
Water-tolerant lithium metal cycling in high lithium concentration phosphonium-based ionic liquid electrolytes†
Robert
Kerr
 a,
Nikhilendra
Singh
b,
Timothy S.
Arthur
b,
Thushan
Pathirana
a,
Fuminori
Mizuno
b,
Kensuke
Takechi
b,
Maria
Forsyth
a and
Patrick C.
Howlett
*a
a,
Nikhilendra
Singh
b,
Timothy S.
Arthur
b,
Thushan
Pathirana
a,
Fuminori
Mizuno
b,
Kensuke
Takechi
b,
Maria
Forsyth
a and
Patrick C.
Howlett
*a
aInstitute for Frontier Materials (IFM), Deakin University, 75 Pigdons Rd, Waurn Ponds, Victoria 3216, Australia. E-mail: Patrick.howlett@deakin.edu.au
bToyota Research Institute of North America, 1555 Woodridge Avenue, Ann Arbor, Michigan 48105, USA
First published on 3rd August 2018
Abstract
Cycling stability at high capacities and water-tolerance are two key properties for the operation of high-capacity lithium (Li) metal–air batteries. Here, we have demonstrated the cycling of Li metal at high rates and high capacities in a newly developed family of ionic liquids based on quaternary alkylphosphonium cations and the bis(fluorosulfonyl)imide anion. A high LiFSI salt concentration of 50 mol% gave the most favourable combination of performance and water-tolerance when compared to 33 mol% and 20 mol%. These high salt content electrolytes exhibited stable cycling at 1 mA cm−2, in 1 h steps for up to 250 cycles at room temperature in the presence of up to 5000 ppm water. The two smallest cations, triethylmethylphosphonium (P1222) and trimethylisobutylphosphonium (P111i4), showed significantly superior cycling capabilities than the larger tributylmethylphosphonium (P1444) and trihexyltetradecylphosphonium (P66614) cations. Furthermore, the two small phosphonium cations supported high current densities and more stable long-term cycling than the N-methyl-N-propylpyrrolidinium cation (C3mpyr), which is of similar molecular weight. Fourier transform infra-red spectroscopy was used to characterise the composition of electrode surface layers, while ex situ X-ray photoelectron spectroscopy showed that the presence of water affects the amount of decomposition products at the electrode surface. These benchmark cycling results represent a significant step forward in the development of water-tolerant electrolytes for Li metal-based batteries.
Introduction
Li–air batteries, when comprising a high-capacity Li metal anode, stand alone as having prospective practical energy densities (500–900 W h kg−1 (ref. 2)) nearly double that of today's benchmark Li-ion batteries (∼250 W h kg−1 (ref. 3)). However, there are a number of specific, yet significant, challenges that remain for a practical Li–air device. The unresolved issues of Li dendrite growth, poor cyclability and the ability to tolerate water that enters the cell when humid air is supplied to the air-cathode remain critical barriers to practical use. Lithium (Li) metal holds enormous promise as an anode material owing to the fact that it has the highest theoretical gravimetric energy density of all candidate materials (3860 mA h g−1). Considerable efforts have been devoted to developing electrolytes that can sustain high rates of Li metal cycling at practical rates without the risk of dendrite-initiated short circuiting that has plagued the commercialisation of Li metal anodes.In practice, limited success has been achieved by implementing a ceramic- or polymer-based (or some combination of both) electrolyte which can act as both a mechanical barrier to dendrite propagation and an impermeable barrier to water. Other methods involving coating or treating the Li metal surface have also demonstrated some level of success.4 A novel approach has been employed by Xu and co-workers whereby the hydrophobicity of SEI-forming additives was exploited in order to separate from the aqueous electrolyte and onto the graphite or lithium metal electrode surface.5 The state-of-the-art in this broad field has been recently summarised in comprehensive reviews by J. J. Zhang and Q. Zhang.6,7 However, these approaches have been unable to overcome the increased cell impedances that lead to limited rate capabilities for practical applications at ambient temperatures.
In a surprising result obtained recently, Li metal cycling was successfully carried out in a liquid electrolyte containing 1 vol% water using the ionic liquid (IL) N-methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl)imide (PP13TFSI) with 0.352 mol kg−1 of LiTFSI.8 Here, stable Li cycling was carried out for ∼120 cycles at 0.1 mA cm−2 with a 34 minute polarisation time. This seminal work has opened the door for researchers to examine ionic liquid electrolytes (ILEs) as a class of water-tolerant electrolytes and to understand the mechanisms that underpin this desired property.
Pyrrolidinium-based ILs have been the most extensively studied family of ILs for Li metal battery ILEs, showing much improved conductivity and electrochemical stability over preceding candidates such as the imidazolium family.9–12 Over the last two years, our group has published the first investigations into the Li metal cycling capability of a new class of ILs based on the family of tetraalkylphosphonium cations.13–15 Whilst the exact role of the cation is not yet clear to us, we have shown that the small phosphonium cation, trimethylisobutylphosphonium (P111i4), imparts superior ionic conductivity and Li metal cycling behaviour when compared to the leading pyrrolidinium counterpart, N-methyl-N-propylpyrrolidinium (C3mpyr).9,15–18 On the anion side, bis(fluorosulfonyl)imide (FSI) has been investigated as an alternative to TFSI due to its contribution to higher fluidity and conductivity – albeit with some sacrifice in thermal19 and oxidative stability.9 Nevertheless, the FSI anion allows for much higher Li+ concentrations than TFSI and also plays a key role in promoting outstanding surface layer properties on both Li and silicon electrodes.16,20–22
Over the past few years, work in our group has turned to the use of ILEs with high concentrations (>50 mol%) of Li salt, having demonstrated an unexpectedly high rate capability of Li metal cycling in C3mpyrFSI23,24 and P111i4FSI.15,18 This emerging class of mixed salt/IL (or solvent-in-salt) electrolytes has been found to benefit from a change in the Li+ transport mechanism at a high salt content leading to a higher Li+ transport number,25 while also forming superior surface layers on Li and silicon electrodes during cycling.18,22,23,25 These benchmark ILEs have achieved Li cycling rates that are approaching practical values at room temperature and are further enhanced at modestly elevated temperatures such as 50 °C.
