. Becerra, F. W. Dahlquist and R. G. Griffin, Science, 1997, 276, 930–932 CrossRef CAS PubMed.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- A. Zagdoun, A. J. Rossini, D. Gajan, A. Bourdolle, O. Ouari, M. Rosay, W. E. Maas, P. Tordo, M. Lelli, L. Emsley, A. Lesage and C. Copéret, Chem. Commun., 2012, 48, 654–656 RSC.
- T.-C. Ong, W.-C. Liao, V. Mougel, D. Gajan, A. Lesage, L. Emsley and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 4743–4747 CrossRef CAS PubMed.
- E. Pump, J. Viger-Gravel, E. Abou-Hamad, M. K. Samantaray, B. Hamzaoui, A. Gurinov, D. H. Anjum, D. Gajan, A. Lesage, A. Bendjeriou-Sedjerari, L. Emsley and J.-M. Basset, Chem. Sci., 2017, 8, 284–290 RSC.
- W. R. Gunther, V. K. Michaelis, M. A. Caporini, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2014, 136, 6219–6222 CrossRef CAS PubMed.
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Núñez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Copéret and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS PubMed.
- P. Wolf, M. Valla, F. Núñez-Zarur, A. Comas-Vives, A. J. Rossini, C. Firth, H. Kallas, A. Lesage, L. Emsley, C. Copéret and I. Hermans, ACS Catal., 2016, 6, 4047–4063 CrossRef CAS.
- F. A. Perras, Z. Wang, P. Naik, I. I. Slowing and M. Pruski, Angew. Chem., Int. Ed., 2017, 56, 9165–9169 CrossRef CAS PubMed.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- F. Blanc, in Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection, ed. M. Van de Voorde and B. Sels, Wiley-Blackwell, 2017, pp. 1003–1028 Search PubMed.
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- T. Gullion, Concepts Magn. Reson., 1998, 10, 277–289 CrossRef CAS.
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- L.-H. Chen, X.-Y. Li, G. Tian, Y. Li, H.-Y. Tan, G. Van Tendeloo, G.-S. Zhu, S.-L. Qiu, X.-Y. Yang and B.-L. Su, ChemSusChem, 2011, 4, 1452–1456 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, D. Gajan, F. Rascón, M. Rosay, W. E. Maas, C. Copéret, A. Lesage and L. Emsley, Chem. Sci., 2012, 3, 108–115 RSC.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- H. Takahashi, D. Lee, L. Dubois, M. Bardet, S. Hediger and G. De Paëpe, Angew. Chem., Int. Ed., 2012, 51, 11766–11769 CrossRef CAS PubMed.
- K. R. Thurber and R. Tycko, J. Chem. Phys., 2014, 140, 184201 CrossRef PubMed.
- F. Mentink-Vigier, S. Paul, D. Lee, A. Feintuch, S. Hediger, S. Vega and G. De Paëpe, Phys. Chem. Chem. Phys., 2015, 17, 21824–21836 RSC.
- S. R. Chaudhari, P. Berruyer, D. Gajan, C. Reiter, F. Engelke, D. L. Silverio, C. Copéret, M. Lelli, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2016, 18, 10616–10622 RSC.
- F. Mentink-Vigier, G. Mathies, Y. Liu, A.-L. Barra, M. A. Caporini, D. Lee, S. Hediger, R. G. Griffin and G. De Paëpe, Chem. Sci., 2017, 8, 8150–8163 RSC.
- S. R. Chaudhari, D. Wisser, A. C. Pinon, P. Berruyer, D. Gajan, P. Tordo, O. Ouari, C. Reiter, F. Engelke, C. Copéret, M. Lelli, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10609–10612 CrossRef CAS PubMed.
- N. J. Brownbill, D. Gajan, A. Lesage, L. Emsley and F. Blanc, Chem. Commun., 2017, 53, 2563–2566 RSC.
