
The biologically mediated water–gas shift reaction: structure, function and biosynthesis of monofunctional [NiFe]-carbon monoxide dehydrogenases
Marila
Alfano
and
Christine
Cavazza
 *
*
Univ. Grenoble Alpes, CEA, CNRS, BIG, CBM, F-38000 Grenoble, France. E-mail: christine.cavazza@cea.fr
First published on 14th May 2018
Abstract
Among the possible renewable energies sources, biomass gasification has been considered one promising alternative to meet the daily growing energy demand. The outcome of this process is a synthetic gas (syngas) mainly composed of carbon monoxide and hydrogen. Syngas can be upgraded by a group of anaerobic micro-organisms, thanks to the biologically mediated water–gas shift (WGS) reaction. In this process, the conversion of CO and H2O into CO2 and H2 is catalyzed by two enzymes: carbon monoxide dehydrogenase (CODH) and hydrogenase. In order to efficiently use micro-organisms as a cost effective and environmentally friendly technology, it is fundamental to deeply understand how this process occurs. In this review paper, an overview on the possible biotechnological uses of the WGS reaction is presented, focusing mainly on the fundamental characterization of the CODH enzyme.
1. Introduction
The intense exploitation of fossil fuels, continuous demographic growth, expanding industrialization and living standard improvements require new sustainable strategies to meet the future demand for commodity products and energy sources. For example, the demand for n-butanol is expected to reach 5.61 Mton per year by 2030 in the manufacture of pharmaceuticals and plastics.1 In the energy sector, liquid, gaseous or solid biofuels hold great promise to deliver an increasing share of the energy required to power a new global green economy. Among them, dihydrogen is an attractive fuel since it can be converted to energy without the production of CO2. Nowadays, its production comes mainly from fossil fuels, with an annual production of more than 60 million metric tons worldwide.2 Biohydrogen production processes through an economically acceptable method are therefore promising alternatives for a possible future H2-based economy.Among available renewable sources, biomass conversion from organic waste is becoming increasingly popular. It is also likely to evolve into the main source of primary energy feedstock for developing countries. Besides bioconversion processes to transform biomass into biofuel or different valuable chemicals, an interesting alternative is the thermal conversion (gasification) of poorly degradable biomass sources like straw and wood.3 Biomass gasification occurs through a combination of complex reactions, including drying, pyrolysis, combustion and reduction, resulting in the production of synthesis gas (syngas). Syngas is mainly composed of a mixture of H2, CO and CO2 and minor amounts of other gasses. It is an inexpensive and versatile substrate generated from any hydrocarbon feedstock, from fossil fuels to biomass. Therefore, a wider application of routes starting from syngas is more appealing since it enables a gradual transition to more sustainable energy. Biomass gasification to provide motor fuel has been in use since the early 1930s. Over the decades, syngas was successfully used in the USA and Europe for heat, electricity and the synthesis of higher alcohols (Fig. 1). This has also prompted the utilization of new isolates and some known anaerobic microorganisms capable of growing with CO and H2 as substrates. CO can be metabolized by four main trophic groups of microorganisms: methanogenic archaea, hydrogenogenic bacteria, sulfate-reducing bacteria and acetogenic bacteria.4 Among the latter group, some Clostridia produce, besides acetate, significant amounts of ethanol, butyrate and butanol.5 Specifically, syngas fermentation to ethanol and isobutanol using Clostridium ljungdahlii is exploited on a relatively small scale by companies like LanzaTech or INEOS Bio. Hydrogenogenic carboxydotrophs directly produce H2 from CO, according to the reaction CO + H2O → H2 + CO2 known as the water–gas shift (WGS) reaction. The conversion of CO into H2 and CO2 involves two key enzymes, a [NiFe]-CO dehydrogenase (CODH), which oxidizes CO to CO2, and an energy-conserving [NiFe]-hydrogenase, which produces dihydrogen.6 Nowadays, the water–gas shift reaction plays a significant role in industrial applications to balance H2/CO ratios of syngas, via a H2-enrichment process.7 The discovery of bacteria capable of growth employing a biological analogue of the WGS reaction holds promise for the design of a biological alternative to this chemical material. In this review, we first discuss the industrial applications of the WGS reaction. In the second part, we describe at the molecular level [NiFe]-CODH, a key enzyme of the WGS reaction.
 | ||
| Fig. 1 Fuel products obtained from synthesis gas transformation. The green squares highlight the process described in this review (adapted from ref. 103). | ||
2. Industrial applications: water–gas shift reaction & Fischer–Tropsch process
Since the beginning of the 20th century, the development and exploitation of the water–gas shift (WGS) reaction have followed the increasing industrial demands for hydrogen.8 This has being achieved progressively, evolving the feedstock and the materials used to meet the production requirement for either pure hydrogen or CO to H2 ratio modulation, the latter being required in the Fischer–Tropsch process or the synthesis of platform molecules such as methanol.7,9 For this reason, WGS facilities can present major differences in the equipment and materials employed and are strongly bounded to the feedstock used for the syngas production, for both economical and practical purposes. In the next paragraphs the different requirements will be described.102.1 The water–gas shift reaction
The WGS reaction, discovered in 1780 by F. Fontana, is a reversible exothermic reaction, described as:| CO + H2O ↔ CO2 + H2 |
| ΔH0298 = −41.09 kJ mol−1 |
In this reaction, the equilibrium constant decreases with increasing temperature. Carbon monoxide and hydrogen conversions are favoured at low temperature but the reaction is kinetically favoured at high temperature.9 Concretely, we face a trade-off between the speed and purity of the products. In this framework, catalysts, specifically their composition and working temperatures, play a predominant role.
When hydrogen production is the main goal, industries traditionally rely on the use of two stage catalytic steps.11 The first step, carried out at high temperatures (HT), between 400 and 500 °C, uses iron oxide stabilized in chromium oxide as a catalyst (Fe2O3–Cr2O3).12 This high temperature reaction is generally extremely fast, but it has the drawback of being thermodynamically limited, resulting in an incomplete CO conversion of the initial syngas composition (almost 90% conversion). To refine the final product, a slower low temperature (LT) step is required, typically between 200 and 220 °C, in order to move the equilibrium towards the hydrogen product. Copper–zinc oxide supported on alumina (Cu–ZnO/Al2O3) catalysts,13 characteristic of this reaction, are able to finally generate the desiderated product, lowering the CO content to less than 1% in the final mixture.8,9,14 The combination of two steps is also necessary since LT catalysts can easily suffer from sulphate poisoning, a substance commonly present in pre-high-temperature mixtures.
Currently, the methods commonly available to perform the WGS reaction are distinguished on the basis of the specific type of reactor used: the traditional reactor, the membrane reactor and the photocatalytic reactor. Concerning the traditional reactor, the WGS reaction can be conducted in a homogeneous state (catalyst and reactant are in the same state) or a heterogeneous state (catalyst and reactant in different states).8 This latter method is by far the most commonly used due to the scarcity of gaseous catalysts. A membrane reactor is able to selectively remove one of the products of a given reaction through a porous membrane.15,16 This allows for a modulation of the outlet gas composition, achieving a complete CO conversion and H2/CO2 separation. In photo-catalytic reactors, Pt/TiO2-based catalysts are irradiated and activated by visible or UV-light.17,18 Although this technology allows for processes to occur at room temperature, its industrialization has been impeded by its many disadvantages for the commercialization.
Comprehensively, in WGS reaction facilities, their construction and their operation are tightly dependent on the syngas: a more controlled syngas composition, having for example CO and H2 as predominant gases, leads to strong advantages in the catalytic process. In addition, the syngas composition strongly depends on the feedstock source and gasification process, resulting in a mixture of gases in which the percentage of each component is strongly variable (Table 1). As mentioned above, the presence of contaminant species, such as sulfur and nitrate compounds, in the inlet gas can lead to catalyst poisoning and a decrease in the global process efficiency. Another important limitation for the HT shift reaction is the H2/CO ratio, which needs to be relatively high to avoid side reactions, such as the formation of metallic iron, methanation, carbon deposition and Fischer–Tropsch reactions, due to the over reduction of the iron oxide.9 In some cases the products of these side reactions can be the real objective, conferring to the WGS reaction a contradistinctive role.
| Source | Composition (vol%) | ||||||
|---|---|---|---|---|---|---|---|
| H2 | CO | CO2 | N2 | CH4 | Other | Ref. | |
| Natural gas steam reforming by air | 56–57 | 10–15 | 7–12 | 22–23 | 0.2–0.3 | — | 22 |
| Water gas | 31.7 | 30 | 3.4 | 13.1 | 12.2 | 9.6 | 24 |
| Coal gasification | 29.4 | 59.4 | 10 | 0.6 | — | 0.6 | 24 |
| Coke oven gas | 55.4 | 5.6 | 1.4 | 4.3 | 28.4 | 4.9 | 24 |
| Charcoal | 29 | 48 | — | — | 48 | — | 20 |
| Grass straw | 2.6 | 12.9 | 17.4 | 64.2 | 2.1 | 0.8 | 37 |
| Demolition wood/paper residue | 6.1 | 9.2 | 16.1 | 63.2 | 2.8 | 2.6 | 37 |
2.2 The use of the WGS reaction in the Fischer–Tropsch process
The Fischer–Tropsch (FT) process is an assembly of different reactions with the aim of converting a CO and H2 mixture into liquid hydrocarbons, using the general reactions shown in Table 2. The process uses syngas as the main source converting it catalytically with a particular inorganic catalyst, working in a range of temperature between 150 and 300 °C and depending upon the syngas nature and desired product.10,19| H2/CO ratio | ||
|---|---|---|
| Main reactions | ||
| Alkanes | nCO + (2n + 1)H2 → CnH2n+2 + nH2O | (2n + 1)/n |
| Alkenes | nCO + 2nH2 → CnH2n + nH2O | 2 |
| Water–gas shift reaction | CO + H2O → CO2 + H2 | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Side reactions | ||
| Alcohols | nCO + 2nH2 → H(–CH2–)OH + (n − 1)H2O | 2 |
| Boudouard reaction | 2CO → CO2 + C | — |
In the FT synthesis the overall process from syngas is commonly named XTL (X-to-liquid), with X depending on the carbon source. Four main classes of carbon sources are the most popular: coal (CTL), natural gas (GTL), biomass (BTL) and waste (WST) (Fig. 2).10
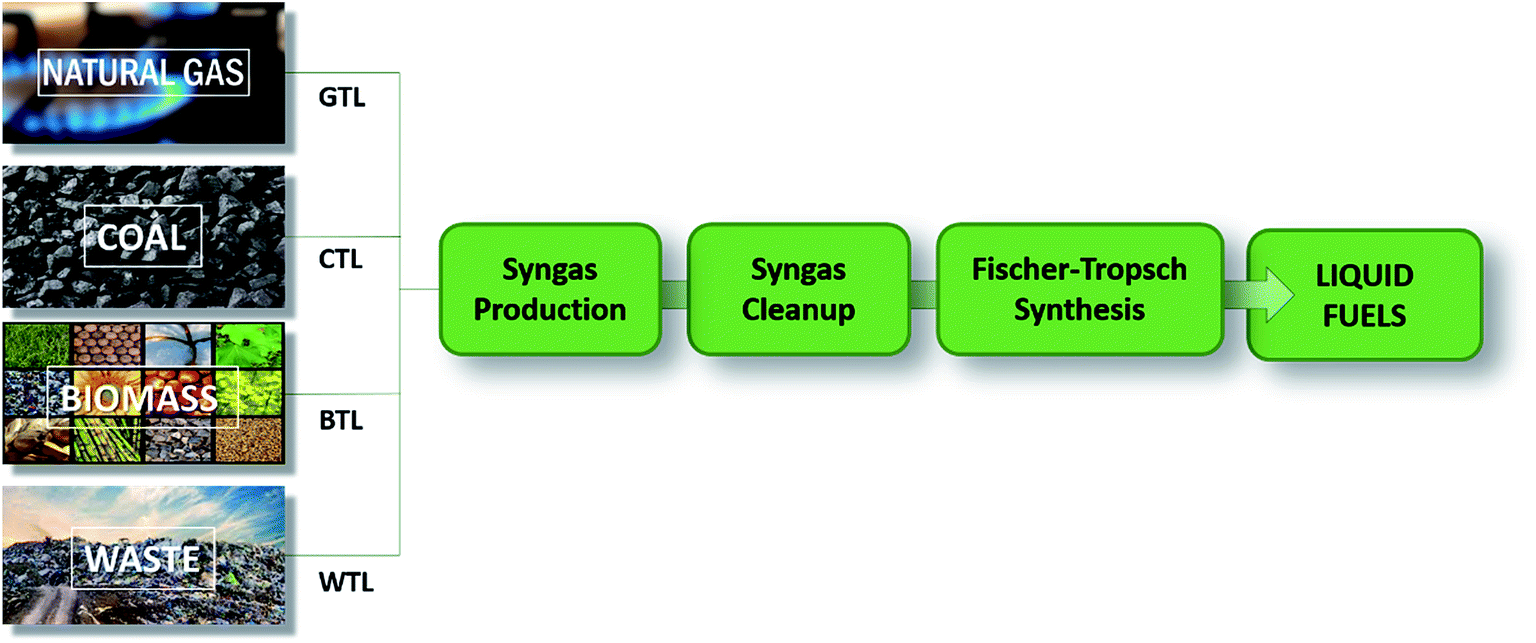 | ||
| Fig. 2 General scheme for describing the XTL process, starting from different carbon sources. X = gas, coal, biomass, and waste (adapted from ref. 10). | ||
The synthesis is, therefore, intrinsically different for the specific carbon sources and an appropriate selection of syngas feed conditions is essential to obtain optimal activity, stability and selectivity in industrial reactors. In general, the H2/CO ratio most suitable for a FT synthesis is 2:1, which changes according to the desired product (Table 2). In a GTL process, the syngas is characterised by a high optimal H2/CO (1.9–2.2) whereas in the CTL and BTL processes, the syngas is characterised by a lower H2/CO (0.6–1.7), which therefore needs to be adjusted.10 This highlights one of the problems that the industry is currently facing: syngas obtained from wood and straw, which is becoming more and more popular due to the low economic and environmental costs, has a H2/CO ratio which is often too low (0.7) for optimal industrial use.20 This is also related to the most currently used industrial catalysts, iron unsupported metal and cobalt deposited on an inert oxide support, which need a specific H2/CO ratio in order to perform efficiently.7 In recent years, new catalysts have been designed to work with a lower H2/CO ratio.21 However, the practicality of this approach is limited by its different drawbacks, which arise from the materials used (working temperatures, fouling, etc.).21 Today, the main route followed is to exploit the WGS reaction that, used to balance the H2/CO ratio, can actively modulate hydrogen concentration at the expense of carbon monoxide reaching the desired feed ratio.7 The employment of such a strategy on an industrial scale is extremely interesting, and its versatility is expanded by the possibility of microbial processes, where the use of inorganic catalysts is set apart in favour of more bio-compatible alternatives.
3. The biologically mediated water–gas shift reaction
An alternative to these inorganic industrial processes is the use of bacteria able to perform the WGS reaction at room temperature and pressure. This biological process has the promise to become a favourable and cost-effective technology for bio-hydrogen production and H2-enriched syngas for the Fischer–Tropsch process.22,23 The key for both these applications is the existence of microorganisms able to convert CO to worthwhile compounds. Among them, anaerobic hydrogenogenic carboxydotrophs use CO as the sole energy source thanks to the biological water–gas shift reaction, fundamental for their growth.24,25 In the following pages, we will focus on hydrogenogenic CO metabolism.In the biologically mediated WGS reaction, the overall reaction is catalyzed by two different metalloenzymes, a monofunctional [NiFe]-carbon monoxide dehydrogenase ([NiFe]-CODH) and an energy-conserving [NiFe]-hydrogenase,6 according to:
| CO + H2O → 2e− + 2H+ + CO2 (CODH) |
| 2e− + 2H+ → H2 (hydrogenase) |
CO is oxidized by CODH, releasing electrons used for the reduction of two protons into bio-hydrogen, catalyzed by an energy-conserving hydrogenase (Fig. 3). CO has a strong tendency to inhibit hydrogenase activity by binding to the Ni ion present in the active site.23 Therefore, the hydrogenase coupled with CODH needs to present a high CO tolerance in order to avoid being inactivated along the catalytic cycle.
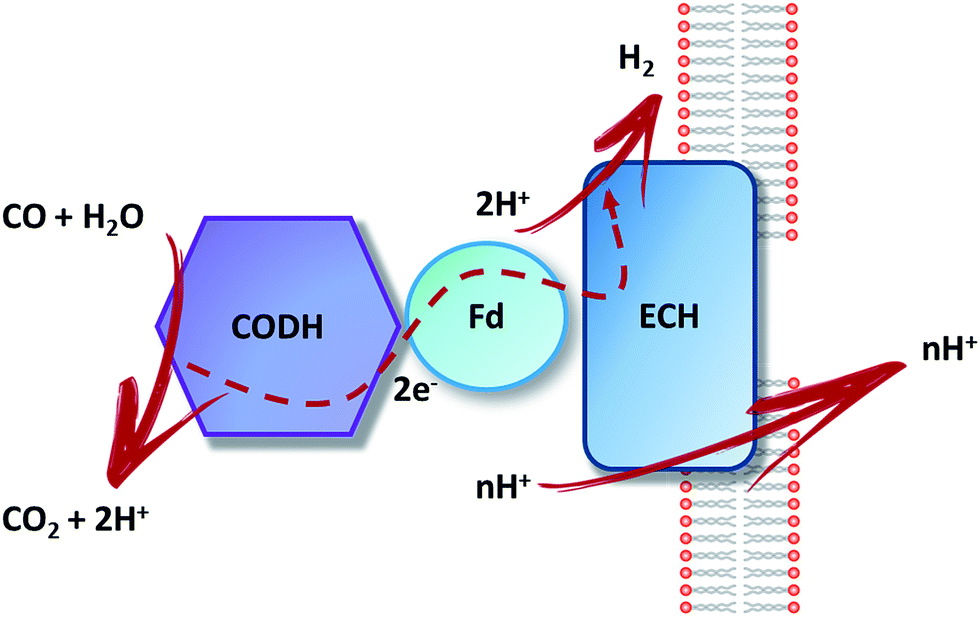 | ||
| Fig. 3 Schematic representation of CO oxidation, electron transfer, H2 production and proton translocation in hydrogenogenic carboxydotrophs. CODH: CO dehydrogenase; Fd: ferredoxin (CooF); ECH: energy-conserving hydrogenase. | ||
Among hydrogenogenic carboxydotrophs, we find thermophilic and mesophilic species (Table 3). Thermophilic species seem to be more abundant than the mesophilic ones, suggesting that high temperature might help hydrogenogenic CO metabolism, by increasing gas diffusion rates.25 The physiology and the utilization for biotechnological applications of two model organisms, Rhodospirillum rubrum and Carboxydothermus hydrogenoformans, are detailed below.
| Species | Origin | Growth conditions | Substrate | Product | H2 Production rate | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| pH | Temp (°C) | mmol g−1 h−1 | ||||||
| a (%mol mol−1 CO). | ||||||||
| Mesophilic bacteria | Rhodospirillum rubrum | Brackish ditch | 6.5 | 30 | CO–H2/CO2 acetate | H2 | 16 | 28,45 |
| Rubrivivax gelatinosus | Lake and pond sediments | 6.7 | 34 | CO–H2/CO2 | H2 | 1.3–33 | 105 | |
| Rhodopseudomonas palustris | Anaerobic sludge digester | n.r. | 30 | CO–H2/CO2 | H2 | 41 | 106 | |
| Thermophilic bacteria | Carboxydothermus hydrogenoformans | Hydrothermal springs on Kunashir island | 6.9–7.8 | 70 | CO | H2 | 83.3a | 38,42 |
| Desulfotomaculum carboxydivorans | Anaerobic bioreactor sludge | 6.8–7 | 55 | CO–H2/CO2 | H2 | — | 107 | |
| Carboxydocella pertinax | Thermal spring of the volcanic Karymskoe lake | 6–6.5 | 65 | CO | H2 | — | 108 | |
| Caldanaerobacter subterraneus pacificus | Submarine hot vent | 6.8–7.1 | 70 | CO–H2/CO2 | Acetate, CO2 and H2 | — | 109 | |
| Carboxydocella sporoproducens | Hot spring Karymskoe lake | 6.8 | 60 | CO–H2/CO2 | H2 | — | 110 | |
| Carboxydocella thermoautotrophica | Terrestrial hot vent | 7 | 58 | CO–H2/CO2 | H2 | — | 111 | |
| Thermoanaerobacter thermohydrosulfuricus | Geothermal spring in Ayas | 6.3–6.8 | 70 | CO | H2 | — | 112 | |
| Moorella stamsii | Digester sludge | 7.5 | 65 | CO | CO2, H2 | — | 113 | |
| Thermincola carboxydiphila | Mud, water and cyanobacterial mat | 7 | 73 | CO–H2/CO2 | H2 | — | 114 | |
| Thermincola ferriacetica | Hydrothermal spring | 7–7.2 | 60 | CO–H2/CO2 | H2 | — | 115 | |
| Calderihabitans maritimus | Sediment from a marine caldera | 7–7.5 | 65 | CO | H2 | — | 116 | |
| Archaea | Thermococcus onnurineus | Deep-sea hydrothermal vent | 6.1–6.2 | 80 | CO | H2 | 151.3 | 117,118 |
| Thermococcus strain AM4 | Hydrothermal vent | 6.8 | 82 | CO | H2, H2S | — | 119 | |
3.1 Rhodospirillum rubrum
R. rubrum is a purple non-sulfur photosynthetic mesophilic bacterium.26 It is a versatile facultative anaerobe, able to obtain energy through a variety of mechanisms. R. rubrum is capable of aerobic or anaerobic respiration, photosynthesis and acid-mixed fermentation, depending on the growth conditions.27 Moreover, it can use CO as the sole energy source during anaerobic growth in the dark, via the WGS reaction.28 It has a higher rate of CO uptake and conversion yield than other similar organisms and can tolerate a small amount of oxygen in the culture medium, making it a particularly attractive strain to investigate for the biohydrogen production from syngas.28,29Focusing on the growth studies made for R. rubrum under syngas, it has been demonstrated that mild-light exposure (1000 m cd) has a slight positive effect on the growth rate and hydrogen production.30R. rubrum also requires an additional carbon source to maintain constant cell growth and efficiently convert CO.31,32 Najafpour et al.33 investigated hydrogen production using different carbon sources, such as malate, formate and acetate, showing that not all the substrates are suitable for hydrogen production. The maximum CO conversion was reached using acetate, resulting in a maximum hydrogen yield of 98% from a syngas composed of 55% CO, 10% CO2, 20% H2 and 15% Ar.30
R. rubrum is thus capable to directly convert syngas into biohydrogen catalysing the WGS reaction. As a proof of concept, Younesi et al.34 studied R. rurbum growth using acetate under a continuous syngas flow in a stirred bioreactor. The study demonstrated a hydrogen production rate of 16 mmol per g of substrate per cell per h and a global conversion yield of 87 ± 2.4%, confirming the practicality of the WGS reaction on a small scale. The feasibility of this method from a more industrial and economical point of view was described by Dadak et al.,35 who studied the photobiological hydrogen production from syngas in industrial continuous bioreactors. The effect of acetate concentration on CO consumption as well as H2 and CO2 production was evaluated, based on both conventional exergy and eco-exergy concepts. This work showed that the eco-exergy concept seems more suitable as a tool for large-scale biohydrogen production using living organisms, highlighting the environmental feedback which could be a sufficient incentive for pushing forward this technology. Concerning the production of high value added products, R. rubrum has shown the ability to produce biopolymers such as polyhydroxyalkanoates (PHA) via syngas fermentation. Revelles et al.36 demonstrated that in the presence of acetate as a carbon source, PHA accumulation in the bacteria was almost 30% of the dried cell weight. As previously described, syngas cannot be used as a sole carbon source for R. rubrum. Moreover, the Calvin–Benson–Bassham cycle plays a minor role in carbon assimilation during syngas fermentation. Instead, CO2 produced from CO via CODH is assimilated into biomass and PHA via carboxylases such as crotonyl-coA carboxylase or pyruvate synthase. This study shed light on the effective utilization of R. rubrum for the production of bioplastics from syngas.
3.2 Carboxydothermus hydrogenoformans
Considering that biomass gasification results in high temperature gas, strictly anaerobic thermophilic bacteria could be better suited for syngas industrial applications.37 Among them, C. hydrogenoformans, isolated from a hot spring on the volcanic island of Kunashir, is a strictly anaerobic gram-positive thermophilic bacterium capable of CO-growth at an optimum temperature of 70 °C.38 It grows fast (doubling time 2 h) in the dark using CO as the sole carbon and energy source by catalysing the WGS reaction, making it an extremely interesting candidate to test as a possible bio-hydrogen production source from syngas.39 However, no data have been published on the ability of this bacterium to convert CO into H2 from syngas, all the studies considering only the conversion from pure CO gas.37 Due to the low aqueous solubility of CO at 70 °C, an optimum substrate/biomass ratio must be used to avoid gas–liquid mass transfer limitation.40C. hydrogenoformans displays a maximal specific activity of 3.5 mol per CO per g VSS (volatile suspended solid) per day at 0.55 mM CO, which compares favourably with those of other hydrogenogenic bacteria. For comparison, the WGS activity of Rubrivivax gelatinosus is optimal at a dissolved CO concentration of 90–100 μM and starts to be inhibited at concentrations greater than 150 μM.40 Considering this remarkable stability, Zhao et al.41 built a working setup using a hollow fibre membrane bioreactor for bio-hydrogen production from CO. C. hydrogenoformans was grown for four months with a hydrogen yield of 92%. Despite the high efficiency, major problems arise from the system used (membrane fouling and low CO transfer rate), which limit the prospective long-term use. Haddad et al.42 obtained a production yield of 95% using a gas-lift reactor with a continuous supply of gas for three months and addition of bacto-peptone in the medium, leading to a remarkable increase of growth, yield and conversion rate regarding CO conversion. However, the cell density remained low, probably due to some deficiency in essential minerals, which restricts the CO feeding-rate and overall CO conversion performance. Another complementary application is the use of a biological WGS reaction to reduce the CO level in outlet gases, which remains the limiting step for some technologies due to catalyst CO poisoning. Henstra et al.43 proved that C. hydrogenoformans can reach levels lower than 2 ppm of CO in outlet gas, making the H2 suitable for CO-sensitive low temperature fuel cells.Besides the use of bioreactors, an alternative consists of the utilization of the two key enzymes involved in the WGSR, i.e., CODH and hydrogenase. Armstrong and collaborators44 successfully co-attached hydrogenase 2 from E. coli and CODH from C. hydrogenoformans to graphite platelets to produce a heterogeneous catalyst. They showed that the amount of H2 produced is equal to the amount of CO consumed and that the enzymes constituting their catalyst react in concert with an efficiency as high as that of the industrial WGS reaction. However, the utilization of this catalyst in applied technology is hindered at the moment because of the difficulty to obtain large amounts of stable active enzymes, which are therefore not competitive with robust synthetic catalysts.
3.3 Prospects of the biologically mediated WGS reaction
The production of hydrogen and chemicals from syngas via the biological WGS reaction and Fischer–Tropsch process offers different advantages compared to conventional chemical metal-based catalysis.4 As already mentioned, biological processes occur at ambient temperature and pressure, lowering energy requirements and costs. In contrast to inorganic catalysts, which can easily be irreversibly poisoned or fouled during utilization, living microorganisms can automatically regenerate via newly replicated cells, leading to a longer lifetime. Furthermore, biological wastes are considered as by-products that can be treated to produce fuel and/or other organic compounds.4 Concerning the potential development for the commercialization of this technology, one of the areas of improvement is bioreactor design and utilization,45 through the optimization of culture media, metabolic or promoter engineering and biodiversity screening. However, several drawbacks need to be overcome for the implement of industrial-scale bioreactors. First, bacterial activity potential is limited by the CO availability to the microorganisms. It is thus necessary to optimize the CO concentration in the feedstock, the gas to liquid mass transfer and the concentration and distribution of bacteria. Important modifications have been done in order to enhance the syngas mass transfer from the gas phase to the liquid phase. Without mass transfer limitation, the biochemical reaction rate should be close to the maximal intrinsic reaction rate. Pressure and mixing conditions also play a role: high pressure (1 MPa) increases mass transfer rates and thus the rate of CO conversion. Second, CO toxicity towards bacteria is another important limiting step. Indeed, CO is a potent inhibitor of growth and catabolic activity due to its high affinity for metalloenzymes, even if intact cells may offer more resistance than purified CO-sensitive enzymes. A better understanding of syngas metabolism would help the design or the selection of CO-resistant strains.45 Finally, the scientific community has not yet established appropriate tools to genetically modify thermophilic carboxydothrophs.37 Despite the latest advances in bio-hydrogen making, the production is still limited to the laboratory scale and improvements in system productivity and hydrogen yield are needed.An alternative consists in the “in vitro” utilization of the two isolated key enzymes involved in the WGS reaction, i.e., CODH and hydrogenase.
4. CO-induced energy-conserving [NiFe]-hydrogenase
Hydrogenases have been classified into three phylogenetically unrelated classes, based on the metal composition of their active site: [Fe]-, [FeFe]- and [NiFe]-hydrogenases. Among them, the two latter types catalyze the reversible oxidation of molecular hydrogen to protons and electrons.46 [NiFe]-hydrogenases, found in bacteria and archaea, have been classified in four main groups. Membrane-associated H2 uptake hydrogenases belong to group 1. Group 2 contains soluble uptake hydrogenases and sensory hydrogenases; group 3 contains heteromultimeric cytoplasmic hydrogenases harbouring a reducible cofactor (F420 or NAD(P)) and group 4, energy-converting hydrogenases.47 CO-induced hydrogenases involved in the WGS reaction belong to the latter group with the Ech hydrogenase from Methanosarcina barkeri and hydrogenase 3 from E. coli (Fig. 4). In E. coli, hydrogenase 3 is part of the formate hydrogen-lyase (FHL) complex and its expression is formate-induced.48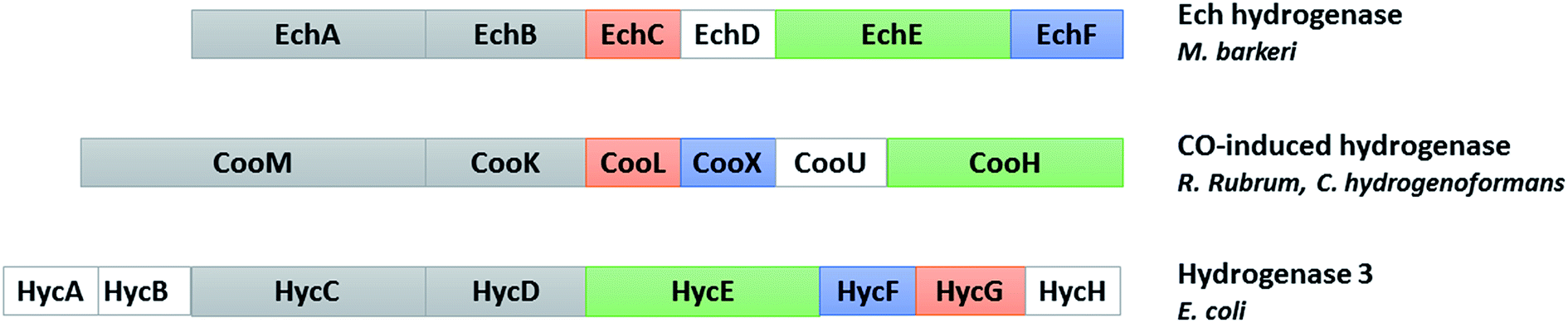 | ||
| Fig. 4 Comparison of the subunits encoded by the cooMKLXUH operon from R. rubrum or C. hydrogenoformans, the Ech operon from Methanosarcina barkeri and the hyc operon from E. coli. Proteins without sequence similarity are shown in white (adapted from ref. 104). | ||
Standard [NiFe]-hydrogenases are heterodimers of 60- and 30-kDa subunits. The dinuclear [NiFe] active site contains additional ligands (1 CO and 2 CN−) terminally bound to the iron ion and is buried in the large subunit, while the small subunit harbors three FeS clusters (named proximal, medial and distal clusters) forming the electron transport chain. Depending on the different groups, additional subunits are present.
In hydrogenogenic carboxydotrophs, the CO-induced hydrogenase is encoded by the cooMKLXUH operon, identified by Ludden and collaborators in R. rubrum,49 present upstream the CODH operon. These membrane-bound hydrogenases contain six subunits: two integral membrane-bound proteins, CooM and CooK corresponding to HycC and HycD in E. coli hydrogenase 3, and four hydrophilic subunits cooLXUH. CooU in R. rubrum has no homologue in E. coli hydrogenase 3. CooX has been postulated to be an Fe4S4 cluster-containing ferredoxin-like protein. CooH and CooL, corresponding to HycE and HycG in E. coli hydrogenase 3, are related to the large and small subunits of standard [NiFe]-hydrogenases, respectively. However, the small subunit is considerably smaller and contains only the cysteine ligands for the proximal FeS cluster. The two subunits are more closely related to the energy-conserving NADH:quinone oxidoreductase (complex I) found in the respiratory chain in mitochondria and aerobic bacteria, and the sequence similarity is also valid for the hydrophobic subunits and CooU.49
Most of the members of this hydrogenase family have been found to be too labile to be purified intact, except the Ech enzyme from M. barkeri which was purified to homogeneity in a high yield.50 However, the characterization of the partly purified R. rubrum hydrogenase highlighted its unique resistance towards CO as well as a very high ratio of H2 evolution to H2 uptake activity.51 In 2002 Hedderich and collaborators successfully purified the whole “CO-oxidizing:H2 evolving” complex from C. hydrogenoformans, containing the monofunctional CODH, the ferredoxin CooF and the CO-induced hydrogenase. They showed that the three partners form a tight complex, accessible to further biochemical studies.52
5. Monofunctional [NiFe]-CODH: structure and mechanism
Ni-dependent CODHs reversibly catalyze the oxidation of CO to CO2, playing a central role in the global carbon cycle in anaerobic microorganisms.6 CODH can form either a monofunctional enzyme, as in the case of R. rubrum, or a bifunctional complex with acetyl-CoA synthase (ACS) such as the one involved in the Wood–Ljungdahl pathway used by acetogens. In this case, the CODH catalyzes the production of CO from CO2 to form acetyl-CoA at the ACS's active site.53 The most commonly described CODHs are the CODH/ACS complex from Moorella thermoacetica (MtCODH/ACS)54 and the monofunctional CODHs from Rhodospirillum rubrum and Carboxydothermus hydrogenoformans. In this chapter, we will focus on monofunctional CODHs. Five genes encoding CODHs have been identified in C. hydrogenoformans with different proposed functions, depending on their genomic context: CODH-I, involved in the WGS reaction; CODH-II, linked to NADH generation; CODH-III, coupled to ACS in the acetyl CoA pathway; and CODH-IV supposed to play a role in the oxidative stress response.55 No function has been found for CODH-V. On the other hand, R. rubrum possesses one CODH involved in the WGS reaction.56 Recently, the monofunctional CODH from Desulfovibrio vulgaris (DvCODH) was described, although its physiological function has not been clearly identified.575.1 Biochemical, structural and spectroscopic characterization
Ni-dependent CODHs are extremely oxygen-sensitive and their purification and manipulation require strictly anaerobic conditions in glove boxes. RrCODH was purified for the first time in 1984,58 followed by the 1st purification of ChCODH-I and ChCODH-II in 2001.59RrCODH and ChCODH-II represent today the best-studied monofunctional enzymes. The three enzymes are membrane-associated proteins that became solubilized upon cell disruption. The two ChCODHs are described as homodimeric NiFeS proteins in solution, with one atom of Ni per molecule of monomer. All described monofunctional CODHs share high specific activity for CO oxidation and high affinity for CO (Table 4). Three X-ray structures of monofunctional CODHs are available in the literature. RrCODH and ChCODH-II structures were solved in 2001 at 2.8 Å (ref. 60) and 1.63 Å (ref. 61) resolution, respectively, while the ChCODH-IV structure was determined at 2.52 Å resolution in 2017.55 The three enzymes display highly similar overall structures (root-mean-square deviation values of 1.16 Å for the ChCODH-IV/RrCODH dimer, considering Cα-atoms of 1206 residues, and 0.82 for the ChCODH-II/RrCODH monomer, considering Cα-atoms of 602 residues) with two identical intertwined subunits covalently bound (Fig. 5A). Each monomer comprises three domains: an N-terminal helical domain, and a middle and a C-terminal α/β (Rossmann-like) domains (Fig. 5B). Despite gel filtration experiments suggesting that RrCODH is a monomer,62 structural analyses evidenced that the enzyme is a homodimer, comparable to ChCODH (Fig. 5C). With an α2 conformation, monofunctional CODHs are considered as prototypes and good models for β2 CODH subunits of the CODH/ACS (αβ)2 tetramer found in acetogens. In 2001, the two structures revealed the unique nature of the active site (called the C-cluster), constituted of a [NiFe3S4] cluster, coordinated to a mononuclear Fe(II) site (Fe1) (Fig. 5D). The X-ray structures highlighted that the Ni ion is an integral constituent of the C-cluster. Initially, a [NiFe3S5] state was observed in the ChCODH-II structure61 but is no longer thought to be physiologically relevant.6 Additional Fe4S4 clusters are present to mediate electron transfer, generated during CO oxidation, from the C-cluster to the protein surface and subsequently to its electron transfer partner. In the WGS reaction, the electrons are mediated to a ferredoxin, CooF, which transfers electrons to the [NiFe]-hydrogenase. A total of five FeS clusters per dimer are present in CODH. In addition to the two C-clusters, the dimer harbors two other types of FeS clusters: two [Fe4–S4] clusters (B-clusters) and a [Fe4–S4] cluster bridging the two subunits (D-cluster) (Fig. 5C). The B-cluster position is in agreement with a role in electron transfer from the C-cluster to the protein surface, towards CooF. In contrast, the role of the D-cluster has not been well established: this cluster is not reducible at potentials as low as −530 mV and may play a structural or oxygen-protective role.63| Molecular weight, kDa | Specific activity (CO oxidation), U mg−1 | K m (CO), μM | k cat (CO), s−1 |
|
CO2 reduction activity, U mg−1 | Ref. | |
|---|---|---|---|---|---|---|---|
| a NR = non-reported, 1 U: 1 μmol of CO oxidized per min. | |||||||
| ChCODH-I native | 62.5 | 15756(70 °C), 1300(20 °C) |
30(70 °C) | 39000(70 °C) |
1.3 × 109 (70 °C) | NR | 59 |
| ChCODH-I recombinant | 8060(70 °C) | NR | NR | NR | NR | 83 | |
| ChCODH-II native | 64.5 | 13828(70 °C), 1000(20 °C) |
18(70 °C) | 31000(70 °C) |
1.7 × 109 (70 °C) | NR | 59 |
| ChCODH-II recombinant | 9600/12229(70 °C) |
NR | NR | NR | 16, 9(25 °C) | 120 | |
| ChCODH-IV recombinant | 68.2 | NR | 0.047(25 °C) | 14(25 °C) | 3 × 108 (25 °C) | NR | 55 |
| RrCODH native | 63.5 | 7700(25 °C) | 32(25 °C) | 8000 | 2.5 × 108 | 30, 1 | 121 |
| DvCODH recombinant | 67 | 1660(37 °C) | NR | NR | NR | 1, 4 | 57 |
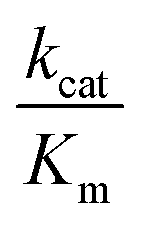
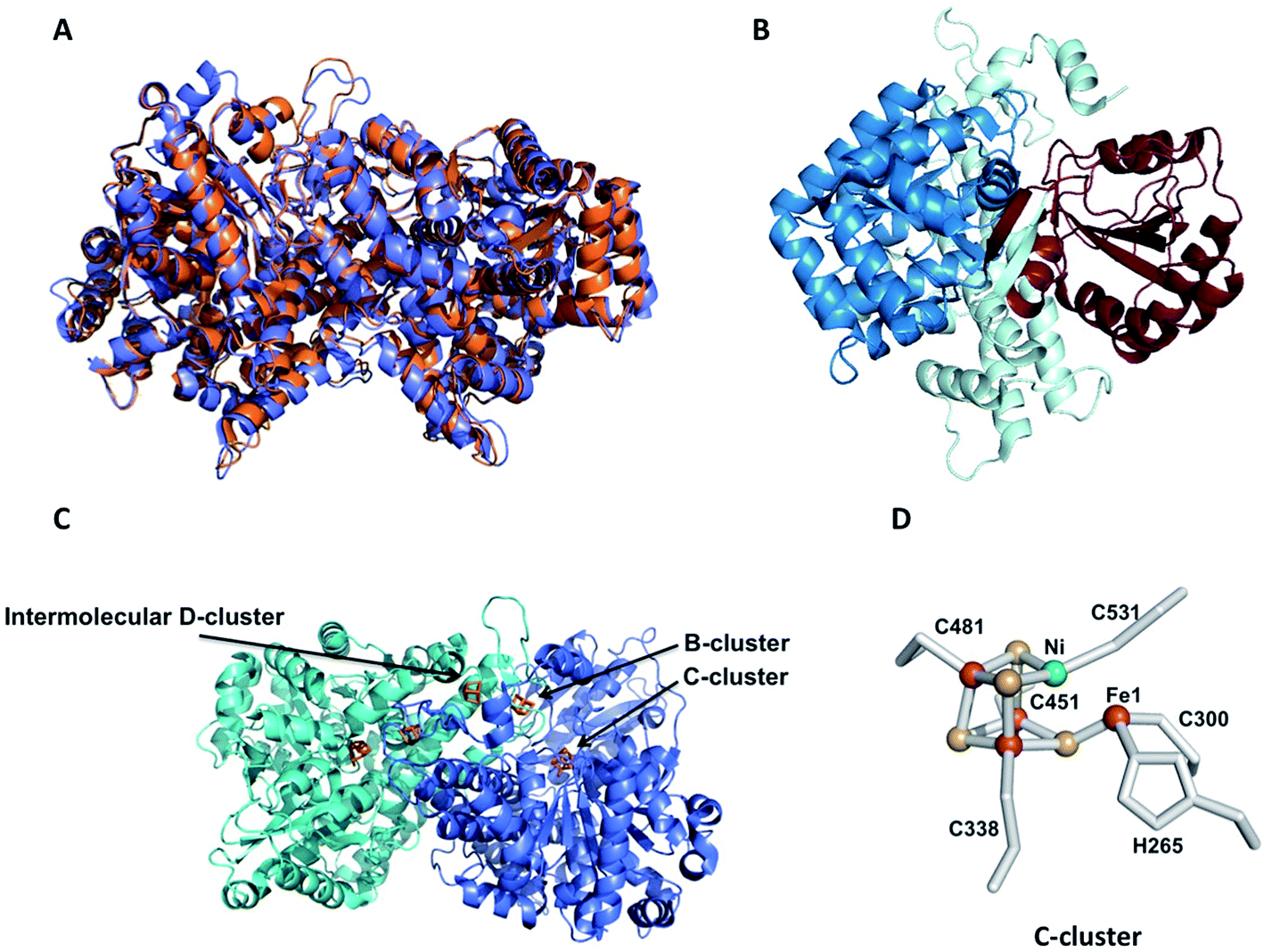 | ||
| Fig. 5 Crystal structures of monofunctional CODHs. (A) Superposition of the crystal structures of RrCODH (PDB code: 1JQK) in orange and ChCODH-IV (PDB code: 6ELQ) in blue; (B) ribbon drawing of the RrCODH monomer: the N-terminal domain is in pale cyan, and the Rossman domains are in blue and brown; (C) homodimer of RrCODH: the two monomers are in blue and cyan, and the Fe4S4 clusters (1 B-cluster per monomer and 1 intermolecular D-cluster) and C-clusters are depicted in orange; (D) C-cluster: nickel is depicted in green, iron in red and sulfur in yellow. | ||
Before the identification of the C-cluster revealed by the first crystal structures of CODH in 2001, extensive spectroscopic studies have been conducted on this enzyme.62,64–67 The presence of five metal centers in the protein dimer rendered the EPR analysis not straightforward. In RrCODH, the B-clusters have usual properties of the [4Fe–4S]2+/1+ cluster with midpoint potential close to −400 mV and gav ∼ 1.94. For the C-cluster, four different oxidation states have been proposed: a catalytically inactive and EPR-silent Cox state; a one-electron reduced Cred1 state, which binds CO and has an EPR signal with g-values at 2.03, 1.88, and 1.71; a two-electron-reduced EPR-silent Cint state; and a three-electron-reduced form, Cred2, which binds CO2 and has a distinct EPR signature with g-values of 1.97, 1.87, and 1.75.68 The electronic structure of these redox states is not clear yet; however, the majority of unpaired electron spin density is localized on Fe in both Cred1 and Cred2, which exhibit large 57Fe and small 61Ni hyperfine values.69
5.2 Reaction mechanism
The reaction mechanism of CODH has been extensively studied in the past decade, thanks to a combination of kinetic, theoretical and spectroscopic approaches. Great support was also given by the determination of several X-ray structures of ChCODH-II,70–73MtCODH/ACS74 and the CODH component of acetyl-CoA decarbonylase/synthase (ACDS)75 from Methanosarcina barkeri (MbCODH), in complex with substrates (CO or CO2) and inhibitors or analogues (CN− and NCO−). They permitted to propose a basic mechanistic model as follows. Several reaction intermediates have been described, postulating that CO binds to nickel and H2O binds to Fe1. After deprotonation of water, a nucleophilic attack of OH− on CO gives a Fe1–Ni-bridging COOH intermediate. A second deprotonation yields an Fe1–Ni-bridging COO− species, before the release of CO2 and the reduction of the cluster by two electrons.6 Electrons are then transferred to an external ferredoxin via Fe4S4 clusters. CODHs are mostly inactive at redox potentials higher than −300 mV. The Cox/Cred1 redox couple is −200 mV and the Cred1/Cred2 redox couple potential was reported to be −530 mV and matches well with the CO/CO2 redox potential. Cred1 (one-electron reduced), competent for CO oxidation, and Cred2 (three-electron reduced) for CO2 reduction are the two catalytically important oxidation states of the C-cluster (Fig. 6). However, some important mechanistic details remain unclear. The inhibitor study showed that CN− inhibits CO oxidation by binding to Cred1, but not CO2 reduction and that NCO− inhibits CO2 reduction by binding to Cred2, but not CO oxidation. The structure of CO− bound C-cluster showed that it binds Ni with bent geometry, completing a distorted tetrahedral geometry, and that the C atom is too far away from water-bound Fe1. Therefore, a “carbon shift” may occur to bring CO close to the Fe1-bound hydroxy group to allow the nucleophilic attack step.6 Moreover, the nature of the CO2 ligand bridged between Ni and Fe1, a key intermediate of the catalysis has not been yet clearly identified. In 2014, Ragsdale and collaborators published a thorough description of the reaction mechanism of CODH.68 Therefore, in this review, we detail only recently published data. In 2015, the determination of ChCODH-II structures in complex with CO2 or its analogue, NCO−, at atomic resolution gave new insights into the reaction mechanism.71 The improvement of the resolution to the atomic level permitted the description of molecular details of the C-cluster and structural analyses with unbiased determination of geometric parameters. To obtain the crystal structures of reduced CO2− and NCO−-bound states, ChCODH-II crystals were anaerobically poised to a redox potential of −600 mV (equivalent to the Cred2 state) by 5 mM titanium(III) citrate soaking, in the presence of NaHCO3 or KNCO, respectively. The 1.03 Å structure of CO2-bound and the 1.09 Å structure of NCO−-bound ChCODH-II showed a similar μ2, η2 coordination for CO2 and NCO− as bent bridging ligands between Ni and Fe1. CO2 is stabilized in the protein pocket by H bonding with His93 and Lys563. The geometry analysis of CO2 binding showed unexpected elongated and identical C–O bond lengths of 1.32 Å (bridging O atom) and 1.30 Å, while in μ2, η2 complexes, CO2 has generally one shorter and one longer C–O bond. The major breakthrough in this study was the observation of a change in the position of the C atom involving a change in the O–C–O angle, 15° smaller than the one previously reported at 130°. The 117° angle suggests a more reduced state of CO2, corresponding to a two-electron, rather than a one-electron, reduced state. Moreover, a short Ni–C bond of 1.81 Å was observed, indicating an unusual electronic structure, likely due to its integration in an FeS scaffold. It also indicates that the CO2 ligand is not protonated. The reaction intermediate trapped in the X-ray structure was then proposed to correspond to a two-electron reduced carboxylate, waiting to be protonated. From a bifunctional attack of a nucleophilic Ni center and an electrophilic Fe center, a flow of electrons from the C-cluster to CO2 occurs, favoring a two-electron transfer process. However, the existence of a transient anionic radical CO2˙− intermediate along the catalysis, reported previously, cannot be ruled out (Fig. 6).76 NCO− also undergoes a 2-electron reduction to yield a carbamoyl fragment (H2NCO+). A shift by 1.36 Å of His93 was observed, compared to free or CO2-bound enzymes, giving the required space for the two hydrogen atoms bound to the N atom, suggesting a protonation coupled with reduction process. However, further reaction of the bound carbamoyl did not occur in cristallo, while the reduction of cyanate to cyanide by ChCODH-II was evidenced in solution, using IR spectroscopy and isotopically labeled N13CO−. The same experiment performed on a single ChCODH-II crystal did not yield any signal corresponding to the CN-bound enzyme, showing that the reaction is impaired in the crystalline state.70 These unprecedented detailed studies are highly challenging in the field of complex multimetallic oxygen-sensitive enzymes and highlight the necessity of combining “in solution” and in cristallo approaches to unravel reaction mechanisms. | ||
| Fig. 6 Proposed reaction mechanism of [NiFe]-CODH (adapted from ref. 6). | ||
5.3 Oxygen inhibition
Iron and sulfur were used in the formation of FeS clusters within ancient proteins when the environment was anaerobic. Today, most of the FeS-containing proteins are recognized to be oxygen-sensitive by the formation of unstable forms of FeS clusters that quickly decompose upon oxygen exposure. In the case of enzymes containing several multimetallic cofactors, the understanding of their oxygen-sensitivity is often complex which may arise for a number of reasons. For example, B- and D-clusters of [NiFe]-CODHs could be directly oxygen-damaged, but the buried active site is also a potential target, since O2 can diffuse through the enzyme, via internal channels dedicated to substrates and product trafficking.The nature of the reactivity of DvCODH, ChCODH-II and ChCODH-IV with dioxygen has been investigated by protein film voltammetry (PFV).55,77 Previous studies have shown that only 10 s-exposure to air is sufficient to inactivate RrCODH and half-lives of 20 min and 60 min were measured for ChCODH-I and ChCODH-II exposed to air, respectively.59 The most O2-resistant enzyme described is ChCODH-IV that retains between 25 and 50% of its original activity after air-exposure. However, while DvCODH and ChCODH-II can be reactivated upon reduction, ChCODH-IV is irreversibly inactivated. Although the mechanism of reactivity of the enzyme towards dioxygen is unknown, three distinct inactive species have been identified thanks to PFV experiments by using a range of O2 concentration at different potentials: one that reactivates spontaneously once oxygen is removed, one that reactivates at reductive potential and one that is irreversibly inactivated.77 Thus, the sensitivity of CODH to O2 is not common to all enzymes: DvCODH reacts more slowly with O2 than ChCODH-II and can be fully reactivated by reduction, unlike ChCODH-II that is partly reactivated and ChCODH-IV that is not reactivated. Moreover, the use of an inhibitor in CO oxidation, which binds the active site, slows down the inhibitory effect of O2, demonstrating that the C-cluster is directly attacked.
6. CODH's active site biosynthesis
Understanding the biosynthesis of complex enzyme-bound heteronuclear metalloclusters, such as those found in nitrogenases, hydrogenases, ACS or CODH, evolved as one of the most challenging topics in the field of metalloenzymology. Given their complexity, the assembly and insertion of the active sites into the target enzyme require specific and elaborated maturation machineries. In the case of CODH, the identification of the dedicated accessory proteins, the characterization of the protein complexes involved in metal insertion and the sequence of the different events leading to the final active enzyme remain unclear. However, the development of the high-level production of fully active CODH for biotechnological applications requires a better understanding of the biosynthesis of the C-cluster.6.1 In vivo activation of CODH
Maintaining the appropriate intracellular concentration of essential metals while avoiding toxicity caused by overload is absolutely critical for cells. If the concentration becomes too low, the cell will suffer from the inactivation of essential metalloenzymes. However, non-physiological high intracellular concentrations of metals can either replace the native metals in essential enzymes, causing their inactivation, or catalyze the formation of highly toxic reactive oxygen species. Transporters, storage proteins and metalloregulators control the intracellular availability of nickel ions and protect cells against free toxic nickel. They work in concert with accessory proteins and metallochaperones that guide nickel to their specific target sites into enzymes. The study of nickel enzyme maturation systems, such as those of urease and hydrogenase, showed that a “nickel delivery complex”, involving the cooperation of several nickel-chaperones, is responsible for nickel insertion into the enzyme's active site.78 In R. rubrum, an operon named cooFSCTJ has been described downstream the hydrogenase operon. The coo operon contains five genes encoding the ferredoxin CooF (mentioned above), CooS, the structural gene of CODH, and three additional nickel proteins, CooC, CooJ and CooT.79 Both operons are regulated by a CO-sensing transcriptional activator, CooA, present downstream of the coo operon.80 CooA co-occurs with CODH operons in most of the CO-oxidizing bacteria. This homodimeric heme protein is a member of the cAMP receptor protein (CRP) family. CooA consists of two heme containing regulatory domains where CO binds and a DNA-binding domain.81 CO binding induces an allosteric conformational change allowing binding to specific target regions resulting in positive gene regulation. Phylogenetic analyses showed that CooC is widely distributed in microorganisms possessing a [NiFe]-CODH, generally in operons with CooS. In contrast, CooT and CooJ are more difficult to identify in genomes. When present, CooC has been shown to be essential for CODH maturation in the absence of high nickel concentration. For example, in R. rubrum, strains containing mutations in the cooC gene displayed minimal CODH activity and required an approximately 1000-fold higher extracellular Ni(II) concentration than the wild-type to restore anaerobic growth under CO and a partial CODH activity.82 Although not essential, CooJ and CooT are nevertheless relevant for nickel delivery to CODH since strains bearing polar insertions in the cooJ gene and polar or non-polar insertions in the cooT gene required 10-fold-higher Ni levels to grow, resulting in a reduced CODH activity. The activation of recombinant ChCODH-II or ChCODH-IV heterologously produced in E. coli does not require CooC: the supply of an excess of Ni(II) (500 μM) in the growth medium is sufficient to correctly activate CODH (Table 4).55,73 However, it does not exclude the involvement of accessory proteins under physiological growth conditions, since the high nickel concentration can compensate for the lack of accessory proteins, as previously shown for other Ni-enzymes. In contrast, when ChCODH-I was heterologously produced in Escherichia coli, its co-production with CooC3 is required and leads to an approximately 4-fold increase in CO oxidation activity. However, with a specific activity of 8060 U mg−1, this value is approximately half the one described for native ChCODH-I which is equal to 15756 U mg−1 (Table 4).83 This could suggest the requirement of other accessory proteins to produce a fully active enzyme. The role of CooC in Ni insertion was also demonstrated in DvCODH, heterologously produced in Desulfovibrio fructosovorans.57 Interestingly, despite similar overall structures, active sites and FeS-cluster organization, the maturation pathways of the different monofunctional CODHs diverge at the molecular level. The respective role of CooC, CooT and CooJ in bacteria needs to be determined (Fig. 7A). Concerning Fe and S insertion into the C-cluster, the co-production of ChCODH-I, II and IV with ISC machinery84 in E. coli leads to an active CODH with the expected 20 Fe and 20 S per dimer. No accessory protein dedicated to the building of the FeS unit of the C-cluster has been identified yet and the classical machinery involved in FeS biogenesis (ISC and/or SUF) seems to participate in the biosynthesis and insertion of the active site (Fig. 7A).
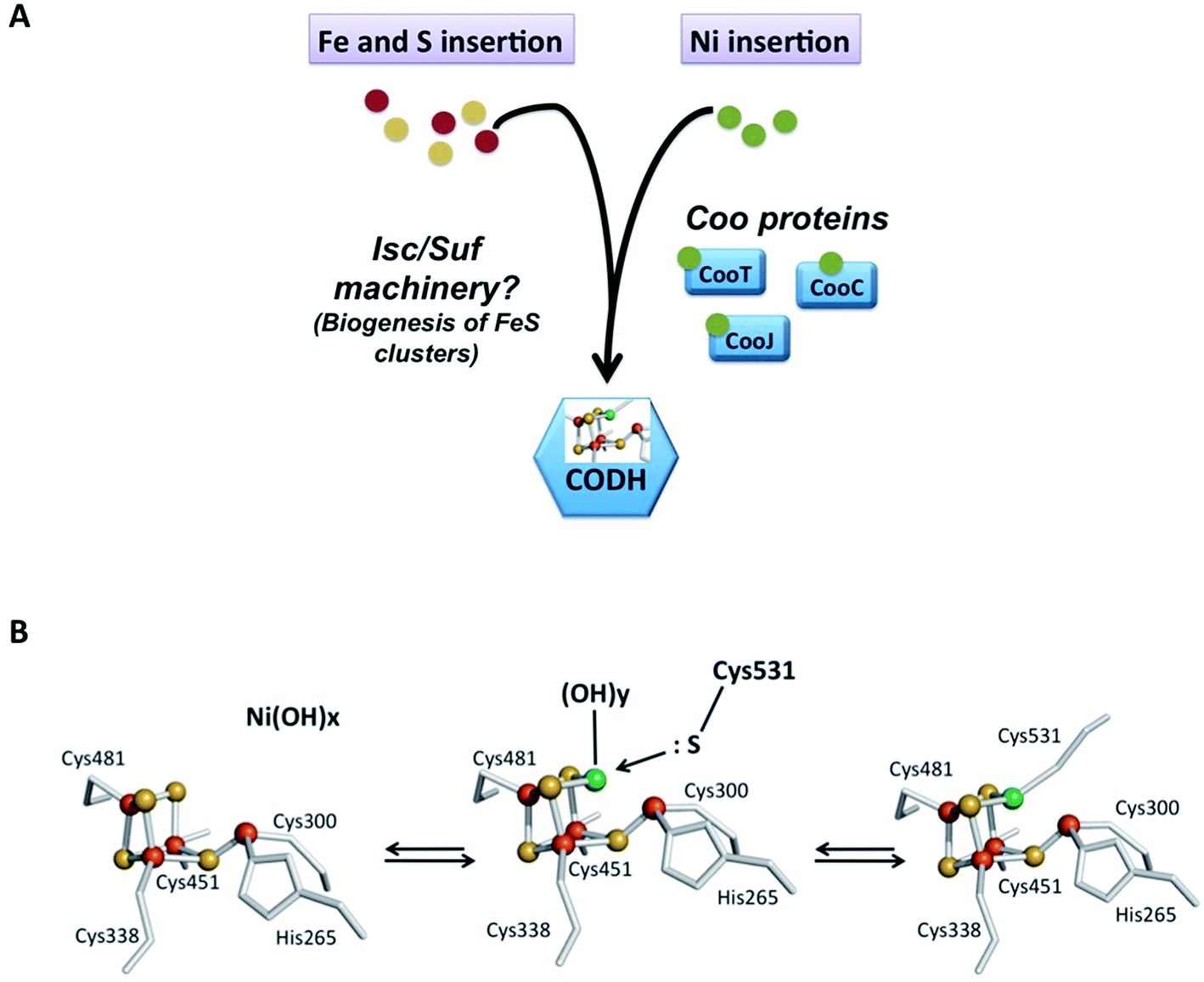 | ||
| Fig. 7 Metal insertion into CODH. (A) Schematic representation of Fe, S and Ni insertion and metallochaperones; (B) proposed model for the mechanism of Ni insertion into Ni free-CODH in R. rubrum, adapted from ref. 85. | ||
6.2 Maturation process
Previous studies showed that the “Fe/S unit” is inserted prior to nickel. Indeed, when RrCODH is produced in R. rubrum in nickel-depleted media, the purified enzyme corresponds to a nickel-free inactive form.65 EPR spectroscopy showed that the redox properties of B-clusters are similar in holo- and nickel-deficient enzymes.66 As expected, the presence of nickel dramatically impacts the midpoint potential of the C-cluster. Mössbauer studies showed that the mononuclear Fe site (Fe1) is not observed if nickel is not present and the spectroscopic properties of the Ni-free C-cluster would be consistent with those of tetracoordinated iron atoms. The addition of nickel salts in the presence of CO under reductive conditions to nickel-deficient CODH is sufficient to convert the Ni-free C-cluster into an active C-cluster. Therefore, nickel incorporation into the C-cluster likely alters the coordination environment of one iron atom. Site-directed mutagenesis of residues coordinating the C-cluster highlighted that the only critical residue required for Ni binding is His265 since the His265Val variant contains 0.24 Ni atoms per monomer.67 Surprisingly, all cysteine mutants, even the Cys531Ala variant affecting the residue directly involved in Ni coordination, contain one Ni per monomer.85 Therefore, cysteine ligands are crucial for CODH activity, but not for C-cluster formation. A model for nickel insertion into the C-cluster has been proposed in the literature but has not been experimentally validated yet (Fig. 7B). This model raised several questions: is an open distorted Fe4S4 cluster (with one iron atom linked with only one inorganic sulfur atom) a real intermediate of the maturation process? How does His265 direct the assembly of the C-cluster? If the present model is wrong with a classical cubic Fe4S4 cluster, nickel atom insertion implicates bond breaking. Whatever the mechanism, nickel insertion is the key step in CODH activation although actual data are insufficient to unravel the molecular mechanism of C-cluster formation, and further deep studies are required.7. Nickel chaperones involved in CODH maturation
7.1 CooC
CooC proteins belong to the SIMIBI-class of NTPases, which also contains UreG86 and HypB,87 involved in urease and [NiFe]-hydrogenase maturation, respectively. Today, NTPases are recognized as common components of maturation pathways of nickel enzymes.78 They potentially play a regulatory role by affecting the interaction of partners in protein complexes or nickel ion affinity, as recently observed in archaea, where the ATPase HypB is described as a metallochaperone enhancer, increasing the affinity of HypA for Ni(II) from 4 μM to 7 nM.88 The CooC family can be divided in two main groups: the “CooC-type” cluster, regrouping proteins involved in monofunctional CODH maturation and the “AcsF-type” cluster, postulated to be involved in either ACS/CODH or CODH activation.89 Three cooC genes were identified in C. hydrogenoformans: cooC1, cooC2 and cooC3. CooC1 and CooC2 are located in the same gene cluster as CODH-III and ACS, while CooC3 is co-expressed with CODH-I. Recently, CooC2 was purified and characterized as an AcsF-type enzyme, with a function associated with the maturation of ACS.89 CooC1 is a nickel-binding enzyme with a stoichiometry of one Ni(II) per dimer, coordinated by two cysteines per dimer within a conserved CXC motif present in all CooC homologues (Fig. 8A). Several X-ray structures of CooC1 were described in the presence or absence of ADP and/or Zn(II) (Fig. 8B).90 Apo-CooC1 is a monomer and dimerizes upon ADP and metal binding. The metal-binding site is located at the dimer interface (Fig. 8C). As observed for HypB, an intricate link between nucleotide and metal binding, metal-induced dimerization of the protein, and the presence of a mixture of monomeric and dimeric states of the NDP-bound form were observed. The only other CooC described in the literature is CooC from R. rubrum (RrCooC). RrCooC was purified directly from R. rubrum cultures.91 The protein is a homodimer in solution and was reported to associate with the membrane. Thus, its purification required a first step of solubilization from the membrane. However, RrCooC is not an integral protein and once released, the protein does not require the presence of detergent to be stable. The protein is an ATPase and its function is directly linked to nickel insertion into CODH.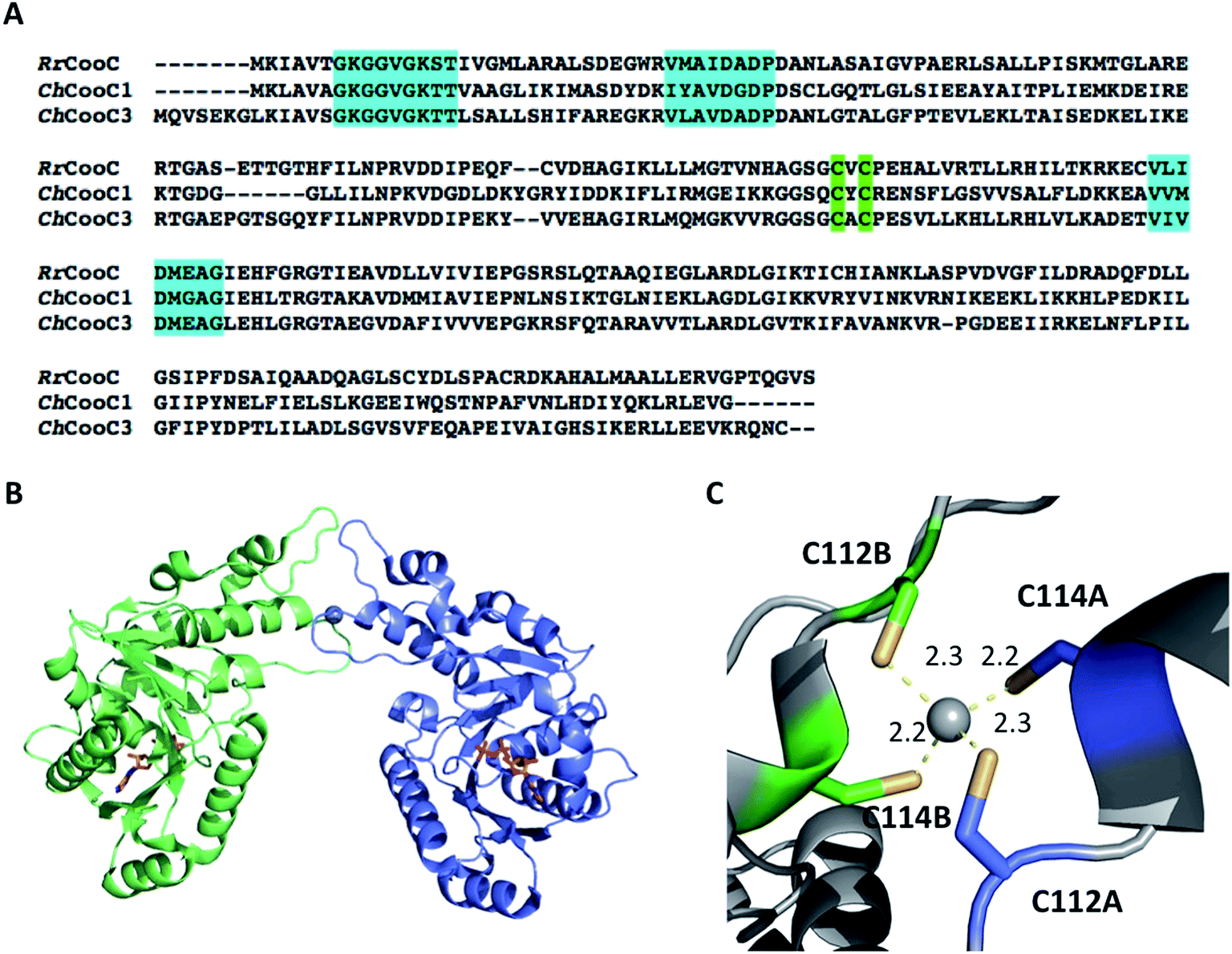 | ||
| Fig. 8 The NTPase CooC. (A) Sequence alignment of RrCooC (accession number: P31897), ChCooC1 (accession number: Q3ACS5) and ChCooC3 (accession number: Q3AB30). The nucleotide-binding site is highlighted in cyan, and the metal-binding site is highlighted in green; (B) overall structure of the ChCoo1 homodimer (PDB code: 3KJI). The two monomers are in green and blue, ADP molecules are depicted as orange sticks and Zn is depicted as a grey sphere; (C) metal-binding site at the dimer interface. | ||
7.2 CooJ
CooJ is a metallochaperone with only a very few biochemical data and no structural information available.92 This 12.5 kDa-protein contains a C-terminal nickel-binding domain with 16 histidines and 2 cysteines in the final 34 amino acids (Fig. 9). The protein was reported to bind four Ni(II) per monomer with a kD value of 4.3 μM, as shown by equilibrium dialysis. When CooJ was purified directly from R. rubrum cultures, the protein co-eluted with CODH, as shown by immunoblotting with an anti-CODH antibody, in association with other proteins, suggesting the formation of a multiprotein complex bound to CODH. Interestingly, Kerby et al. were able to show the RrCooJ's nickel-binding domain to be partially dispensable for its physiological function: a truncated version of CooJ with only 6 histidines and no cysteine in the C-terminal part is still able to activate CODH in vivo, suggesting that the His-tail could play another role in the bacterium, in addition to CODH maturation. Although not widely distributed, CooJ possesses common features with other nickel chaperones involved in Ni-enzyme maturation (Fig. 9). Some HypB and UreE93 (urease maturation) homologues possess a histidine-rich motif proposed to be involved in nickel storage, conferring them a second function in addition to their role in enzyme maturation. As for CooJ, the role of histidines in SlyD94 (hydrogenase maturation) and LarC95 (lactate racemase maturation) remains to be determined. The two unique proteins Hpn and Hpn-2 that contribute to nickel storage are also related to urease maturation in Helicobacter pylori. Both required and essential for H. pylori colonization, phylogenetic analyses revealed that they are restricted to gastric strains of Helicobacter.96 | ||
| Fig. 9 Histidine-rich proteins related to Ni-enzyme maturation. Histidines and cysteines are depicted in green and blue, respectively. Histidine-rich motifs are underlined. UniprotKB accession numbers: RrCooJ: P72321, RrHypB: Q2RXN8, UreE from Klebsiella aerogenes: P18317, SlyD from Escherichia coli: P0A9K9, LarC from Lactobacillus fermentum: V4XLJ5, Hpn from Helicobacter pylori: P0A0V6, and Hpn-2 from Helicobacter pylori: A3RDS2. | ||
7.3 CooT
RrCooT is a homodimeric all-β protein of 14 kDa with seven strands forming two bent anti-parallel β-sheets made of β6, β7, β1 and β2 (β-sheet I), and β3, β4 and β5 (β-sheet II), respectively, tightly packed on top of each other (Fig. 10A).97 The protein binds specifically one Ni(II) per dimer with a high affinity in the nanomolar range. In its amino acid sequence, two potential Ni(II)-binding residues are present, a cysteine in position 2 and a histidine in position 41 (Fig. 10B). Site-directed mutagenesis showed that the replacement of cysteine 2 by serine prevents Ni(II)-binding to CooT. Concerning Histidine 41, the X-ray structure of apo-RrCooT revealed that the residue is too far away from cysteine 2 to contribute together to a common nickel-binding motif (Fig. 10A). Moreover a conformational change in the β-sheet RrCooT's structure upon Ni(II) binding, to bring His41 close to Cys2, is highly unlikely. In turn, the two cysteines A and B from each monomer are very close to each other, suggesting the presence of a nickel-binding site formed by the two N-termini of the homodimer. In addition to cysteine 2, the amino terminal and/or the backbone amide group of close residues, such as Met3, are the putative ligands to complete the coordination of Ni(II) in a square-planar geometry, as shown by spectroscopic characterization. | ||
| Fig. 10 The nickel-binding protein CooT. (A) Crystal structure of the RrCooT homodimer; (B) amino-acid sequence of RrCooT; (C) occurrence of CooT in anaerobic archaea and bacteria. | ||
Initially, CooT was only identified in R. rubrum and Rhodopseudomonas palustris. However, the analysis of 2775 complete prokaryotic proteomes performed in 2017 allowed the identification of 111 CooT homologues, mainly present in anaerobic bacteria, but also in anaerobic archaea such as methanogens and hyperthermophilic species.97 In 83 proteomes, CooT co-occurs with CooS, suggesting that CooT should be considered as a potential actor with CooC in the maturation pathway of monofunctional CODHs. Moreover, it is worth mentioning that in the remaining 28 proteomes, CooT co-occurs with CooC and ACDS, mainly found in methanogens (22 proteomes), or with CooC and ACS/CODH in Thermodesulfovibrio yellowstonii and Butyrivibrio proteoclasticus. In the four remaining proteomes, the role of CooT needs to be elucidated (Fig. 10C). In any case, the presence of CooT is related to an anaerobic life style and to nickel metabolism. Based on their amino acid sequences, all CooT homologues harbor a single conserved RNA_bind_2 domain, predicting a role in RNA metabolism. However, electrophoretic mobility shift assay experiments with nucleic acids purified from R. rubrum showed that RrCooT could bind to neither RNA nor DNA, refuting this hypothesis.