Here, we present an electrochemical investigation into a series of tetraalkylphosphonium-FSI ILEs, namely methyltriethylphosphonium-FSI (P1222FSI), trimethylisobutylphosphonium-FSI (P111i4FSI), methyltributylphosphonium-FSI (P1444FSI), and trihexyltetradecylphosphonium-FSI (P66614FSI) and compare their ability to cycle Li metal. During the course of this study, we observed exceptional performance of these ILEs both with and without the presence of water. Thereafter, we sought to determine the key parameters that influence this ability such as Li+ concentration and the cation structure. Preliminary examination and comparison of the Li metal electrodes after cycling was carried out using FTIR and XPS to detect compositional or structural differences and relate them to either the differing cations or the presence of water.
Experimental
Three new ILs were synthesised for the initial series of electrochemical testing, each comprising a tetraalkylphosphonium cation and the bis(fluorosulfonyl)imide (FSI) anion: methyltriethylphosphonium (P1222FSI), tributylmethylphosphonium (P1444FSI), and trihexyltetradecylphosphonium (P66614FSI). The structures of the constituent ions are shown in Fig. 1. P1222FSI and P1444FSI were synthesised by anion exchange from the corresponding tosylate compounds (Cytec Industries). Trimethylisobutylphosphonium-FSI (P111i4FSI) was provided by Cytec and used without further purification,15 while the P66614 was exchanged from the chloride form (Cytec). N-Methyl-N-propylpyrrolidinium-FSI (C3mpyrFSI, Solvionic, 99.9%) was used as received. Purity was confirmed by bromide and potassium ion selective electrode measurements, differential scanning calorimetry, nuclear magnetic resonance spectroscopy, and mass spectroscopy. All ILs were dried under vacuum on a Schlenk line and the water content was measured using Karl Fischer titration to ensure that the levels were below 50 ppm.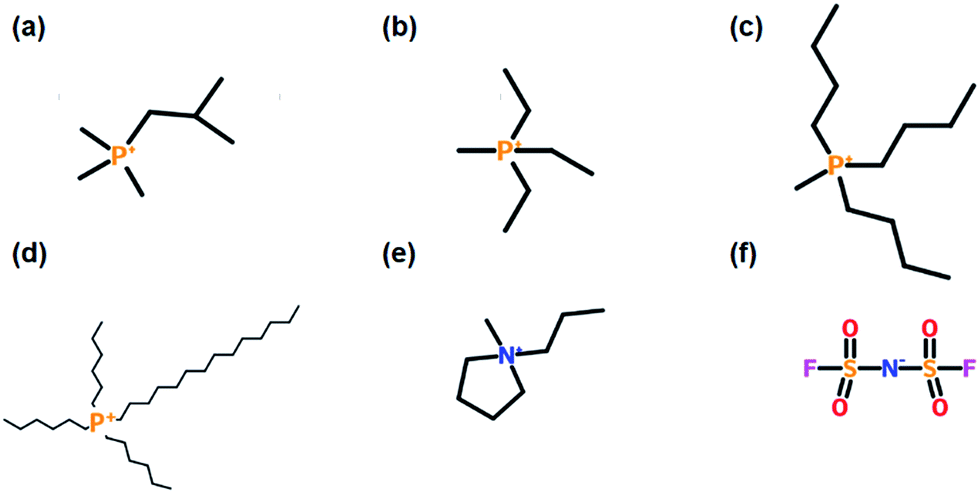 | ||
| Fig. 1 Chemical structures of the five cations used in this work: (a) P111i4, (b) P1222, (c) P1444, (d) P66614, and (e) C3mpyr, and (f) the FSI anion. | ||
LiFSI (Coorstek) was added to reach the desired molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, and 1:5 for Li+:P+ using Table 1. For each electrolyte composition, a working solution of a desired water concentration was prepared by diluting a stock ‘wet’ electrolyte (∼10000 ppm water) with the ‘dry’ electrolyte. The water content was then measured after electrochemical testing using Karl-Fischer titration. All Karl-Fischer measurements were within 10% of the quoted values throughout the text (within the experimental uncertainty of the Karl-Fischer measurement). The viscosity (rolling ball method: 2.5 mm diameter capillary) and density (oscillating U-tube principle) were recorded simultaneously using a DMA5000/Lovis 2000ME unit (Anton Paar) at a preset temperature interval. The setup allowed characterization under hermetically sealed conditions.
:1, 1:2, and 1:5 for Li+:P+ using Table 1. For each electrolyte composition, a working solution of a desired water concentration was prepared by diluting a stock ‘wet’ electrolyte (∼10000 ppm water) with the ‘dry’ electrolyte. The water content was then measured after electrochemical testing using Karl-Fischer titration. All Karl-Fischer measurements were within 10% of the quoted values throughout the text (within the experimental uncertainty of the Karl-Fischer measurement). The viscosity (rolling ball method: 2.5 mm diameter capillary) and density (oscillating U-tube principle) were recorded simultaneously using a DMA5000/Lovis 2000ME unit (Anton Paar) at a preset temperature interval. The setup allowed characterization under hermetically sealed conditions.