- P. Fricke, D. Mance, V. Chevelkov, K. Giller, S. Becker, M. Baldus and A. Lange, J. Biomol. NMR, 2016, 65, 121–126 CrossRef CAS PubMed.
- C. Copéret, W. C. Liao, C. P. Gordon and T.-C. Ong, J. Am. Chem. Soc., 2017, 139, 10588–10596 CrossRef PubMed.
- A. H. Linden, W. T. Franks, Ü. Akbey, S. Lange, B. J. Van Rossum and H. Oschkinat, J. Biomol. NMR, 2011, 51, 283–292 CrossRef CAS PubMed.
- W. Song, J. B. Nicholas, A. Sassi and J. F. Haw, Catal. Lett., 2002, 81, 49–53 CrossRef CAS.
- W. Dai, C. Wang, M. Dyballa, G. Wu, N. Guan, L. Li, Z. Xie and M. Hunger, ACS Catal., 2015, 5, 317–326 CrossRef CAS.
- M. Bjørgen, U. Olsbye, S. Svelle and S. Kolboe, Catal. Lett., 2004, 93, 37–40 CrossRef.
- M. Bjørgen, S. Akyalcin, U. Olsbye, S. Benard, S. Kolboe and S. Svelle, J. Catal., 2010, 275, 170–180 CrossRef.
- K. Lammertsma and H. Cerfontain, J. Am. Chem. Soc., 1979, 101, 3618–3624 CrossRef CAS.
- K. Lammertsma, J. Am. Chem. Soc., 1981, 103, 2062–2069 CrossRef CAS.
- K. Hemelsoet, A. Nollet, V. Van Speybroeck and M. Waroquier, Chem.–Eur. J., 2011, 17, 9083–9093 CrossRef CAS PubMed.
- E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2015, 5, 992–1003 CrossRef CAS.
- W. Song, H. Fu and J. F. Haw, J. Phys. Chem. B, 2001, 105, 12839–12843 CrossRef CAS.
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221, 1–10 CrossRef.
- J. Li, Y. Wei, J. Chen, P. Tian, X. Su, S. Xu, Y. Qi, Q. Wang, Y. Zhou, Y. He and Z. Liu, J. Am. Chem. Soc., 2012, 134, 836–839 CrossRef CAS PubMed.
- M. Bjørgen, U. Olsbye and S. Kolboe, J. Catal., 2003, 215, 30–44 CrossRef.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed.
- N. M. Szeverenyi, M. J. Sullivan and G. E. Maciel, J. Magn. Reson., 1982, 47, 462–475 CAS.
- K. Takegoshi, S. Nakamura and T. Terao, Chem. Phys. Lett., 2001, 344, 631–637 CrossRef CAS.
- C. R. Morcombe, V. Gaponenko, R. A. Byrd and K. W. Zilm, J. Am. Chem. Soc., 2004, 126, 7196–7197 CrossRef CAS PubMed.
- T. I. Korányi and J. B. Nagy, J. Phys. Chem. B, 2005, 109, 15791–15797 CrossRef PubMed.
- J. Pérez-Pariente, J. Sanz, V. Fornés and A. Corma, J. Catal., 1990, 124, 217–223 CrossRef.
- M. Bertmer and H. Eckert, Solid State Nucl. Magn. Reson., 1999, 15, 139–152 CrossRef CAS PubMed.
- R. Y. Brogaard, B. M. Weckhuysen and J. K. Nørskov, J. Catal., 2013, 300, 235–241 CrossRef CAS.
- R. Rungsirisakun, B. Jansang, P. Pantu and J. Limtrakul, J. Mol. Struct., 2005, 733, 239–246 CrossRef CAS.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- J. F. Haw, J. B. Nicholas, W. Song, F. Deng, Z. Wang, T. Xu and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 4763–4775 CrossRef CAS.
- M.-H. Sun, S.-Z. Huang, L.-H. Chen, Y. Li, X.-Y. Yang, Z.-Y. Yuan and B.-L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03848a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |