
Experimental and first-principles study of a metal–organic framework with sulfur embedding cathode for enhanced performance lithium–sulfur battery†
Yan
Feng
 *a,
Yuliang
Zhang
a,
Guixiang
Du
a,
Jingbo
Zhang
a and
Xiaohui
Qu
*bc
*a,
Yuliang
Zhang
a,
Guixiang
Du
a,
Jingbo
Zhang
a and
Xiaohui
Qu
*bc
aCollege of Chemistry, Tianjin Normal University, Tianjin 300387, P. R. China. E-mail: hxxyfy@mail.tjnu.edu.cn
bEnergy Storage and Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: xqu1981@gmail.com
cCollege of Engineering Research, University of California, Berkeley, California 94720, USA
First published on 20th June 2018
Abstract
Lithium–sulfur (Li–S) batteries, which are well-known and much studied rechargeable batteries, are very promising because of their low cost, environmental friendliness, very high specific capacity and superior energy density. However, applications of Li–S batteries have been obstructed by their fast capacity fading and low coulombic efficiency due to soluble polysulfide migration during charge/discharge cycling. Herein, we present a strategy utilizing cobalt metal–organic framework (CoMOF) with rough porous surface and defective structure as a host material for sulfur accommodation, which implements a CoMOF and S composite (CoMOF–S) as a cathode. Capacity retention of CoMOF–S cathode surpasses that of pure sulfur by 87.18% after 100 cycles at 0.1C, while achieving coulombic efficiency above 98% at a high rate of 2.0C. We reveal structural features by combining X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations, which confirm the covalent bond connection between CoMOF and S. We attribute the excellent cycling performance to covalent bond immobilization of sulfur.
1. Introduction
Lithium–sulfur (Li–S) battery, as a high-specific energy rechargeable battery, is very attractive because it provides high theoretical specific capacity of 1675 mA h g−1 and theoretical specific energy of 2600 W h Kg−1, which is based on complete reaction of Li with S to form Li2S.1–5 Moreover, sulfur is environmentally benign and abundant in the earth's crust, providing cheap and promising cathode material for Li–S batteries. Despite these considerable advantages, there are still many challenges in the use of Li–S batteries. One of the main issues is the high solubility of polysulfide formed during charge/discharge cycling. The soluble intermediate Li polysulfides can diffuse through the organic electrolyte and migrate between cathode and anode (shuttle effect), which leads to low active materials utilization, low coulombic efficiency, and drastic capacity fading.6–8To address the abovementioned challenges, metal–organic frameworks (MOFs) have been used to accommodate S and reduce dissolution of Li polysulfides. MOFs are formed by linking organic and inorganic groups through covalent coordination linkages. In addition, other types of interactions such as H-bonding, π–π stacking and van der Waals forces may also play important roles in shaping the three-dimensional (3D) structures of the MOFs.9 Because of their tunable chemical composition and highly porous frameworks, in which fast mass transportation of related species is favorable, MOFs have been proved to be particularly suitable for electrochemical applications.10–12 A pioneering study focused on the utilization of MOFs as a confined matrix for sulfur impregnation was performed by Tarascon et al.13 The group employed a highly porous and hydrothermally stable Cr-MOF (MIL-100(Cr)) to encapsulate sulfur via melt diffusion process. Capacity retention was improved by using Cr-MOF compared with mesoporous carbon as host. Subsequently, Qian et al. utilized Cu-MOF (HKUST-1) as host material to trap sulfur and thus diminish the dissolution problem.14 Following these studies, many other MOFs, such as Zn-MOF (ZIF-8),15,16 Al-MOF (MIL-53, NH2-MIL-53),15 Cr-MOF (MIL-101),17 Ni-MOF,18 and Cu-MOF (MOF-525)19 have been used as sulfur confinements. Nevertheless, there is still a need to find suitable MOFs as sulfur hosts to further improve the electrochemical performance to meet the demands of high-capacity and high-power energy Li–S batteries. Moreover, physical and chemical interactions between MOF and S still need to be explored.
In this study, we report the preparation of cobalt MOF (CoMOF) with rough porous surface and defective structure as a matrix host to accommodate and confine sulfur. By sulfur-melt-diffusion method,20,21 sulfur effectively impregnates and absorbs into the CoMOF structure. Electrochemical performance results show that the CoMOF and S composite exhibits high specific capacity, improved cycle life, and high coulombic efficiency during high rate charge/discharge process. In addition, we investigated the chemical interaction/connection between the CoMOF host and sulfur both by X-ray photoelectron spectroscopic (XPS) analysis and density functional theory (DFT) calculations.
2. Experimental section
2.1 Chemicals and reagents
Co(NO3)2·6H2O was purchased from Tianjin Fuchen Chemical Reagents Factory, China. Benzene-1,3,5-tricarboxylic acid and sulfur were purchased from Tianjin Jiangtiantongyi Technology Co. Ltd., China. Dimethylformamide (DMF) and KOH were purchased from Tianjin Fengchuan Chemical Reagent Technology Co. Ltd., China. Polyvinylidene fluoride (PVDF) was purchased from Kureha America Corporation, USA. N-Methyl-2-pyrrolidone (NMP, anhydrous, 99.5 wt%) was purchased from Sigma-Aldrich, USA. Acetylene black was purchased from Denka, Japan. The separator (Celgard 2400) was obtained from Celgard, US. Lithium foil was purchased from FMC-Lithium Corporation, USA.2.2 Synthesis of CoMOF
A mixture of 5.08 g of Co(NO3)2·6H2O, 3.83 g of benzene-1,3,5-tricarboxylic acid, 0.76 g KOH, and 50 mL of deionized water was added to a flask and stirred for 60 min at room temperature. Then, the flask was tightly sealed and refluxed at 180 °C for 5 h. Following this, the mixture was cooled naturally to room temperature. The precipitate was filtered and washed with deionized water and then dried at 40 °C overnight in a vacuum oven to obtain a pink powder labeled as CoMOF.2.3 Synthesis of CoMOF–S
CoMOF powder and sulfur powder were mixed in weight ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, and 1:3. First, they were ground by hand in an argon filled glovebox. Then, the three mixtures were transferred into separate Al2O3 crucibles in an alumina tube furnace. The mixture samples were heated at 155 °C for 10 h under argon flow in the furnace. After cooling naturally, homogeneous dark-pink powders were obtained. The powders were ground in mortars and labeled as CoMOFS1, CoMOFS2, and CoMOFS3 for CoMOF:S weight ratios of 1:1, 1:2, and 1:3, respectively.
:1, 1:2, and 1:3. First, they were ground by hand in an argon filled glovebox. Then, the three mixtures were transferred into separate Al2O3 crucibles in an alumina tube furnace. The mixture samples were heated at 155 °C for 10 h under argon flow in the furnace. After cooling naturally, homogeneous dark-pink powders were obtained. The powders were ground in mortars and labeled as CoMOFS1, CoMOFS2, and CoMOFS3 for CoMOF:S weight ratios of 1:1, 1:2, and 1:3, respectively.
2.4 Fabrication of cathode materials
CoMOF–S cathodes were fabricated by mixing CoMOF–S powders with acetylene black and PVDF binder in the weight ratio of 8:1:1. Dissolved in NMP, mixtures were stirred at room temperature into a uniform slurry. Then, they were coated on an aluminum foil current collector with a Mitutoyo doctor blade and an Elcometer motorized film applicator. Mass loading of CoMOF–S is ∼1.5 mg cm−2. After the NMP dried, the laminated electrode was further dried in vacuum at 50 °C for 24 h. For comparison, pure sulfur cathode was prepared by the same procedure.
2.5 Cell assembly and testing
2325 coin cells (Panasonic) were assembled in an argon filled glovebox with oxygen concentration of 0.1 ppm and dew point of −70 °C. CoMOF–S electrode was punched to 1/2 inch OD and the counter electrode was a lithium metal disk of 11/16 inch OD. The separator used was polypropylene film (Celgard 2400). The electrolyte for cell testing was composed of 1 M lithium salt bis(trifluoromethane sulfonyl)imide (LiTFSI) dissolved in a mixture of 1,3-dioxolane (DOL) and dimethoxyethane (DME) (1:1 in volume).
2.6 Cell testing
Galvanostatic charge–discharge cycling tests were carried out on a Maccor Series 4000 battery test system in a thermal chamber at 30 °C. Voltage window for cell test is 1.5–3.0 V. Cyclic voltammetry (CV) test was performed on a VMP galvanostat/potentiostat workstation (Bio-Logic Corporation, USA) at a scan rate of 0.02 mV s−1 at potential interval between 1.5 and 3.0 V. Electrochemical impedance spectroscopy (EIS) was conducted on a CHI660A workstation (Shanghai Chenhua, China) with a frequency range of 100 kHz to 10 mHz. Cells were cycled at 0.1C (1C = 1675 mA h g−1) for the first cycle, and then brought to half lithiation for the second cycle and left undisturbed for 4 h before EIS measurements.2.7 Material characterization
X-ray diffraction (XRD) measurements were recorded on a Rigaku D8A X-ray diffractometer (Bruker, Germany) using CuKα radiation (λ = 0.154 nm) in the 2θ range of 5–70°. Thermogravimetric analyses (TGA) were performed under N2 using a TGA Q600 system (TA Instruments, US). The samples were heated from room temperature to 600 °C at a heating rate of 10 °C min−1. N2 adsorption–desorption isotherms were collected at liquid nitrogen temperature using a NOVA surface area analyzer (Quantachrome, USA). Pore size distributions were determined by the Barrett–Joyner–Halenda (BJH) method. XPS measurements were performed on an X-ray photoelectron spectrometer (PHI-5000, USA) using a monochromatic Al Kα (1486.7 eV) source at a voltage of 15 kV and emission current of 10 mA. Morphologies of samples were characterized with a scanning electron microscopy system (SEM, FEI Nova Nano 2300) with accelerating voltage of 15 kV. For post-cell-test analysis, cycled Li–S cells were opened in an argon-filled glovebox and the electrodes were washed thoroughly with DOL/DME with a volume ratio of 1:1.
2.8 Computation details
XPS binding energy (BE) was predicted using eqn (1):| BE = EC 1s + Ecorr | (1) |
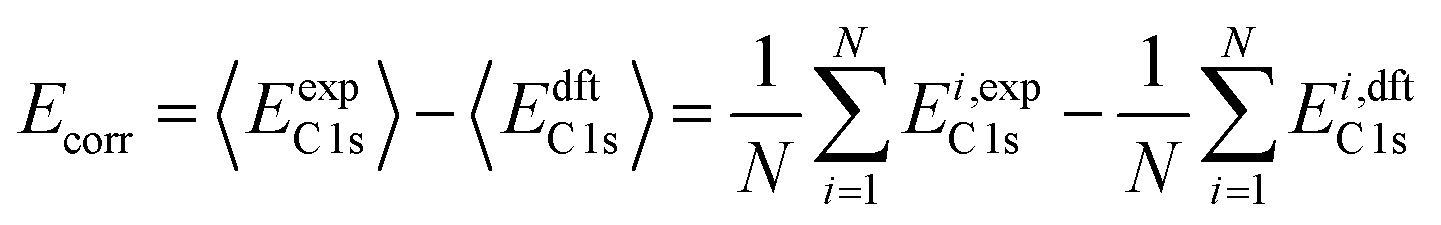 | (2) |
The structures of all the species were fully optimized using TPSSh density functional in combination with the 6-31+G(d,p) basis set.24 A larger basis set 6-311++G(d,p) was used in the single point energy calculation and orbital energy analysis. Orbital energies were extracted from canonical molecular orbitals. All quantum mechanical calculations were carried out using Gaussian 09.25
3. Results and discussion
Three CoMOF–S composite materials with different CoMOF:S weight ratios (1:1, 1:2, and 1:3) were prepared by sulfur-melt-diffusion method and are denoted as CoMOFS1, CoMOFS2, and CoMOFS3, respectively, in this study. Preparation details are described in methods section. SEM images are shown in Fig. 1 to characterize microscopic morphology and structural information of CoMOF–S composites. In case of pristine CoMOF (Fig. 1a and b), the as-synthesized product mainly consists of irregular rod-shape crystal structures. The lengths of CoMOF crystals are distributed in the range from 20 to 50 μm with diameters within 5–10 μm. In the enlarged SEM image of CoMOF (Fig. 1c), which is the rectangular area of Fig. 1b, many macroscopic pores and crystal defects exist on the surfaces of the crystals. In case of CoMOFS2 (Fig. 1d), the crystal sizes are much smaller than those of pristine CoMOF crystals. This is caused by the preparation process. Before sulfur melting, CoMOF and sulfur mixtures were ground by hand in the argon filled glovebox. Thus, crystal sizes of CoMOF–S composite become smaller compared with those of pristine CoMOF. Fig. 1e shows the enlarged area of Fig. 1d, in which many smaller crystal rods adhere to the surface of large CoMOF crystals. Sulfur is filled in the pores and tightly aggregates on the defects of CoMOF crystals (Fig. 1f, enlarged rectangular area of Fig. 1e). From the corresponding element energy diffraction spectrum (EDS) mapping of CoMOFS2 (Fig. 1g), the homogeneous distribution of Co, C, O, and S elements can be clearly observed, which further confirms that sulfur particles embedded uniformly into the MOF structure, exhibiting close contact between the two.
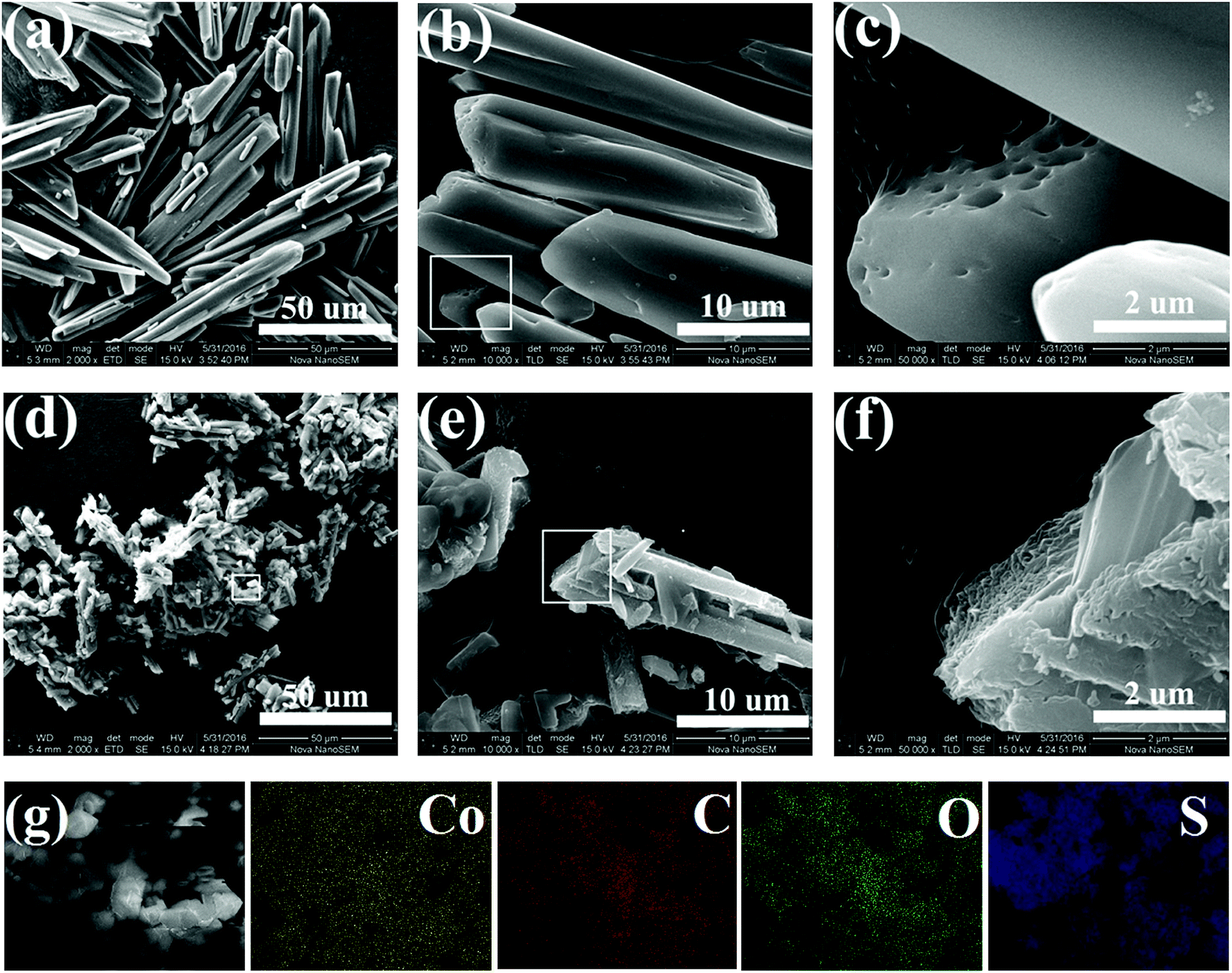 | ||
| Fig. 1 SEM images of CoMOF (a–c) and CoMOFS2 (d–f) materials; (g) elemental mapping images of CoMOFS2. | ||
Fig. 2a shows the XRD patterns of CoMOF and CoMOF–S composite materials. After sulfur impregnation at 155 °C, the diffraction peaks of crystalline sulfur are clearly seen in the CoMOF–S composite materials. However, intensity of crystalline CoMOF peaks is low, mainly because a large number of sulfur crystal diffraction peaks are covered up by the MOF structure. Sulfur ratios in CoMOF–S composite materials were determined by TGA under N2 atmosphere, as shown in Fig. 2b. Below 200 °C, the observed mass loss most likely corresponds to the loss of physically absorbed water on the CoMOF material. From 200 °C to nearly 300 °C, there is almost no weight loss for CoMOF, while pure sulfur exhibits one-step weight loss. This indicates that the weight loss at 200–300 °C can only be attributed to the content of sulfur in the composite materials. Based on these findings, the calculated weights of sulfur in CoMOF–S composites are 39.0 wt%, 56.6 wt%, and 65.6 wt%, corresponding to CoMOFS1, CoMOFS2, and CoMOFS3, respectively.
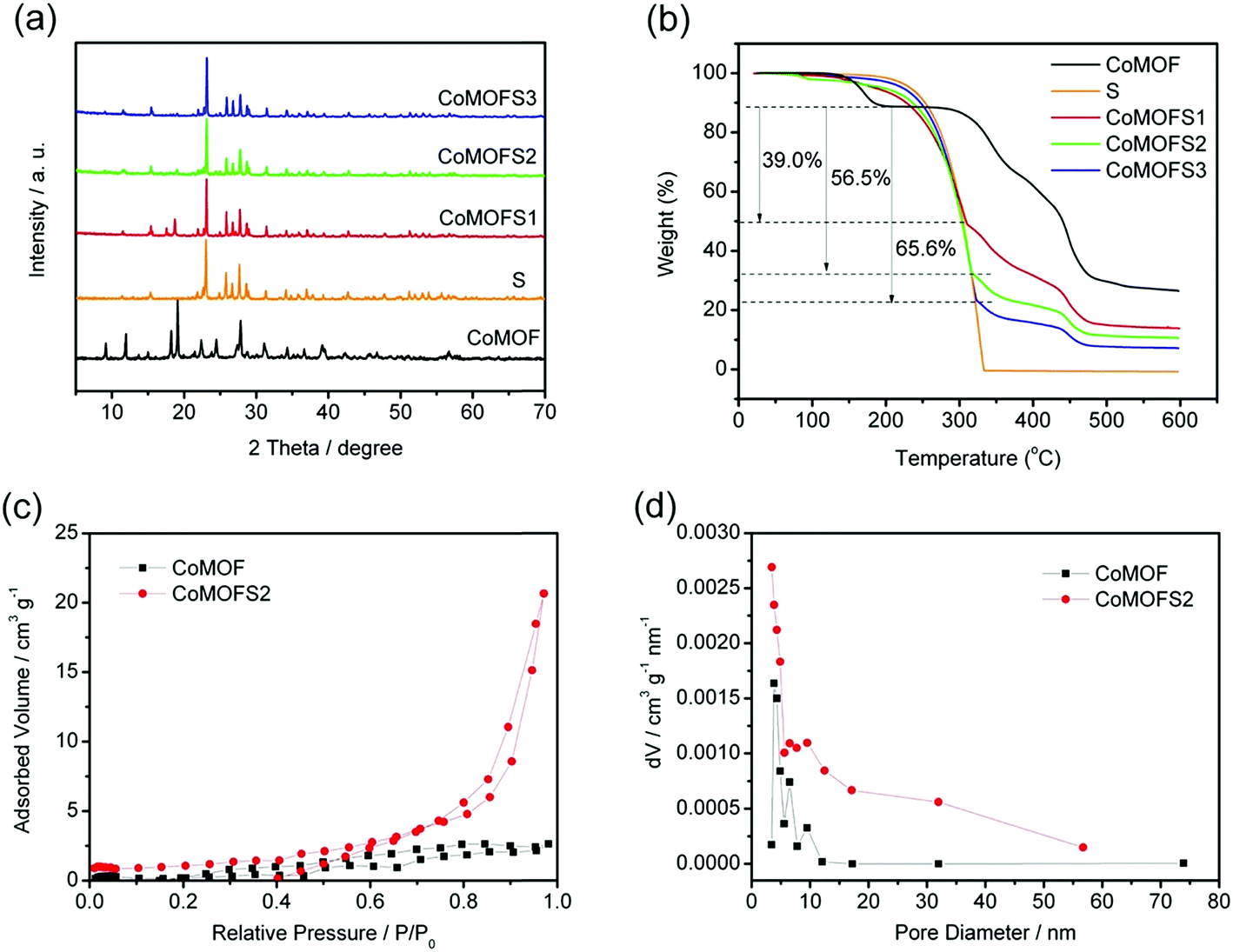 | ||
| Fig. 2 Characteristics of CoMOF and CoMOF–S materials: (a) XRD patterns; (b) TGA curves in nitrogen at a heating rate of 10 °C min−1; (c) N2 adsorption and desorption isotherms of CoMOF and CoMOFS2; and (d) BJH pore-size distribution plots of CoMOF and CoMOFS2. | ||
Porosity of CoMOF and CoMOFS2 was estimated by N2 adsorption–desorption analysis, as shown in Fig. 2c. CoMOF and CoMOFS2 exhibit type-IV isotherms with a hysteresis loop in the relative pressure range of 0.8 to 1.0, indicative of inhomogeneous mesoporous structure. The pore size distribution curves of CoMOF and CoMOFS2, as shown in Fig. 2d and calculated from the adsorption branch by the BJH method, display mesoporous pores with typical sizes of 3–10 nm (CoMOF) and 3–30 nm (CoMOFS2). Both the BET surface area (4.106 m2 g−1) and BJH surface area (13.077 m2 g−1) of CoMOFS2 became larger than those of pristine CoMOF (0.874 m2 g−1 and 3.095 m2 g−1, respectively), while the average pore diameter of CoMOFS2 (3.428 nm) became slightly smaller than that of CoMOF (3.823 nm). This phenomenon may be caused by sulfur impregnation. In the heating process at 155 °C, some part of the sulfur is embedded into the CoMOF matrix, resulting in the decrease in pore diameter of CoMOF; the rest of the sulfur may cover the surface of CoMOF crystals, leading to amplification of surface area.
Fig. 3a shows the cycle performances of CoMOF–S cathodes at charge/discharge current density of 0.1C with cut-off voltage of 1.5–3.0 V. CoMOF–S cathodes display much better cycling performance than the pure sulfur cathode; among them, the CoMOFS2 cathode exhibits the best cycle stability and reversible capacity. Initial delithiation capacity of CoMOFS2 is 1522 mA h g−1, which is higher than those of CoMOFS1 (1429.1 mA h g−1) and CoMOFS3 (1515.5 mA h g−1). The reversible capacity of CoMOFS2 is above 800 mA h g−1 at 0.1C rate and capacity decay of CoMOFS2 cathode is 34.66% lower than that of pure sulfur cathode under the same charge/discharge conditions. All the above results indicate that CoMOF–S with appropriate sulfur content (56.6 wt%) improves electrochemical performance of sulfur cathodes. However, all CoMOF–S cathodes suffer severe irreversible capacity fading in the first 20 cycles, which is typically observed in other types of cathode materials.26,27 After 20 cycles, their capacity decay drastically decreased. Cycling stability of the CoMOF–S cathodes is much higher than that of pure sulfur cathodes. Fig. 3b shows the specific capacity–potential curves of the CoMOFS2 cathode at the 1st, 5th, 10th, 25th, 50th, and 100th cycles. A typical discharge process with two plateaus at around 2.3 and 2.1 V is observed. The upper voltage plateau at 2.3 V corresponds to conversion of S8 to long chain polysulfides (Li2S8, Li2S6, or Li2S4), while the lower voltage plateau at 2.1 V reflects the further reduction of high-order polysulfides to low-order polysulfides (Li2Sn, n < 4) and finally to insoluble lithium sulfides (Li2S2/Li2S).28 The CoMOFS2 cathode manifests excellent high-rate performance (Fig. 3c and d), decreasing slowly from the reversible 1290 mA h g−1 at 0.1C to 970, 840, 710, and 610 mA h g−1 at 0.2C, 0.5C, 1.0C, and 2.0C, respectively. In addition, a capacity of 853 mA h g−1 is obtained when current is recovered from 2.0C to 0.1C, indicating a highly reversible low fading in capacity. From Fig. 2c, we can clearly see that both the rate performances and coulombic efficiency of CoMOFS2 are remarkably higher than those of the pure sulfur cathode at the same charge/discharge rate. The coulombic efficiency is ∼93% at low rate of 0.1C, which may be caused by the severe “shuttle effect”, where polysulfides diffuse through the electrolyte and migrate between the cathode and anode. However, the coulombic efficiency is ∼98% and ∼97% at high rate of 1.0C and 2.0C, respectively, which indicates that the CoMOF structure can stabilize some of the sulfur species and sulfur inside the MOF can be accessed and utilized efficiently at high current rate. In addition, the role of CoMOF structure is only as the sulfur host; it provides almost no capacity in the CoMOF–S cathode, which can be proven by the cycling performance of pristine CoMOF, as shown in Fig. S4 of the ESI.†
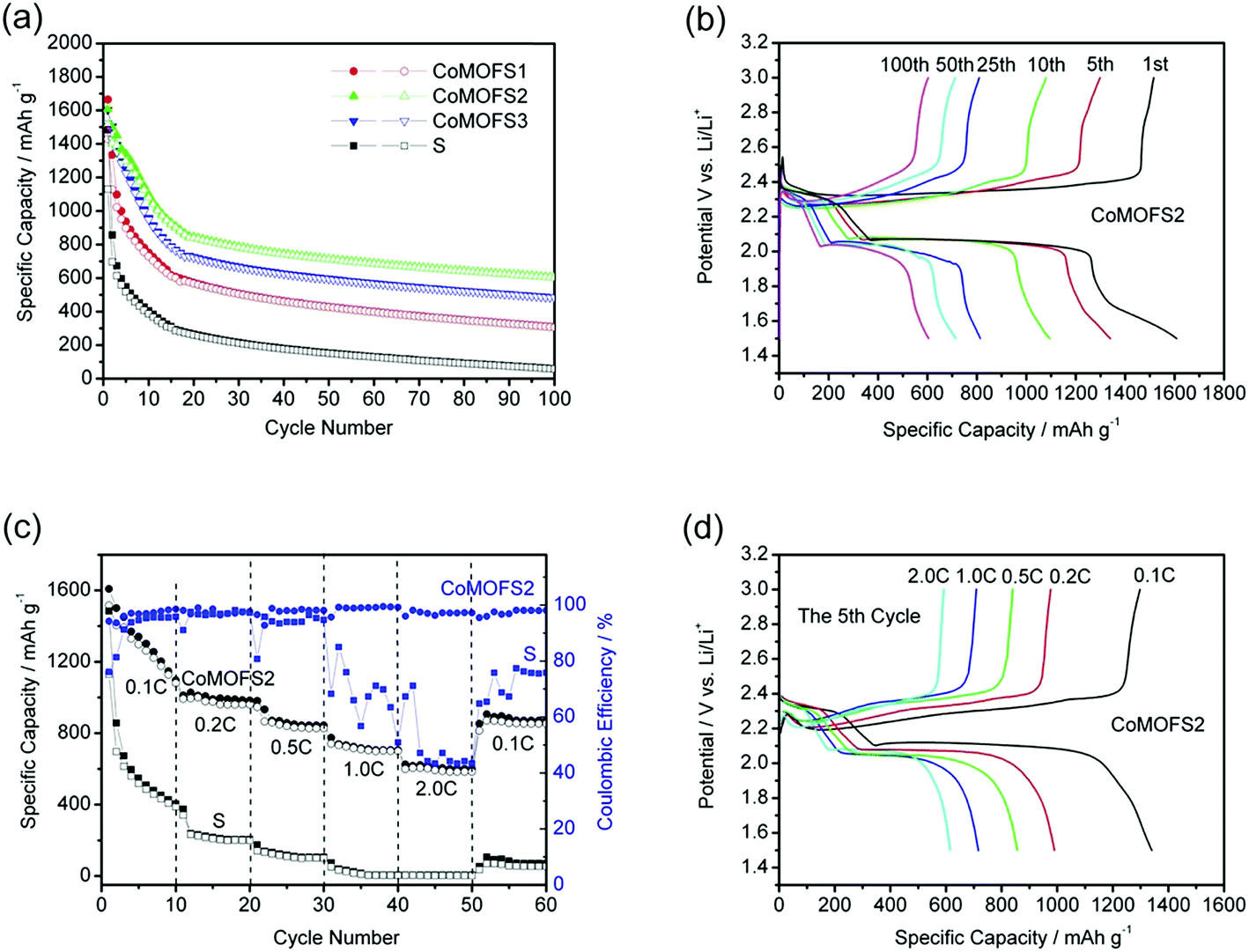 | ||
| Fig. 3 (a) Cycle performances of CoMOF–S cathodes at charge/discharge current density of 0.1C. (b) The specific capacity–potential curves of CoMOFS2 cathode at the 1st, 5th, 10th, 25th, 50th, and 100th cycle. (c) High-rate performances and coulombic efficiency of CoMOFS2 and pure S cathodes at charge/discharge densities of 0.1C, 0.2C, 0.5C, 1.0C, and 2.0C. (d) The specific capacity–potential curves of CoMOFS2 cathode at high-rate charge/discharge current densities of 0.1C, 0.2C, 0.5C, 1.0C, and 2.0C. (1C = 1675 mA h g−1) based on 5th cycle's data. | ||
Cyclic voltammetry (CV) profiles of CoMOFS2 cathodes at a scan rate of 0.02 mV s−1 for the first three cycles are shown in Fig. 4a. The curve shapes of the first cycle are different from the subsequent cycles, which is attributed to the irreversible reaction involved in the first cycle. During the positive scan in the second scanning, two anodic peaks are observed at ∼2.5 and ∼2.7 V. The former peak indicates the oxidation reaction to form sulfur species is ∼2.5 V and the latter peak indicates some side reaction, such as oxidation of the carbon black at ∼2.7 V. During the negative scan, two cathodic peaks appear at ∼2.4 and ∼2.0 V, showing that the reduction reaction is around these two potentials. The peak at ∼2.4 V indicates the transformation of S to higher-order Li2Sx (4 ≤ x ≤ 8) and the peak at ∼2.0 V was caused by further reduction to higher-order Li2Sx (x ≤ 4) and finally to Li2S.29,30 After scanning downward to 1.5 V, one clear reduction peak at ∼1.6 V is found, which may be caused by reaction of LiNO3 in the electrolyte. The same situations are also found in the second and third cycles.
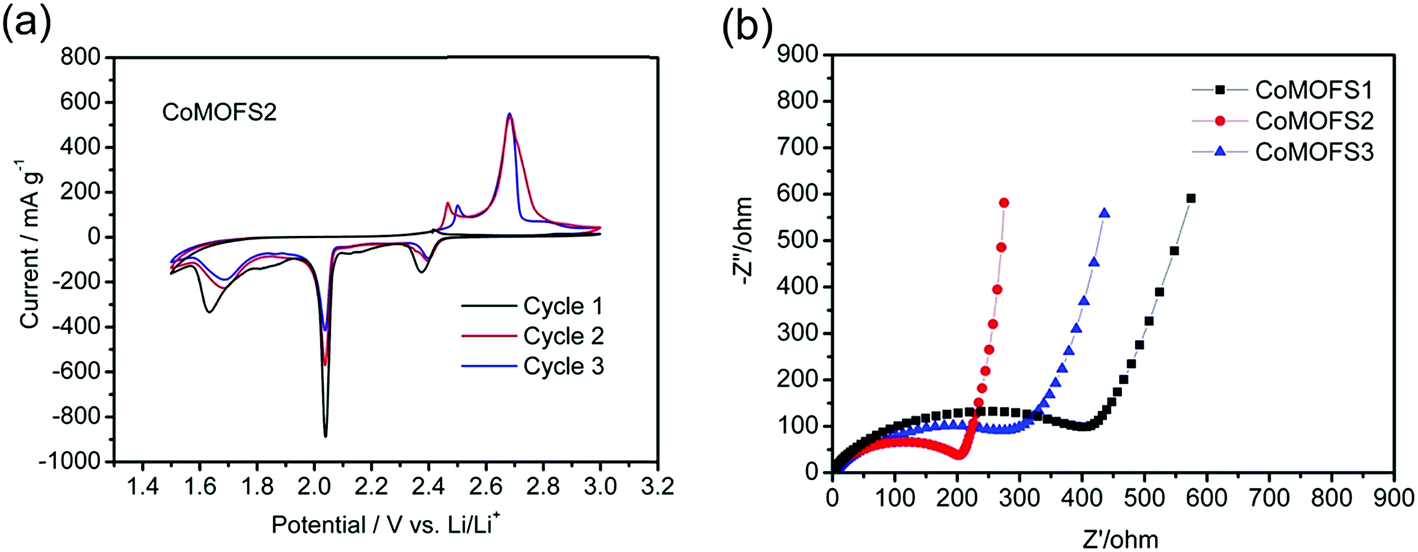 | ||
| Fig. 4 (a) Cyclic voltammetry curves of CoMOFS2 cathodes at a scan rate of 0.02 mV s−1 for the first three cycles; (b) EIS Nyquist plots of experimental data of CoMOF–S cathodes (insert: equivalent circuit for EIS data fitting). | ||
As shown in Fig. 4b, EIS was conducted to evaluate the electrochemical kinetic properties of the CoMOF–S cathodes and their equivalent circuit for EIS data fitting. The Nyquist plots for all the electrodes consist of one semicircle and a straight line, which are respectively associated with charge transfer and solid state diffusion process of lithium ions in the electrode and the Warburg region.31,32 EIS data were fitted to an electrochemical model using Zview module software, as shown in the inset in Fig. 4b. Rs at the high-frequency intercept on the real axis represents bulk resistance, and is dependent on electrolyte solution, active material, current collector and so on. Rp is the charge transfer resistance at the electrode–electrolyte interface, and is associated with the interface between electrolyte and active particles. CPE is the constant phase element and represents the conventional double-layer and passivation film capacitance. W is the finite length Warburg impedance with an open circuit terminus.33,34Table 1 shows the EIS fitting results of CoMOF–S cathodes. No distinct difference exists in the Rs values of the four samples, which proves that the cells have the same test conditions. Rp value of the CoMOFS2 cathode is the smallest among the CoMOF–S cathodes, which indicates the highest electrochemical activity and best utilization of electrodes among them. Comparing the Rp values in Table 1, CoMOFS2 cathode has the lowest Rp value, which indicates that lithium diffusion in CoMOFS2 cathode is the fastest among the CoMOF–S cathodes. This further demonstrates that the CoMOFS2 cathode has good lithium intercalation kinetics, corresponding to improved rate performance.
| Parameter | CoMOFS1 | CoMOFS2 | CoMOFS3 |
|---|---|---|---|
| R s/Ω | 2.81 | 1.09 | 0.34 |
| R p/Ω | 278.90 | 139.20 | 201.60 |
| CPE-T/μF | 23.55 | 10.58 | 8.64 |
| CPE-P/F | 0.67 | 0.70 | 0.73 |
| W-R/Ω | 143.60 | 147.70 | 65.92 |
| W-T/Ω | 0.39 | 0.38 | 0.11 |
| W-P/Ω | 0.72 | 0.71 | 0.47 |
To further investigate the capacity decay during charge/discharge cycling, SEM images of CoMOFS2 cathode before cycling and after 10, 20, 50, and 100 cycles were recorded, as shown in Fig. 5. Before cycling, the CoMOFS2 particles are in tight contact with carbon black powder to form a homogenous and smooth electrode surface (Fig. 5a). With increasing cycling, the layer on the outside of CoMOFS2 cathode may undergo deposition of polysulfides (Fig. 5b–e). From the EIS analysis, as shown in Fig. 5f, we can draw the same conclusion. The single depressed semicircles in the high-to-medium frequency region can be ascribed to the charge-transfer resistance Rp, which represents the electrode reaction kinetics. After extended cycling from 1 to 20 cycles, the charge transfer resistance of CoMOFS2 decreases tremendously. This decrease is ascribed to loss of active materials upon cycling, which leads to capacity decay. With continued cycling, charge transfer resistance remains unchanged, which evidences the stability of the electrode after 20 cycles. This phenomenon matches well with the charge/discharge cycling performance shown in Fig. 3a.
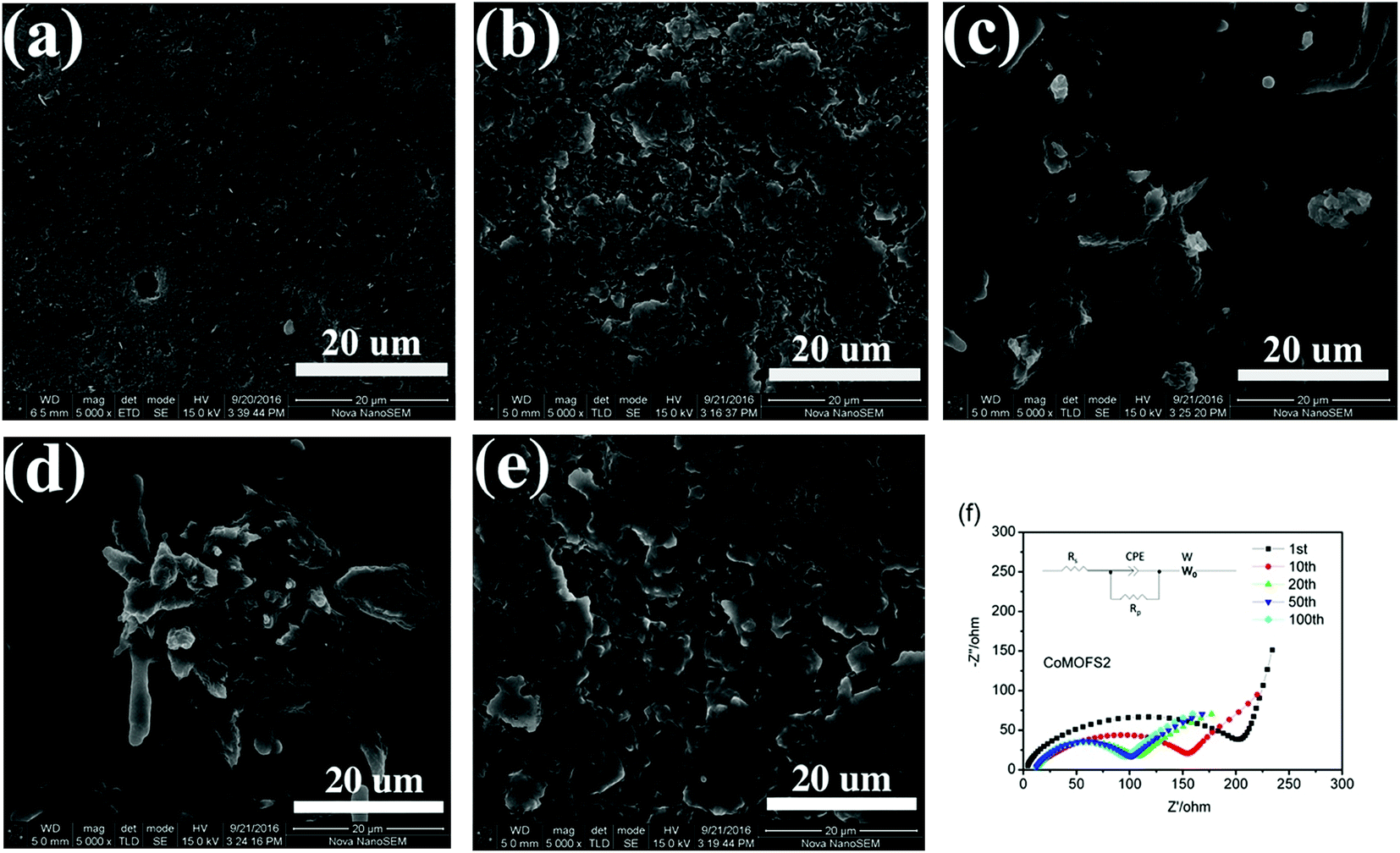 | ||
| Fig. 5 SEM images of CoMOFS2 cathode (a) before and after (b) 10, (c) 20, (d) 50, and (e) 100 charge/discharge cycles; (f) EIS Nyquist plots of experimental data of the CoMOFS2 cathode after 1, 10, 20, 50, and 100 charge/discharge cycles (insert: equivalent circuit for EIS data fitting). | ||
To further identify the chemical status of the CoMOF–S material and reveal the reason for its improvement in electrochemical performance, X-ray photoelectron spectroscopy (XPS) measurements and density functional theory (DFT) calculations were performed. As shown in Fig. 6a, the C 1s spectrum of the CoMOFS2 material exhibits three peaks with binding energies of 284.5, 285.7, and 288.6 eV that correspond to carbon atoms in different functional groups of the MOF structure.35 Evidently, the fourth peak with binding energy of 286.5 eV is related to the structure of MOF as a result of sulfur binding, which is absent from the XPS of pristine CoMOF (Fig. 6b). To match the XPS peaks with the corresponding carbon atoms, DFT calculations were used to predict binding energy for each carbon. In agreement with four distinct XPS peaks, there are four unique carbon atoms, as shown in Fig. 6c. The ortho and piso aromatic carbons contribute two theoretical peaks at 284.7 and 285.2 eV, respectively, and are responsible for the 284.5 and 285.7 eV peaks observed experimentally. Ortho and piso aromatic carbons are also the primary carbon types in pristine CoMOF, which explains the duplication with pristine CoMOF for these two peaks. The other two carbons, namely, [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–S]− and OC–O–S carbons, are associated with peaks at 286.8 and 288.7 eV, respectively. The OC–O–S carbon is responsible for the peak at 288.6 eV, which overlaps with the OC–O–H carbon peak in pristine CoMOF. The additional peak at 286.5 eV emitted by CoMOF–S is assigned to the [OC–S]− group, which deviates from theoretical value (286.8) by only 0.3 eV. Deviations of the DFT predictions for all CoMOF–S XPS peaks are within 0.5 eV. It is worth noting that the predicted accuracy is better for pristine CoMOF. Compared with the experimental values (284.7, 285.5 and 288.7 eV) in Fig. 6b, the maximum deviation from theoretical values (284.9, 285.3 and 288.4 eV) is only 0.3 eV. This excellent consistency is strong evidence that the proposed structure model in Fig. 5c exemplifies the connection mode between CoMOF and S. It is unambiguous that sulfur bonds with carbon to form strong covalent bonds.
C–S]− and OC–O–S carbons, are associated with peaks at 286.8 and 288.7 eV, respectively. The OC–O–S carbon is responsible for the peak at 288.6 eV, which overlaps with the OC–O–H carbon peak in pristine CoMOF. The additional peak at 286.5 eV emitted by CoMOF–S is assigned to the [OC–S]− group, which deviates from theoretical value (286.8) by only 0.3 eV. Deviations of the DFT predictions for all CoMOF–S XPS peaks are within 0.5 eV. It is worth noting that the predicted accuracy is better for pristine CoMOF. Compared with the experimental values (284.7, 285.5 and 288.7 eV) in Fig. 6b, the maximum deviation from theoretical values (284.9, 285.3 and 288.4 eV) is only 0.3 eV. This excellent consistency is strong evidence that the proposed structure model in Fig. 5c exemplifies the connection mode between CoMOF and S. It is unambiguous that sulfur bonds with carbon to form strong covalent bonds.
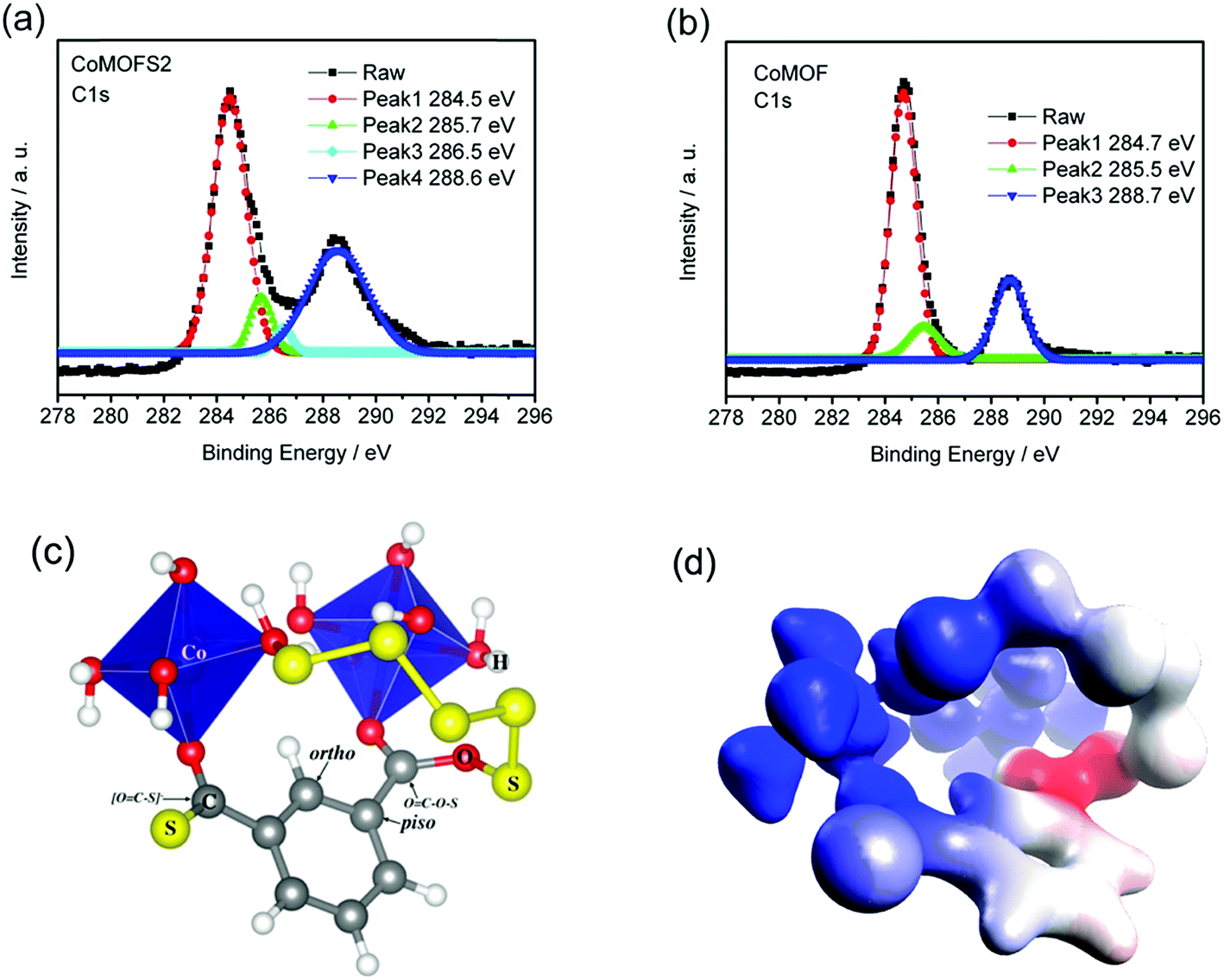 | ||
| Fig. 6 (a) XPS C 1s spectrum of CoMOFS2; (b) XPS C 1s spectrum of CoMOF; (c) CoMOF–S composite model. Blue polyhedrons illustrate coordination environment of cobalt cation. OH− group simulates coordination from neighboring benzene-1,3,5-tricarboxylic acid. The text with arrow labels the name of the positions; (d) the molecular electrostatic potential of CoMOF–S projected on the electron density = 0.08 a.u. isodensity surface. The colors form a continuum from most negative (blue) to most positive potential (red). | ||
We want to emphasize that we enumerated all possible structures for CoMOF and S composite, including covalent bond, hydrogen bond and van der Waals structures. The structure presented in Fig. 5c is the one with the best agreement with experiment. We computed the C 1s orbital energy using Koopmans' theorem22,36 at TPSSh/6-311++G(d,p) level,24 which was then corrected by an empirical parameter to obtain the XPS binding energy. The detailed computational methods are described in the methods section. As shown in Fig. 6c, we proposed two schemes for sulfur connecting to CoMOF. (1) Substitution for carboxylic –OH group, which changes a OC–O–H to a OC–S–H or a deprotonated [OC–S]− group. DFT simulation reveals that the energy of the deprotonated state is lower than that of the protonated state by 12.33 kJ mol−1. Therefore, the structure with [OC–S]− group is more energetically favorable and will be used in the XPS predictions. (2) Substitution for H atom of carboxyl group or H2O ligand. The combination of two substitutions results in a cross-link inside the pores of CoMOF. The cross-links tilt the carboxylic group plane slightly due to spatial constraints. In addition to direct O atom substitution, the structure distortion will also contribute to atomic charge redistribution. Natural bond orbital (NBO)37 population analysis shows that the atomic charge on carbon atom of OC–S–H is 0.85, which is close to that of the carbon of OC–O–H (0.82) in pristine CoMOF. However, the charge on carbon atom of [OC–S]− is sharply decreased to 0.28, which indicates higher electron density. Consequently, binding energy drops as a result of increased shielding of the nuclear charge. This can be seen more intuitively from the electrostatic potential in Fig. 6c. The [OC–S]− group resides in an area with significantly more negative potential than OC–S–H. As a consequence, it exerts less attraction to the electrons, which leads to lower binding energy. Both substitution schemes immobilize the sulfur atoms to the CoMOF pores by strong covalent bond. We expect that these schemes will facilitate sulfur confinement in the CoMOF framework.
4. Conclusion
In summary, we utilized a cobalt metal–organic framework (MOF) with rough porous surface and defective structure, denoted as CoMOF–S, as a host material for sulfur immobilization. CoMOF was obtained by simple chemical synthesis using cobalt salt and benzene-1,3,5-tricarboxylic acid as the ligand. Sulfur was incorporated into CoMOF pores by melt-diffusion method at 155 °C. X-ray photoelectron spectroscopy results and DFT predictions are in excellent agreement. We are confident that sulfur is connected to the carboxylic functional group of the MOF via chemical bonding, which provides a strong anchor for robust sulfur confinement and effective sulfur utilization. The CoMOF–S composite materials are successfully implemented as cathodes for Li–S batteries. Among them, the CoMOFS2 cathode, which absorbs two-fold higher sulfur in weight, delivers best performance in both cycle stability and high-rate charging/discharging. After 100 charge/discharge cycles, capacity retention of CoMOFS2 cathode is 87.18% higher than that of pure sulfur cathode at a current rate of 0.1C. At a high-rate of 2.0C, the CoMOFS2 cathode can still maintain a specific capacity of above 600 mA h g−1. The excellent cycling performance is attributed to the strong covalent bonds with sulfur and suitable pore sizes in MOF structure for sulfur confinement, which significantly mitigate the loss of soluble polysulfides from the pores.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Natural Science Foundation of Tianjin (No. 18JCYBJC21800) and the Academic Innovation Funding of Tianjin Normal University (No. 52XC1502). Prof. Yan Feng was sponsored by an Exchange Scholarship Funding of Tianjin Normal University for Overseas Academic Research. The authors appreciate Dr Gao Liu and Dr Vincent S Battaglia in Lawrence Berkeley National Laboratory for their useful discussion and valuable consultation.References
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Designing high-energy lithium–sulfur batteries, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- B. D. McCloskey, Attainable gravimetric and volumetric energy density of Li–S and Li-ion battery cells with solid separator-protected Li metal anodes, J. Phys. Chem. Lett., 2015, 6, 4581–4588 CrossRef PubMed.
- L. Ma, K. E. Hendrickson, S. Wei and L. A. Archer, Nano Today, 2015, 10, 315–338 CrossRef.
- A. Manthiram, Y. Fu and Y.-S. Su, Challenges and prospects of lithium–sulfur batteries, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef PubMed.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Lithium–sulfur batteries: electrochemistry, materials, and prospects, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Review-the importance of chemical interactions between sulfur host materials and lithium polysulfides for advanced lithium–sulfur batteries, J. Electrochem. Soc., 2015, 162, A2567–A2576 CrossRef.
- L. Ji, M. Rao, H. M. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Carins and Y. Zhang, Graphene oxide as a sulfur immobilizer in high performance lithium/sulfur cells, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef PubMed.
- Y.-S. Su, Y. Fu, T. Cochell and A. Manthiram, A Strategic approach to recharge lithium–sulphur batteries for long cycle life, Nat. Commun., 2013, 4, 2985 CrossRef PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Modular chemistry: secondary building units as a basis for the design of highly porous and robust metal–organic carbonxylate frameworks, Acc. Chem. Res., 2001, 34, 319–330 CrossRef PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Metal–organic frameworks for energy storage: batteries and supercapacitors, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Metal–organic frameworks and their derived nanostructures for electrochemical energy storage and conversion, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- S. L. Li and Q. Xu, Metal–organic frameworks as platforms for clean energy, Energy Environ. Sci., 2013, 6, 1656–1683 RSC.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J.-M. Tarascon, Cathode composites for Li–S batteries via the use of oxygenated porous architectures, J. Am. Chem. Soc., 2011, 133, 16154–16160 CrossRef PubMed.
- Z. Wang, X. Li, Y. Gui, Y. Yang, H. G. Pan, Z. Y. Wang, C. Wu, B. Chen and G. Qian, A metal–organic framework with open metal sites for enhanced confinement of sulfur and lithium–sulfur battery of long cycling life, Cryst. Growth Des., 2013, 13, 5116–5120 CrossRef.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Rational design of a metal–organic framework host for sulfur storage in fast, long-cycle Li–S batteries, Energy Environ. Sci., 2014, 7, 2715–2724 RSC.
- J. Zhou, X. Yu, X. Fan, X. Wang, H. Li, Y. Zhang, W. Li, J. Zheng, B. Wang and X. Li, The impact of the particle size of a metal–organic framework for sulfur storage in Li–S batteries, J. Mater. Chem. A, 2015, 3, 8272–8275 RSC.
- Z. Zhao, S. Wang, R. Liang, Z. Li, Z. Shi and G. Chen, Graphene-wrapped chromium-MOF (MIL-101) sulfur composite for performance improvement of high-rate rechargeable Li–S batteries, J. Mater. Chem. A, 2014, 2, 13509–13512 RSC.
- J. Zheng, J. Tian, D. Wu, D. Gu, M. Gu, W. Xu, C. Wang, F. Gao, M. H. Engelhard, J.-G. Zhang, J. Liu and J. Xiao, Lewis acid–base interactions between polysulfides and metal organic framework in lithium sulfur batteries, Nano Lett., 2014, 14, 2345–2352 CrossRef PubMed.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Y. Wang, B. Chen and G. Qian, Mixed-metal–organic framework with effective Lewis acidic sites for sulfur confinement in high-performance lithium–sulfur batteries, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef PubMed.
- Z. Li, C. Li, X. Ge, J. Ma, Z. Zhang, Q. Li, C. Wang and L. Yin, Reduced graphene oxide wrapped MOFs-derived cobalt-doped porous carbon polyhedrons as sulfur immobilizers as cathodes for high performance lithium sulfur batteries, Nano Energy, 2016, 23, 15–26 CrossRef.
- R. Fang, S. Zhao, S. Pei, X. Qian, P.-X. Hou, H.-M. Cheng, C. Liu and F. Li, Toward more reliable lithium–sulfur batteries: an all-graphene cathode structure, ACS Nano, 2016, 10, 8676–8682 CrossRef PubMed.
- T. Koopmans, Über Die Zuordnung von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms, Physica, 1934, 1, 104–113 CrossRef.
- M. Giesbers, A. T. M. Marcelis and H. Zuilhof, Simulation of XPS C 1s Spectra of Organic Monolayers by Quantum Chemical Methods, Langmuir, 2013, 29, 4782–4788 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- S. Xin, L. Gu, N.-H. Zhang, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, Smaller molecules promise better lithium–sulfur batteries, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, A highly ordered nanostructured carbon–sulphur cathode for lithium–sulphur batteries, Nat. Mater., 2009, 8, 500–506 CrossRef PubMed.
- W. Li, J. Hicks-Garner, J. Wang, J. Liu, A. F. Gross, E. Sherman, J. Graetz, J. J. Vajo and P. Liu, V2O5 polysulfide anion barrier for long-lived Li–S batteries, Chem. Mater., 2014, 26, 3403–3410 CrossRef.
- Z. Lin, C. Nan, Y. Ye, J. Guo, J. Zhu and E. J. Cairns, High-performance lithium/sulfur cells with a bi-functionally immobilized sulfur cathode, Nano Energy, 2014, 9, 408–416 CrossRef.
- M.-K. Song, Y. Zhang and E. J. Cairns, A long-life, high-rate lithium/sulfur cell: a multifaceted approach to enhancing cell performance, Nano Lett., 2013, 13, 5891–5899 CrossRef PubMed.
- Y. Feng, N. Feng, Y. Wei and Y. Bai, Preparation and improved electrochemical performance of SiCN-graphene composite derived from poly(silylcarbondiimide) as Li-ion battery anode, J. Mater. Chem. A, 2014, 2, 4167–4177 Search PubMed.
- N. Feng, Y. Feng, Y. Z. Wei and X. Zhou, Preparation and electrochemical performance of a porous polymer-derived silicon carbonitride anode by hydrofluoric acid etching for lithium-ion batteries, RSC Adv., 2014, 4, 23694–23702 RSC.
- Y. Feng, Y. Wei, Z. Jia, Y. Zhang, V. Battaglia and G. Liu, Polymer-derived and sodium hydroxide-treated silicon carbonitride material as anodes for high electrochemical performance Li-ion batteries, ChemistrySelect, 2016, 2, 309–317 CrossRef.
- Y. Feng, Y. Zhang, Y. Wei, X. Song, Y. Fu and V. S. Battaglia, A ZnS nanocrystal/reduced graphene oxide composite anode with enhanced electrochemical performances for lithium-ion batteries, Phys. Chem. Chem. Phys., 2016, 18, 30630–30642 RSC.
- R. Chen, T. Zhao, T. Tian, S. Cao, P. R. Coxon, K. Xi, D. Fairen-Jimenez, R. V. Kumar and A. Cheetham, Graphene-wrapped sulfur/metal organic framework-derived microporous carbon composite for lithium sulfur batteries, APL Mater., 2014, 2, 124109 CrossRef.
- M. Giesbers, A. T. M. Marcelis and H. Zuilhof, Simulation of XPS C 1s Spectra of Organic Monolayers by Quantum Chemical Methods, Langmuir, 2013, 29, 4782–4788 CrossRef PubMed.
- F. Weinhold and C. R. Landis, Natural Bond Orbitals and Extensions Of Localized Bonding Concepts, Chem. Educ.: Res. Pract. Eur., 2001, 2, 91–104 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of pristine CoMOF after heating at 155 °C. XRD patterns of pristine CoMOF before and after heating at 155 °C. SEM images of CoMOFS1 and CoMOFS3. Cycle performance of pristine CoMOF (without S) cathode at a charge/discharge current density of 167.5 mA h g−1. See DOI: 10.1039/c8se00195b |
| This journal is © The Royal Society of Chemistry 2018 |