:P+ molar ratio expressed in mol kg−1 of LiFSI for three different molar ratios
| Molar ratio (Li+:P+) |
LiFSI concentration (mol kg−1) | ||||
|---|---|---|---|---|---|
| P1222FSI | P111i4FSI | P1444FSI | P66614FSI | C3mpyr | |
| 1:5 |
0.64 | 0.64 | 0.50 | 0.30 | 0.66 |
| 1:2 |
1.60 | 1.60 | 1.26 | 0.75 | 1.65 |
| 1:1 |
3.19 | 3.19 | 2.52 | 1.5 (insoluble) | 3.3 |
Electrochemical experiments were performed with a Biologic SP-200 potentiostat inside an Ar glovebox using a standard 3-electrode set-up consisting of a Li metal strip counter electrode, a Li metal strip reference electrode and a Ni working electrode (1.5 mm diameter, ALS Co., Ltd. Japan). Cyclic voltammetry was performed at 20 mV s−1, with room temperature measured as 20 ± 1 °C. The 10th cycle is presented throughout in order to compare values once stable cycling was achieved.
Symmetrical Li|Li 2032 coin cells were prepared using a Li strip (Sigma) lightly rolled and mechanically cleaned and then rinsed in hexane which had itself been dried extensively using calcium hydride and molecular sieves. 11 mm diameter electrodes were then punched out for assembly with a 14 mm diameter Celgard 3501 separator that was soaked in an ionic liquid electrolyte on a glass microscope slide sitting atop a hot plate set to 50 °C. A 0.5 mm spacer layer and a 1.4 mm spring were used in assembly, with final sealing performed using a Hohsen coin cell crimper. Long-term cell cycling was performed on a Neware BTS-3000 battery cycler. EIS measurements on Li|Li symmetrical cells were performed on a Biologic VMP3 multi-channel potentiostat (EC-Lab v10.44) in the frequency range of 500 kHz to 50 mHz (6 steps per decade) with an amplitude of 10 mV.
Scanning electron microscopy was performed on a JSM IT 300 series microscope and energy dispersive X-ray spectroscopy with an Oxford X-Max 50 mm2 EDX detector. FTIR was performed on a Perkin-Elmer Frontier spectrometer operating in ATR mode. 16 scans were recorded for each spectrum acquired at a resolution of 4 cm−1, with measurements taken inside an argon glovebox.
X-ray photoelectron spectroscopy (XPS) was performed using a Phi 5600 and analyzed using Multipack software. Spectra were aligned to the C1s signal at 284.7 eV. After electrochemical measurements, the lithium foil was washed thoroughly in dimethyl carbonate and allowed to dry. All processes were performed in an Ar-filled glove box. The samples were loaded into the XPS chamber in a sealed transfer vessel.
Results and discussion
Three-electrode electrochemical measurements
The deposition and stripping of Li metal through cyclic voltammetry on a Ni working electrode was performed in P1222FSI, P1444FSI, and P66614FSI electrolytes at three concentrations of LiFSI corresponding to Li+:P+ ratios of 1:1, 1:2, and 1:5 (the 1:1 concentration was above the solubility limit for LiFSI in P66614FSI). The resulting voltammograms (Fig. 2) show that the rate of plating and stripping is approximately five times higher for P1222FSI when compared to P1444FSI, presumably the result of the higher viscosity of the 1:1 P1444FSI electrolyte (391 mPa s for 1:1 P1222FSI compared to 813 mPa s for 1:1 P1444FSI at 20 °C). In both cases, the rate of deposition is observed to decrease with increasing salt concentration. However, lower Li+ concentrations result in Li+ transport limitations, especially in the case of Fig. 2(a), where a second reduction peak corresponding to the electroreduction of the electrolyte is observed at potentials lower than −0.2 V for 0.64 mol kg−1 LiFSI (red curve).
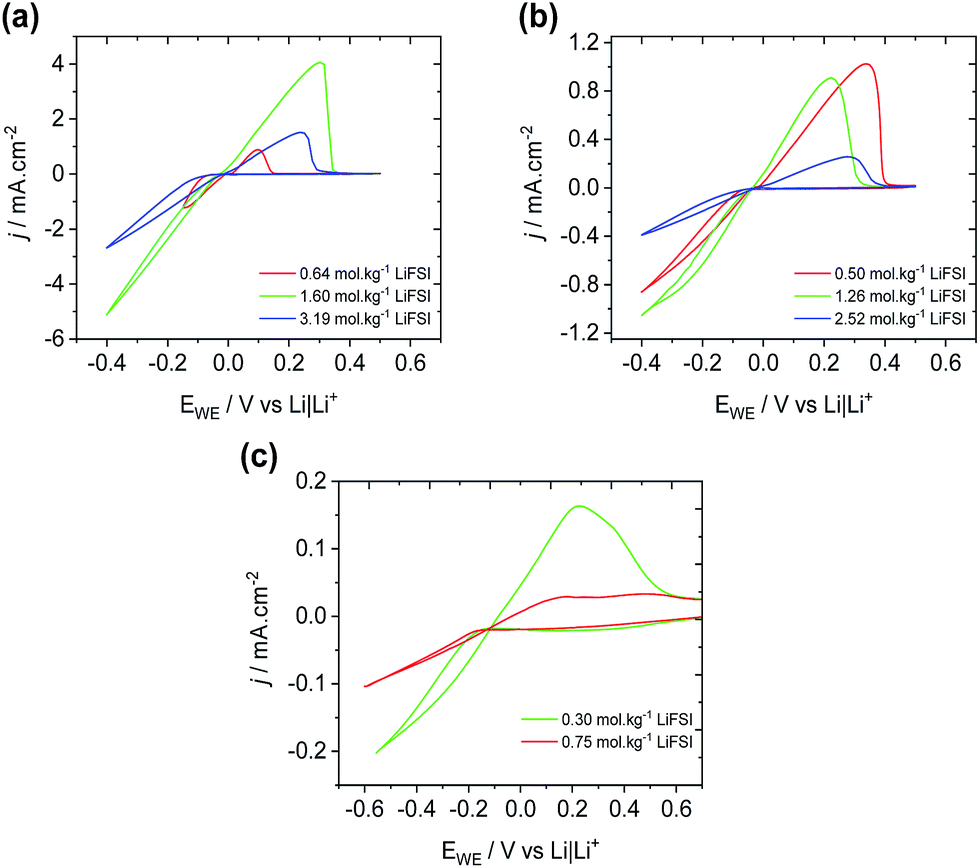 | ||
| Fig. 2 Cyclic voltammograms (10th cycle) of the deposition and stripping of Li from a nickel working electrode in (a) P1222FSI, (b) P1444FSI, and (c) P66614FSI with various amounts of LiFSI. Scan rate is 20 mV s−1 at room temperature. | ||
A series of CVs is presented in Fig. 3 where the cycling of Li is carried out in electrolytes with increasing water concentrations. Fig. 3(a) shows lithium cycling in P1444FSI with 2.52 mol kg−1 of LiFSI and an increasing amount of water up to 10000 ppm. The stability of the Li deposition over the 10 cycles was confirmed by the relatively stable currents upon deposition and stripping. Furthermore, it can be seen that the rate of Li deposition goes through a maximum at around 2000 ppm of water, after which both the deposition rate and the cycling efficiency drop. The deposition rate and cycling efficiency can be plotted versus the water content to show this trend more clearly for the different P1222FSI and P1444FSI ILE compositions (Fig. S1†). The surprising trend in performance can also be seen for the other P1222FSI and P1444FSI systems as shown in Fig. 3b–d, and is also observed for these same systems at 50 °C (Fig. S2†).