In all CooT homologues, cysteine 2, a key residue in Ni(II)-binding, is strictly conserved, highlighting the existence of an novel Ni(II)-binding protein family linked to anaerobic metabolism in bacteria and archaea.
8. Summary and prospects
The development of next generation biofuel and bio-based chemical technologies, respecting green chemistry's principles, is a real challenge. In this field, synthetic biology has the potential to shift biology from a research field centered on the observation of nature to a discipline that highlights the design and construction of novel biological systems and organisms.In this review, we focused on the biologically mediated WGS reaction, involving two amazing enzymes, CODH and hydrogenase, containing remarkable active sites. These enzymes are involved in energy conversion processes and are often the most highly active and selective catalysts operating under mild conditions with small molecules that are known to be difficult to activate (such as H2 or CO2). Interest in hydrogenases and CODH has dramatically increased in recent years due to the hydrogen fuel initiatives and the development of new methods for CO2 sequestration. In the field of hydrogenases, these concerns led to the profusion in the literature of deep studies on the deciphering of their reaction mechanism, the synthesis of bio-inspired catalysts, and the assembly of their active site in the protein scaffold. The combination of all these approaches has been really successful in recent years with the development of new bio-inspired processes, including combining accessory proteins with active site synthetic analogues.98
In the case of CODH, the mechanism, the structure and the biosynthesis of its active site, unique in biological systems, intrigue both chemists and biologists. The major difficulties are related to the complexity of its multimetallic cofactor, and the attempts to synthesize bio-inspired models of CODH have failed. However, since CODH catalyzes reversibly the oxidation of CO, the full understanding of the C-cluster synthetic pathway would open new prospects for the synthesis of metal catalysts for both syngas upgrading and CO2 valorization. Today, the most active catalysts described are based on palladium phosphine complexes99,100 and there is still a need to explore bio-inspired metallic catalysts to efficiently and economically reduce CO2.
Besides the use of inorganic catalysts, the optimization of microorganisms capable of performing the WGS reaction and the engineering of CODH to enhance its activity will represent a great opportunity for biotechnological applications. However, bacteria that naturally produce CODH require complex growth conditions (temperature, strict anaerobic conditions, high CO concentrations, defined growth media, etc), limiting their use in bioreactors on an industrial scale. On the other hand, heterologous production of complex metalloenzymes is often limited because of the requirement of specific maturation machinery. However, recent advances highlighted the possibility to produce several fully active CODH in the easy-to-grow bacterium Escherichia coli. E. coli is a robust microorganism,101 able to grow easily and quickly, under aerobic or anaerobic conditions (using aerobic/anaerobic respirations or fermentation102), with a wide variety of energy sources, and is widely used in modern biotechnology. Therefore, the use of optimized genetically modified E. coli strains to perform a biological WGS reaction becomes worth considering.
To conclude, the combination of inorganic chemistry, biochemistry and microbiology would have great impacts on the design of new methods to optimize biological WGS reaction efficiency and its integration into biomass gasification devices.
Conflicts of interest
There are no conflicts to declare.References
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- D. Stefan, M. Michael, J. Holger, H. Marta, B. Andreas, P. Regina and W. Peter, ChemSusChem, 2017, 10, 42–47 CrossRef PubMed.
- O. Tirado-Acevedo, M. S. Chinn and A. M. Grunden, in Advances in Applied Microbiology, Academic Press, 2010, vol. 70, pp. 57–92 Search PubMed.
- A. M. Henstra, J. Sipma, A. Rinzema and A. J. Stams, Curr. Opin. Biotechnol., 2007, 18, 200–206 CrossRef PubMed.
- B. Lagoa-Costa, H. N. Abubackar, M. Fernández-Romasanta, C. Kennes and M. C. Veiga, Bioresour. Technol., 2017, 239, 244–249 CrossRef PubMed.
- Y. Kung and C. L. Drennan, Curr. Opin. Chem. Biol., 2011, 15, 276–283 CrossRef PubMed.
- D. B. Bukur, B. Todic and N. Elbashir, Catal. Today, 2016, 275, 66–75 CrossRef.
- G. K. Reddy and P. G. Smirniotis, Water Gas Shift Reaction, Elsevier, 2015, vol. 83 Search PubMed.
- B. Smith RJ, M. Loganathan and M. S. Shantha, Int. J. Chem. React. Eng., 2010, 8, 85201 Search PubMed.
- P. M. Maitlis and A. de Klerk, Greener Fischer-Tropsch Processes for Fuels and Feedstocks, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325–440 Search PubMed.
- G. C. Chinchen, R. H. Logan and M. S. Spencer, Appl. Catal., 1984, 12, 69–88 CrossRef.
- J. L. Ayastuy, M. A. Gutiérrez-Ortiz, J. A. González-Marcos, A. Aranzabal and J. R. González-Velasco, Ind. Eng. Chem. Res., 2005, 44, 41–50 CrossRef.
- D. C. Grenoble, M. M. Estadt and D. F. Ollis, J. Catal., 1981, 67, 90–102 CrossRef.
- D. Mendes, A. Mendes, L. M. Madeira, A. Iulianelli, J. M. Sousa and A. Basile, Asia-Pac. J. Chem. Eng., 2010, 5, 111–137 CrossRef.
- R. Koc, N. K. Kazantzis and Y. H. Ma, J. Loss Prev. Process Ind., 2011, 24, 852–869 CrossRef.
- S. Sato and J. M. White, J. Am. Chem. Soc., 1980, 102, 7206–7210 CrossRef.
- C. Yixuan, W. Zhaobin, C. Yanxin, L. Huaxin, H. Zupei, L. Huiqing, D. Yonglei, Y. Chuying and L. Wenzhao, J. Mol. Catal., 1983, 21, 275–289 CrossRef.
- D. Selvatico, A. Lanzini and M. Santarelli, Fuel, 2016, 186, 544–560 CrossRef.
- S. K. Sansaniwal, K. Pal, M. A. Rosen and S. K. Tyagi, Renewable Sustainable Energy Rev., 2017, 72, 363–384 CrossRef.
- Y. Lu, Q. Yan, J. Han, B. Cao, J. Street and F. Yu, Fuel, 2017, 193, 369–384 CrossRef.
- O. Bičáková and P. Straka, Int. J. Hydrogen Energy, 2012, 37, 11563–11578 CrossRef.
- S. K. M. R. Rittmann, H. S. Lee, J. K. Lim, T. W. Kim, J. H. Lee and S. G. Kang, Biotechnol. Adv., 2015, 33, 165–177 CrossRef PubMed.
- J. Sipma, A. M. Henstra, S. M. Parshina, P. N. Lens, G. Lettinga and A. J. M. Stams, Crit. Rev. Biotechnol., 2006, 26, 41–65 CrossRef PubMed.
- M. Diender, A. J. M. Stams and D. Z. Sousa, Front. Microbiol., 2015, 6, 1–18 Search PubMed.
- R. L. Kerby, S. S. Hong, S. A. Ensign, L. J. Coppoc, P. W. Ludden and G. P. Roberts, J. Bacteriol., 1992, 174, 5284–5294 CrossRef PubMed.
- J. E. Schultz and P. F. Weaver, J. Bacteriol., 1982, 149, 181–190 Search PubMed.
- R. L. Kerby, P. W. Ludden and G. P. Roberts, J. Bacteriol., 1995, 177, 2241–2244 CrossRef PubMed.
- H. Zhu, T. Wakayama, Y. Asada and J. Miyake, Int. J. Hydrogen Energy, 2001, 26, 1149–1154 CrossRef.
- G. D. Najafpour and H. Younesi, World J. Microbiol. Biotechnol., 2007, 23, 275–284 CrossRef.
- K. T. Klasson, K. M. O. Lundbäck, E. C. Clausen and J. L. Gaddy, J. Biotechnol., 1993, 29, 177–188 CrossRef.
- G. Najafpour, H. Younesi and A. R. Mohamed, Biochem. Eng. J., 2004, 21, 123–130 CrossRef.
- G. Najafpour, H. Younesi and A. R. Mohamed, Energy Sources, Part A, 2006, 28, 1013–1026 CrossRef.
- H. Younesi, G. Najafpour, K. S. Ku Ismail, A. R. Mohamed and A. H. Kamaruddin, Bioresour. Technol., 2008, 99, 2612–2619 CrossRef PubMed.
- A. H. Y. Ali Dadak, M. Aghbashlo, M. Tabatabaei and G. Najafpour, Environ. Sci. Technol., 2014, 33, 482–489 Search PubMed.
- O. Revelles, N. Tarazona, J. L. García and M. A. Prieto, Environ. Microbiol., 2016, 18, 708–720 CrossRef PubMed.
- S. M. Tiquia-Arashiro, Thermophilic Carboxydotrophs and Their Applications in Biotechnology, 2014, vol. 10 Search PubMed.
- V. A. Svetlichny, T. G. Sokolova, M. Gerhardt, M. Ringpfeil, N. A. Kostrikina and G. A. Zavarzin, Syst. Appl. Microbiol., 1991, 14, 254–260 CrossRef.
- M. Wu, Q. Ren, A. S. Durkin, S. C. Daugherty, L. M. Brinkac, R. J. Dodson, R. Madupu, S. A. Sullivan, J. F. Kolonay, D. H. Haft, W. C. Nelson, L. J. Tallon, K. M. Jones, L. E. Ulrich, J. M. Gonzalez, I. B. Zhulin, F. T. Robb and J. A. Eisen, PLoS Genet., 2005, 1, e65 Search PubMed.
- Y. Zhao, R. Cimpoia, Z. Liu and S. R. Guiot, Appl. Microbiol. Biotechnol., 2011, 91, 1677–1684 CrossRef PubMed.
- S. R. G. Ya Zhao, M. Haddad, R. Cimpoia and Z. Liu, Int. J. Hydrogen Energy, 2013, 38, 2167–2175 CrossRef.
- M. Haddad, R. Cimpoia and S. R. Guiot, Int. J. Hydrogen Energy, 2014, 39, 2543–2548 CrossRef.
- A. M. Henstra and A. J. M. Stams, Int. J. Microbiol., 2011, 2011, 1–4 CrossRef PubMed.
- O. Lazarus, T. W. Woolerton, A. Parkin, M. J. Lukey, E. Reisner, J. Seravalli, E. Pierce, S. W. Ragsdale, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14154–14155 CrossRef PubMed.
- K. S. K. Ismail, G. Najafpour, H. Younesi, A. R. Mohamed and A. H. Kamaruddin, Biochem. Eng. J., 2008, 39, 468–477 CrossRef.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814 CrossRef PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef PubMed.
- J. S. McDowall, B. J. Murphy, M. Haumann, T. Palmer, F. A. Armstrong and F. Sargent, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3948–E3956 CrossRef PubMed.
- J. D. Fox, H. E. Yiping, D. Shelver, G. P. Roberts and P. W. Ludden, J. Bacteriol., 1996, 178, 6200–6208 CrossRef PubMed.
- M. Jörn, B. Stefan, K. Jürgen, K. Andreas and H. Reiner, Eur. J. Biochem., 2001, 265, 325–335 Search PubMed.
- J. D. Fox, R. L. Kerby, G. P. Roberts and P. W. Ludden, J. Bacteriol., 1996, 178, 1515–1524 CrossRef.