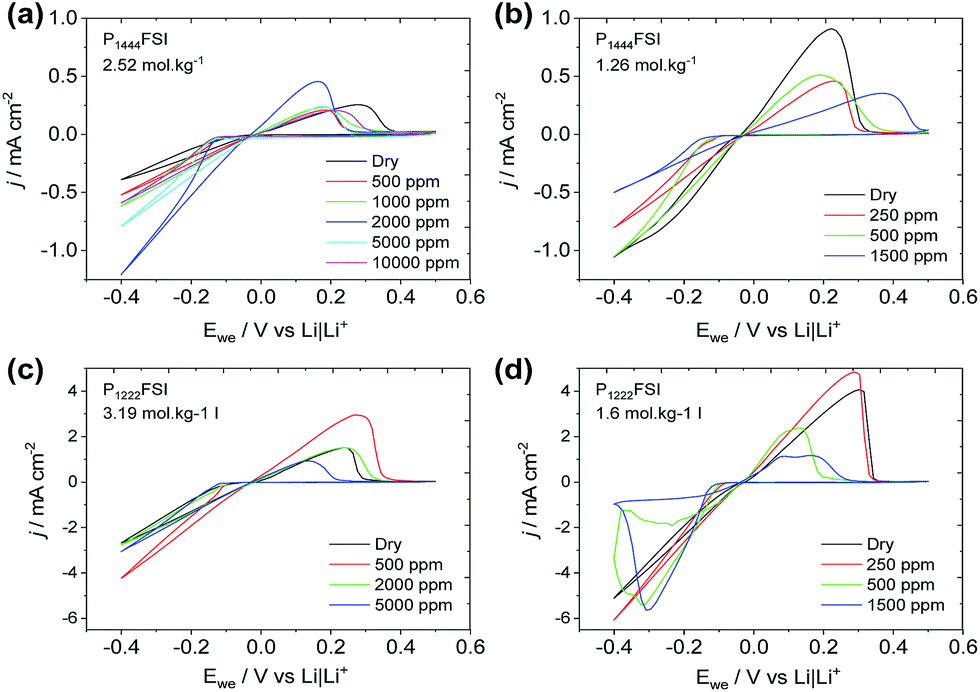 | ||
| Fig. 3 Cyclic voltammograms (10th cycle) of the deposition and stripping of Li from a nickel working electrode in P1444FSI with (a) 2.52 mol kg−1 or (b) 1.26 mol kg−1 of LiFSI, and P1222FSI with (c) 3.19 mol kg−1 or (d) 1.6 mol kg−1 of LiFSI at various concentrations of water. Scan rate is 20 mV s−1 at room temperature. | ||
For Li cycling CVs in Fig. 2 and 3, the maximum rate of deposition with respect to water concentration occurs at significantly higher water concentrations at the highest Li+ concentration (∼2000 ppm for 1:1 Li+:P+vs. ∼500 ppm for 1:2). There are two possible contributing factors for this observation; the high Li+ concentration reduces the electrochemical activity of water and thus prevents it from being reduced at the Li metal electrode, and the higher salt content aids in forming a denser, compact and protective SEI that blocks water from reaching the fresh underlying Li. These observations are consistent with the water-in-salt-electrolyte concept that has been developed previously26 and observed for the N-methyl-N-propylpiperidinium TFSI ionic liquid,8 where the drop in cycling efficiency at high water contents is presumably due to unbound water reaching the lithium metal surface where it can react chemically to generate LiOH at the electrode surface and hydrogen gas.
An analogous scenario has been shown to occur in the Li–S system where dissolved polysulfides are able to both participate in forming a stable SEI layer when present in low concentrations, while at high concentrations they etch the lithium metal surface.27 Thus, the role and mechanisms by which additives can influence the reactive lithium metal electrode are complex and worthy of further studies.
Symmetrical cell cycling
The upper limit of cycling performance for the two best-performing electrolytes (P1222 and P111i4) was determined using Li|Li symmetrical cell cycling. From the three-electrode measurements, it was found that Li cycling using the highest salt concentration (1:1) for each electrolyte gave the most favourable combination of cycling stability and water tolerance. P66614FSI was not investigated in symmetrical cell cycling experiments as the 1:1 electrolyte could not be prepared. It is also noted that the 1:1 P1444FSI electrolyte was unable to cycle at 1 mA cm−2 at room temperature.
Fig. 4(a) shows the cell cycling of P1222FSI and P111i4FSI with 1:1 LiFSI at 1 mA cm−2 with increasing step times from 1 h to 8 h. In both cases, stable cycling is achieved with overpotentials under 0.08 V up to a 4 h step time for 10 cycles. At an 8 h step time, the peak overpotentials increase significantly. After 4–5 cycles, evidence of soft short-circuiting is observed in both electrolytes as a sudden partial drop in overpotentials.28 This occurs when the dendrites have poor electrical contact with the Li metal electrodes, giving rise to erratic voltage profiles such as those seen here. The complementary experiment is shown in Fig. 4(b) where the current density is increased from 1 mA cm−2 to 8 mA cm−2 while holding the total charge per step constant (decreasing step time) at 1 mA h cm−2. Here, stable cycling is achieved up to 2 mA cm−2 (30 min step time). At 4 mA cm−2, a high demand for Li+ transport results in a large concentration gradient across the electrolyte and high overpotentials approaching 0.5 V. This type of behaviour promotes Li dendrite formation and ultimately leads to the short-circuiting that is observed at 8 mA cm−2.
 | ||
| Fig. 4 Symmetrical Li|Li cell cycling using (a) increasing step time at a constant current density of 1 mA cm−2 and (b) increasing current density at a constant 1 mA h cm−2 step capacity. | ||
The long-term cycling shown in Fig. 5 was performed at 1 mA cm−2 with a 1 h step time; parameters that fit comfortably within the performance limits for these electrolytes were chosen (based on Fig. 4). Here, we again focused on testing the electrolytes with a high salt content (1:1 Li+:P+) as they showed the most favourable combination of high cycle performance and high water tolerance. The cycling behaviour was recorded for 250 cycles and a comparison was made upon addition of 1000 ppm and 5000 ppm water. In order to ascertain whether the phosphonium cation plays an influential role in the stability of these electrolytes in the presence water, identical measurements were performed using the C3mpyr cation (which also has a very similar molecular weight to these small phosphoniums).
 | ||
| Fig. 5 Long-term symmetrical Li|Li cell cycling using a step time of 1 h at 1 mA cm−2 at ambient temperature. Cells were prepared using (a) P1222FSI, (b) P111i4FSI, and (c) C3mpyrFSI with 1:1 LiFSI and at various concentrations of water. (d) Coulombic efficiency measurements of the electrolytes with various concentrations of water in a Li|Cu cell cycled according to a protocol described in ref. 29. | ||
The cycling of the cells with dry P1222FSI and P111i4FSI in Fig. 5(a and b) continued beyond the 500 h shown here until at 1100 h (1100 mA h cm−2 cycled charge per electrode) the cycling was discontinued with no noticeable instability in the cycling behaviour (Fig. S3†). The overpotentials continued to grow in a steady manner until approximately 1000 h when they reached a plateau of 100 mV. For the similarly sized phosphonium cations, P111i4FSI (Fig. 5(b)), the overpotentials increased to nearly double that of the P1222 within the first 500 h. The overpotentials continued to increase up to 180 mV after nearly 1000 h of cycling at which point they started to decrease before undergoing short-circuit at 1100 h. The C3mpyr also displayed stable cycling over the first 500 h (after some brief periods of initial instability), albeit with a higher final overpotential of 130 mV.
The addition of 1000 ppm water had a relatively minor effect on the symmetrical cycling behaviour for the three electrolytes. Aside from the brief instances of what we believe to be SEI reorganisation (where the overpotentials are erratic in places but then regain stability) in the case of P111i4, the overpotentials generally remain steady. While the water content was not recorded after cycling, it is our expectation that only negligible amounts of water are consumed during cycling, as previously shown, and does not contribute to the initial instabilities.5 At 5000 ppm, however, a much more severe effect was seen on the long-term stability of the cells. The P1222 still shows stable cycling after 500 h, although there are signs of instability such as a sudden decrease in overpotentials. The P111i4 short circuits completely after 300 h, while the C3mpyr only manages stable cycling for approximately 10 cycles. It is noted here that the poor water-tolerance of C3mpyr is suggestive of the idea that the cation may play a role towards the water-tolerance of these electrolyte systems. The involvement of the cation in the surface structure of the lithium electrode surface is thus a question well worth exploring.
Li|Cu coin cells were constructed and cycled in accordance with Method 3 described in a recent paper by Adams et al. in order to determine the impact of water on the average coulombic efficiency (CE), which is a measure of the stability of the lithium electrode surface under cycling conditions.29 In this procedure, 4 mA h cm−2 of lithium was deposited onto the copper working electrode and then completely stripped. This pre-conditioning step is performed in order to condition the copper surface through the deposition of SEI products which may minimise further parasitic electrolyte reactions upon subsequent cycling. 4 mA h cm−2 was then re-deposited and a fraction of this (0.25) was then cycled at relatively modest current densities (0.4 mA h cm−2). The CE measurement using a 4 M LiFSI in dimethoxyethane (DME, Sigma) electrolyte was first carried out in order to directly compare our results against those in the original work where an average CE of 99.4% was achieved. Here, we obtained an average CE of 91.9% which indicates that the lithium metal deposition is not fully stabilised for the chosen copper foil substrate. While further work is required to refine our procedure, a comparison of the average CE values relative to each other is reported here. The P1222FSI with 3.2 mol kg−1 LiFSI achieved an average CE of 97.7% – significantly higher than that of the DME-based electrolyte. The addition of 1000 ppm water sees the average CE drop to 95.7%, while at 5000 ppm it drops further to 86.7%. A lower LiFSI content of 0.64 mol kg−1 lowered the average CE to 94.9%, with the addition of 1000 ppm water decreasing it further to 91.1%. This measurement supports the notion that a higher lithium salt content (i) improves cycling efficiency in a dry electrolyte and (ii) improves the water tolerance of the system. These results do, however, indicate that the presence of water does lower the cycling efficiency. As the parameters of this experiment are chosen well within the performance limits of these electrolytes, combined with the surface-sensitive nature of this measurement, we can conclude that the presence of water impacts the surface layer on the lithium electrode.
Surface characterisation
SEM images and ATR-FTIR spectra of the Li electrode surfaces were used to investigate the morphology and composition of these electrolytes. SEM images show that a compact and homogeneous surface layer is formed in the dry electrolyte. However, the addition of water sees the homogeneous surface become lightly covered with a dispersed layer of the deposit. This patchy, non-compact surface layer suggests that water may disrupt the formation of the deposited layer. While the initial cell overpotentials, in some cases, decreased upon addition of water (Fig. S4a†), the tendency of these cells to undergo failure by short-circuiting at such high water contents suggests that either the formed surface layers are detrimentally affected by the presence of water, or not as effective at preventing dendrite formation. The comparison of the patchy surface layers formed at low Li+ concentrations in both dry and wet electrolytes (Fig. S4b and c†) confirms that the structure of this layer is affected by both the Li+ and water concentrations.FTIR spectra were recorded for cycled lithium metal electrode surfaces having undergone 10 cycles at 1 mA cm−2, in 1 h steps in the ILEs with 1:1 LiFSI (the final step is deposition). As has previously been reported for pyrrolidinium-FSI and phosphonium-FSI based IL electrolytes, the formed surface layer is composed primarily of FSI anion reduction products (e.g., Li2S and Li2NSO2F) as well as Li2O, LiOH, Li2CO3 and LiF.30–32 Li surface spectra for C3mpyrFSI, P111i4FSI and P1222FSI (Fig. S5b†) do not show any differences in composition when comparing the most prominent peaks around 1359 cm−1, 1376 cm−1, and 1172 cm−1 for SO2 and SO2–N–SO2, respectively. The strong symmetric SO2 peak at 1170–1180 cm−1 (and asymmetric peaks at 1360–1370 cm−1) generally sits approximately 10 cm−1 higher for the cycled Li surfaces when compared to the corresponding ILE (Fig. S5a†). For the dry P1222FSI, however, the SO2 peaks do match those for the corresponding ILE. A closer examination of the fingerprint region for P1222FSI:LiFSI 1:1 in Fig. 6 can be used to probe any changes that occur upon addition of 5000 ppm water. The most pronounced change is a positive shift of the SO2 peaks by 10 cm−1, so that they lie in the same position as for the cycled Li metal surfaces in the other ILEs. The FTIR data here suggest that the P1222FSI may induce an –SO2– surface layer environment that is different to both the P111i4FSI and C3mpyrFSI. The addition of water affects this layer in such a way that the –SO2– reverts to an environment that is similar to the other ILEs.
 | ||
| Fig. 6 (a and b) SEI images and (c) FTIR-ATR spectra comparing the plated Li electrode surface after Li|Li cycling for 10 cycles at 1 mA cm−2 with a 1 h step time. Electrolyte is P1222FSI:LiFSI 1:1 (a) dry and (b) with 5000 ppm water. Vacc = 5 kV; scale bar = 200 μm. Dashed lines in the FTIR spectrum correspond to previously reported peak assignments for Li cycled in P111i4FSI.1 | ||
X-ray photoelectron spectroscopy was performed on select samples to further probe the effect of water on the composition of the surface layers. This technique was utilized to obtain information on the possible role of the ILE cation in surface layer formation by studying the F1s, S2p, P2p and Li1s spectra at various sputtering times (i.e. various depths into the surface layers formed). The samples were tested either after soaking in an ILE for 12 h or after cycling for 10 cycles at 1 mA cm−2 for 1 h in a select ILE as shown in Fig. 7. It should be noted that the atomic concentration of lithium is the deconvoluted peak-area for the metallic component of the Li1s spectra.
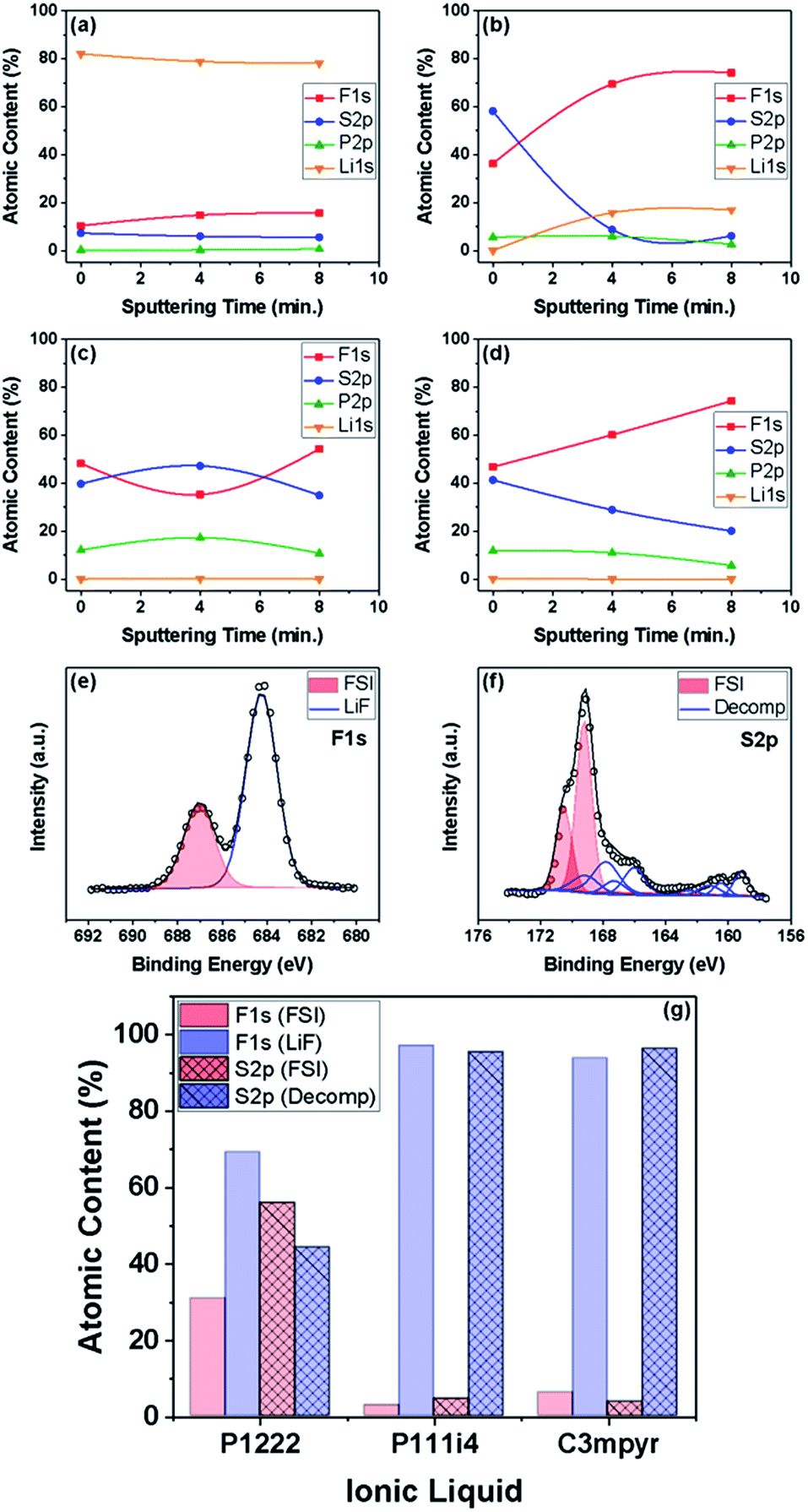 | ||
| Fig. 7 Compositional depth profile of a lithium metal electrode surface as determined from XPS spectra recorded at different Ar milling times: (a) soaked in 1:1 P1222FSI:LiFSI, (b) soaked in wet (5000 ppm water) P1222FSI:LiFSI, (c) cycled in 1:1 P1222FSI:LiFSI, and (d) cycled in wet (5000 ppm water) 1:1 P1222FSI:LiFSI. The F1s and S2p spectra for the sample cycled in dry 1:1 P1222FSI:LiFSI are shown in (e) and (f), respectively. Shown in (g) is a comparison between electrodes cycled in dry P1222FSI, P111i4FSI, and C3mpyrFSI with 1:1 LiFSI. | ||
In Fig. 7(a), spectra were recorded for a Li sample soaked in dry 1:1 P1222FSI:LiFSI. Here, no surface layer was observed to form, while the surface-residual presence of Li-based compounds (e.g. Li2S and LiF) was noted along with the absence of the phosphonium cation. In Fig. 7(b), we observed a sample soaked in wet 1:1 P1222FSI:LiFSI. Here, in contrast to Fig. 7(a), ILE presence on the surface was detected, suggesting a possible role played by water in promoting the decomposition of the ILE on Li metal surfaces. However, metallic lithium was observed after 4 minutes of sputtering. In Fig. 7(c) and (d), we observed a sample cycled in dry 1:1 P1222FSI:LiFSI and wet P1222FSI:LiFSI, respectively. The absence of metallic Li and the persistent phosphorous content with sputtering were evidence of a uniform surface layer formed by electrochemical cycling. The cation was found present in all samples other than the dry soaked sample where a surface layer was not observed to form. This suggests the possible involvement of the cation in the surface layer formed from 1:1 P1222FSI:LiFSI.
Peak deconvolution of high-resolution P2p and S2p peaks was performed to investigate chemical species present at the interface. The F1s and S2p spectra for the sample cycled in dry 1:1 P1222FSI:LiFSI are shown in Fig. 7(e) and (f), respectively. On closer inspection of the deconvoluted spectra, FSI and LiF characteristics were both found to be present in the total F content as seen in Fig. 7(e). Furthermore, the inspection of the deconvoluted S2p spectra in Fig. 7(f) confirmed the formation of a mixture of the FSI anion and decomposition products such as SOx and S2− species. The presence of these products in similar ILEs has been well studied in the literature,12,33 and the quantity, layering and mixture of the species have been suggested to affect the Li–metal cycling capability. A comparative quantification of surface components is provided in Fig. 7(g). Here, 1:1 P1222FSI:LiFSI was found to be the only ILE which highlighted a FSI/decomposed species mixture present in the surface layer. In comparison, 1:1 P111i4:LiFSI and 1:1 C3mpyr:LiFSI showcased a high content of LiF and other decomposed species with a lack of ILEs. Indeed, the presence and nature of the cation play a pivotal role in the observed surface layer formation and subsequent lithium cycling from ILEs.
These observations are consistent with the trends found in the EIS measurements (Fig. S6†) for the cycled Li|Li symmetrical cells using the P1222FSI and P111i4FSI with 3.2 mol kg−1 LiFSI electrolytes with various amounts of added water. Here, the spectra show an increase in the charge-transfer component (R2 − R1) upon addition of water, as summarised below in Table 2. This increase suggests that the presence of water results in a surface deposit that is more resistive for Li+ transport.
| Electrolyte | R1 (Ω) | R2 (Ω) | R2 − R1 (Ω) |
|---|---|---|---|
| P111i4FSI 1:1 dry |
20 | 48 | 28 |
| P111i4FSI 1:1 1000 ppm |
26 | 76 | 50 |
| P111i4FSI 1:1 5000 ppm |
36 | 110 | 74 |
| P1222FSI 1:1 dry |
18 | 29 | 9 |
| P1222FSI 1:1 1000 ppm |
22 | 83 | 61 |
| P1222FSI 1:1 5000 ppm |
23 | 86 | 63 |
While cycling a Li metal electrode in these ILEs in the presence of water may lead to the build-up of decomposition products at the electrode surface during cycling and result in a surface layer that is more resistive, the promising performances suggest that the modified surface film still offers similar or superior cycling stability, creating the opportunity to develop a truly water-tolerant system through traditional approaches such as modification of the initial surface layer through electrolyte additives and/or surface pre-treatments.
Conclusions
A recently developed class of ionic liquids based on tetraalkylphosphonium cations and the bis(fluorosulfonyl)imide anion were investigated and compared with a pyrrolidinium IL for their ability to cycle Li metal at high rates and charge. Cyclic voltammetry in these electrolytes showed that not only could the smallest alkyl chain lengths sustain higher cycling rates due to higher fluidity, but also the stable cycling of Li metal could be conducted in the presence of a small amount of water. The amount of water that could be added to these electrolytes was mostly dependent on the Li+ concentration, with the 1:1 Li+:P+ concentration showing the best combination of performance and water tolerance. The P1222 cation showed the highest degree of stability in symmetrical cell cycling studies, with a stable cycling of 1 mA h cm−2 for 250 cycles in the presence of 5000 ppm water. The P111i4 cation displayed similarly exceptional behaviour, while C3mpyr also performed well for the dry electrolyte but failed to cycle in the presence of 5000 ppm water. Coulombic efficiency measurements using Li|Cu cells revealed that the Li cycling efficiency is lowered upon addition of 1000 ppm water, suggesting that water does play a role in the surface stability of the lithium electrode. FTIR and XPS analyses of cycled electrodes suggest that the composition of the surface deposit is not impacted by the presence of water. Instead, the SEM images and XPS depth profiling suggest that water may facilitate a more rapid decomposition of ILEs with cycling leading to a more heterogeneous and less uniform surface layer. The resistivity, as measured by electrochemical impedance spectroscopy, of these surface layers was found to increase more rapidly than that of the electrodes cycled in the corresponding dry electrolytes.
Perhaps most interesting is the evidence provided through comparisons between phosphonium cations and a pyrrolidinium cation as to the role of the cation in forming a stable surface layer structure. XPS measurements were able to detect a higher involvement of the P1222 cation in the composition of the electrode surface layer, which may be a factor in its increased stability towards water.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. M. A. Girard, M. Hilder, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, J. Phys. Chem. C, 2017, 121, 21087–21095 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef PubMed.
- S. T. Myung, F. Maglia, K. J. Park, C. S. Yoon, P. Lamp, S. J. Kim and Y. K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef.
- J. Zhao, L. Liao, F. F. Shi, T. Lei, G. X. Chen, A. Pei, J. Sun, K. Yan, G. M. Zhou, J. Xie, C. Liu, Y. Z. Li, Z. Liang, Z. N. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 11550–11558 CrossRef PubMed.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2018, 1, 122–132 CrossRef.
- C. W. Sun, J. Liu, Y. D. Gong, D. P. Wilkinson and J. J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef PubMed.
- F. Mizuno, T. S. Arthur and K. Takechi, ACS Energy Lett., 2016, 1, 542–547 CrossRef.
- Q. Zhou, W. A. Henderson, G. B. Appetecchi, M. Montanino and S. Passerini, J. Phys. Chem. B, 2008, 112, 13577–13580 CrossRef PubMed.
- A. I. Bhatt, A. S. Best, J. H. Huang and A. F. Hollenkamp, J. Electrochem. Soc., 2010, 157, A66–A74 CrossRef.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97–A101 CrossRef.
- P. C. Howlett, N. Brack, A. F. Hollenkamp, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2006, 153, A595–A606 CrossRef.
- K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309–321 CrossRef.
- K. Tsunashima, Y. Sakai and M. Matsumiya, Electrochem. Commun., 2014, 39, 30–33 CrossRef.
- G. M. A. Girard, M. Hilder, H. Zhu, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 8706–8713 RSC.
- R. Kerr, D. Mazouzi, M. Eftekharnia, B. Lestriez, N. Dupre, M. Forsyth, D. Guyomard and P. C. Howlett, ACS Energy Lett., 2017, 2, 1804–1809 CrossRef.
- M. Hilder, G. M. A. Girard, K. Whitbread, S. Zavorine, M. Moser, D. Nucciarone, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Electrochim. Acta, 2016, 202, 100–109 CrossRef.
- M. Forsyth, G. M. A. Girard, A. Basile, M. Hilder, D. R. MacFarlane, F. Chen and P. C. Howlett, Electrochim. Acta, 2016, 220, 609–617 CrossRef.
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 162, 658–662 CrossRef.
- K. Yamaguchi, Y. Domi, H. Usui, M. Shimizu, K. Matsumoto, T. Nokami, T. Itoh and H. Sakaguchi, J. Power Sources, 2017, 338, 103–107 CrossRef.
- R. Bhattacharyya, B. Key, H. L. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef PubMed.
- J. F. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef PubMed.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef.
- H. Yoon, A. S. Best, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 4656–4663 RSC.
- L. M. Suo, Y. S. Hu, H. Li, M. Armand and L. Q. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef PubMed.
- C. Yan, X. B. Cheng, C. Z. Zhao, J. Q. Huang, S. T. Yang and Q. Zhang, J. Power Sources, 2016, 327, 212–220 CrossRef.
- W. Y. Li, H. B. Yao, K. Yan, G. Y. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef PubMed.
- B. D. Adams, J. M. Zheng, X. D. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2018, 8(7), 1702097 CrossRef.
- A. Budi, A. Basile, G. Opletal, A. F. Hollenkamp, A. S. Best, R. J. Rees, A. I. Bhatt, A. P. O'Mullane and S. P. Russo, J. Phys. Chem. C, 2012, 116, 19789–19797 CrossRef.
- A. Basile, A. I. Bhatt and A. P. O'Mullane, Aust. J. Chem., 2012, 65, 1534–1541 CrossRef.
- G. M. A. Girard, M. Hilder, N. Dupre, D. Guyomard, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, ACS Appl. Mater. Interfaces, 2018, 10, 6719–6729 CrossRef PubMed.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255–19269 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00159f |
| This journal is © The Royal Society of Chemistry 2018 |