- B. Soboh, D. Linder and R. Hedderich, Eur. J. Biochem., 2002, 269, 5712–5721 CrossRef PubMed.
- S. W. Ragsdale, L. Yi, G. Bender, N. Gupta, Y. Kung, L. Yan, T. A. Stich, T. Doukov, L. Leichert, P. M. Jenkins, C. M. Bianchetti, S. J. George, S. P. Cramer, R. D. Britt, U. Jakob, J. R. Martens, G. N. Phillips and C. L. Drennan, Biochem. Soc. Trans., 2012, 40, 501–507 CrossRef PubMed.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567–572 CrossRef PubMed.
- L. Domnik, M. Merrouch, S. Goetzl, J. H. Jeoung, C. Léger, S. Dementin, V. Fourmond and H. Dobbek, Angew. Chem., Int. Ed., 2017, 56, 15466–15469 CrossRef PubMed.
- D. Bonam, L. Lehman, G. P. Roberts and P. W. Ludden, J. Bacteriol., 1989, 171, 3102–3107 CrossRef PubMed.
- J. Hadj-Saïd, M. E. Pandelia, C. Léger, V. Fourmond and S. Dementin, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1574–1583 CrossRef PubMed.
- D. Bonam, S. A. Murrell and P. W. Ludden, J. Bacteriol., 1984, 159, 693–699 Search PubMed.
- V. Svetlitchnyi, C. Peschel, G. Acker and O. Meyer, J. Bacteriol., 2001, 183, 5134–5144 CrossRef PubMed.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11973–11978 CrossRef PubMed.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281–1285 CrossRef PubMed.
- D. Bonam and P. W. Ludden, J. Biol. Chem., 1987, 262, 2980–2987 Search PubMed.
- J. Feng and P. A. Lindahl, Biochemistry, 2004, 43, 1552–1559 CrossRef PubMed.
- P. Stephens, M. McKenna, S. A. Ensign, D. Bonam and P. W. Ludden, J. Biol. Chem., 1989, 264, 16347–16350 Search PubMed.
- D. Bonam, M. C. Mckennat, P. J. Stephenst and P. W. Ludden, Biochemistry, 1988, 85, 31–35 Search PubMed.
- N. J. Spangler, P. A. Lindahl, V. Bandarian and P. W. Ludden, J. Biol. Chem., 1996, 271, 7973–7977 CrossRef PubMed.
- N. J. Spangler, M. R. Meyers, K. L. Gierke, R. L. Kerby, G. P. Roberts and P. W. Ludden, J. Biol. Chem., 1998, 273, 4059–4064 CrossRef PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef PubMed.
- P. A. Lindahl, S. W. Ragsdale and E. Munck, J. Biol. Chem., 1990, 265, 3880–3888 Search PubMed.
- A. Ciaccafava, D. Tombolelli, L. Domnik, J. H. Jeoung, H. Dobbek, M. A. Mroginski, I. Zebger and P. Hildebrandt, Angew. Chem., Int. Ed., 2017, 56, 7398–7401 CrossRef PubMed.
- J. Fesseler, J. H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560–8564 CrossRef PubMed.
- J. H. Jeoung and H. Dobbek, J. Am. Chem. Soc., 2009, 131, 9922–9923 CrossRef PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef PubMed.
- Y. Kung, T. I. Doukov, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Biochemistry, 2009, 48, 7432–7440 CrossRef PubMed.
- W. Gong, B. Hao, Z. Wei, D. J. Ferguson, T. Tallant, J. A. Krzycki and M. K. Chan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9558–9563 CrossRef PubMed.
- M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 54, 8337–8339 CrossRef PubMed.
- M. Merrouch, J. Hadj-Saïd, L. Domnik, H. Dobbek, C. Léger, S. Dementin and V. Fourmond, Chem.–Eur. J., 2015, 21, 18934–18938 CrossRef PubMed.
- C. J. Zeer-Wanklyn and D. B. Zamble, Curr. Opin. Chem. Biol., 2017, 37, 80–88 CrossRef PubMed.
- R. L. Kerby and P. W. Ludden, J. Bacteriol., 1997, 179, 2259–2266 CrossRef PubMed.
- Y. He, D. Shelver, R. L. Kerby and G. P. Roberts, J. Biol. Chem., 1996, 271, 120–123 CrossRef PubMed.
- G. P. Roberts, R. L. Kerby, H. Youn and M. Conrad, J. Inorg. Biochem., 2005, 99, 280–292 CrossRef PubMed.
- W. B. Jeon, J. Cheng and P. W. Ludden, J. Biol. Chem., 2001, 276, 38602–38609 CrossRef PubMed.
- T. Inoue, K. Takao, F. Yuto, T. Yoshida and Y. Sako, Biosci., Biotechnol., Biochem., 2014, 78, 582–587 CrossRef PubMed.
- B. Roche, L. Aussel, B. Ezraty, P. Mandin, B. Py and F. Barras, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 923–937 CrossRef PubMed.
- W. B. Jeon, S. W. Singer, P. W. Ludden and L. M. Rubio, J. Biol. Inorg Chem., 2005, 10, 903–912 CrossRef PubMed.
- B. Zambelli, M. Stola, F. Musiani, K. De Vriendt, B. Samyn, B. Devreese, J. Van Beeumen, P. Turano, A. Dikiy, D. A. Bryant and S. Ciurli, J. Biol. Chem., 2005, 280, 4684–4695 CrossRef PubMed.
- A. M. Sydor, H. Lebrette, R. Ariyakumaran, C. Cavazza and D. B. Zamble, J. Biol. Chem., 2014, 289, 3828–3841 CrossRef PubMed.
- S. Watanabe, T. Kawashima, Y. Nishitani, T. Kanai, T. Wada, K. Inaba, H. Atomi, T. Imanaka and K. Miki, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7701–7706 CrossRef PubMed.
- C. M. Gregg, S. Goetzl, J. H. Jeoung and H. Dobbek, J. Biol. Chem., 2016, 291, 18129–18138 CrossRef PubMed.
- J. H. Jeoung, T. Giese, M. Grünwald and H. Dobbek, J. Mol. Biol., 2010, 396, 1165–1179 CrossRef PubMed.
- W. B. Jeon, J. Cheng and P. W. Ludden, J. Biol. Chem., 2001, 276, 38602–38609 CrossRef PubMed.
- R. K. Watt and P. W. Ludden, J. Biol. Chem., 1998, 273, 10019–10025 CrossRef PubMed.
- S. B. Mulrooney, S. K. Ward and R. P. Hausinger, J. Bacteriol., 2005, 187, 3581–3585 CrossRef PubMed.
- H. Kaluarachchi, J. W. Zhang and D. B. Zamble, Biochemistry, 2011, 50, 10761–10763 CrossRef PubMed.
- B. Desguin, P. Soumillion, P. Hols and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5598–5603 CrossRef PubMed.
- D. Vinella, F. Fischer, E. Vorontsov, J. Gallaud, C. Malosse, V. Michel, C. Cavazza, M. Robbe-Saule, P. Richaud, J. Chamot-Rooke, C. Brochier-Armanet and H. De Reuse, PLoS Pathog., 2015, 11, e1005312 Search PubMed.
- J. Timm, C. Brochier-Armanet, J. Perard, B. Zambelli, S. Ollagnier-de-Choudens, S. Ciurli and C. Cavazza, Metallomics, 2017, 9, 575–583 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef PubMed.
- A. Miedaner, C. J. Curtis, R. M. Barkley and D. L. DuBois, Inorg. Chem., 1994, 33, 5482–5490 CrossRef.
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345–3351 CrossRef.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef PubMed.
- G. Unden and J. Bongaerts, Biochim. Biophys. Acta, Bioenerg., 1997, 1320, 217–234 CrossRef.
- P. L. Spath and D. C. Dayton, Preliminary Screening – Technical and Economic Assessment of Synthesis Gas to Fuels and Chemicals with Emphasis on the Potential for Biomass-Derived Syngas, Golden, CO, United States, 2003 Search PubMed.
- A. Kunkel, J. A. Vorholt, R. K. Thauer and R. Hedderich, Eur. J. Biochem., 1998, 252, 467–476 Search PubMed.
- E. J. Wolfrum and A. S. Watt, Appl. Biochem. Biotechnol., 2002, 98–100, 611–625 CrossRef PubMed.
- Y.-K. Oh, Y.-J. Kim, J.-Y. Park, T. H. Lee, M.-S. Kim and S. Park, Biotechnol. Bioprocess Eng., 2005, 10, 270 CrossRef.
- S. N. Parshina, J. Sipma, Y. Nakashimada, A. M. Henstra, H. Smidt, A. M. Lysenko, P. N. L. Lens, G. Lettinga and A. J. M. Stams, 2018, 2159–2165.
- Y. Yoneda, T. Yoshida, S. Kawaichi, T. Daifuku, K. Takabe and Y. Sako, Int. J. Syst. Evol. Microbiol., 2012, 62, 1692–1697 CrossRef PubMed.
- T. G. Sokolova, J. M. González, N. A. Kostrikina, N. A. Chernyh, T. P. Tourova, C. Kato, E. A. Bonch-Osmolovskaya and F. T. Robb, Int. J. Syst. Evol. Microbiol., 2001, 51, 141–149 CrossRef PubMed.
- T. V. Slepova, T. G. Sokolova, A. M. Lysenko, T. P. Tourova, T. V. Kolganova, O. V. Kamzolkina, G. A. Karpov and E. A. Bonch-Osmolovskaya, Int. J. Syst. Evol. Microbiol., 2006, 56, 797–800 CrossRef PubMed.
- T. G. Sokolova, N. A. Kostrikina, N. A. Chernyh, T. P. Tourova, T. V. Kolganova and E. A. Bonch-Osmolovskaya, Int. J. Syst. Evol. Microbiol., 2002, 52, 1961–1967 Search PubMed.
- M. Balk, H. G. H. J. Heilig, M. H. A. van Eekert, A. J. M. Stams, I. C. Rijpstra, J. S. Sinninghe-Damsté, W. M. de Vos and S. W. M. Kengen, Extremophiles, 2009, 13, 885–894 CrossRef PubMed.
- J. I. Alves, A. H. van Gelder, M. M. Alves, D. Z. Sousa and C. M. Plugge, Int. J. Syst. Evol. Microbiol., 2013, 63, 4072–4076 CrossRef PubMed.
- T. G. Sokolova, N. A. Kostrikina, N. A. Chernyh, T. V. Kolganova, T. P. Tourova and E. A. Bonch-Osmolovskaya, Int. J. Syst. Evol. Microbiol., 2005, 55, 2069–2073 CrossRef PubMed.
- D. G. Zavarzina, T. G. Sokolova, T. P. Tourova, N. A. Chernyh, N. A. Kostrikina and E. A. Bonch-Osmolovskaya, Extremophiles, 2007, 11, 1–7 CrossRef PubMed.
- Y. Yoneda, T. Yoshida, H. Yasuda, C. Imada and Y. Sako, Int. J. Syst. Evol. Microbiol., 2013, 63, 3602–3608 CrossRef PubMed.
- G. J. Schut, G. L. Lipscomb, D. M. N. Nguyen, R. M. Kelly and M. W. W. Adams, Front. Microbiol., 2016, 7, 1–9 Search PubMed.
- M.-S. Kim, S. S. Bae, Y. J. Kim, T. W. Kim, J. K. Lim, S. H. Lee, A. R. Choi, J. H. Jeon, J.-H. Lee, H. S. Lee and S. G. Kang, Appl. Environ. Microbiol., 2013, 79, 2048–2053 CrossRef PubMed.
- T. G. Sokolova, C. Jeanthon, N. A. Kostrikina, N. A. Chernyh, A. V. Lebedinsky, E. Stackebrandt and E. A. Bonch-Osmolovskaya, Extremophiles, 2004, 8, 317–323 CrossRef PubMed.
- T. Inoue, T. Yoshida, K. Wada, T. Daifuku, K. Fukuyama and Y. Sako, Biosci., Biotechnol., Biochem., 2011, 75, 1392–1394 CrossRef PubMed.
- S. A. Ensign and P. W. Ludden, J. Biol. Chem., 1991, 266, 18395–18403 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |