
A surfactant-assisted strategy to tailor Li-ion charge transfer interfacial resistance for scalable all-solid-state Li batteries†
Chengtian
Zhou
,
Alfred Junio
Samson
,
Kyle
Hofstetter
and
Venkataraman
Thangadurai
 *
*
Department of Chemistry, University of Calgary, 2500 University Dr, NW Calgary AB T2N 1N4, Canada. E-mail: vthangad@ucalgary.ca
First published on 18th July 2018
Abstract
Solid-state batteries with Li anode present a promising design to achieve high energy density and safe batteries that meet today's growing demands for portable electronics, electric vehicles, and grid-scale energy storage. Garnet-type solid Li-ion electrolytes exhibit desired physical and chemical properties, including high total (bulk + grain-boundary) Li-ion conductivity, chemical stability with elemental Li, and high electrochemical stability window (6 V vs. Li+/Li), which make them a unique candidate membrane for all-solid-state batteries. A significant challenge with all-solid-state batteries is the high area specific resistance (ASR) in the solid electrolyte/Li anode interface. Although a substantial reduction in interfacial ASR has been achieved recently with Li-stuffed garnets and Li anode, the equipment and techniques used present massive challenges in both cost-effectiveness and scalability. Here, we show a surfactant-assisted wet chemical method to deposit a ZnO layer on Li-stuffed Li6.5La2.9Ba0.1Zr1.4Ta0.6O12 (LLBZT) that increases the contact area between Li/LLBZT and reduces interfacial ASR from 70 to 10 Ω cm2 at room temperature. Microstructural analysis reveals the uniform distribution of nano ZnO, which causes an excellent Li wetting on the garnet electrolyte and improvement in the contact area between the electrolyte and electrode. Electrochemical characterization and galvanostatic cycling confirm stable Li plating/stripping for more than a hundred cycles at 0.1 mA cm−2, demonstrating a compelling strategy to solve the Li/solid electrolyte interface problem in all-solid-state Li batteries.
Rechargeable lithium-ion batteries (LIBs) have become the power source of choice for portable electronic devices due to their higher gravimetric and volumetric energy densities compared to other commercial rechargeable systems such as Ni-metal hydride and Ni–Cd batteries.1 A significant part of the success of LIBs can be attributed to the use of liquid organic carbonate electrolytes, which allows for a higher operating voltage of about 4 V. In contrast, aqueous electrolyte-based batteries are typically limited to an operating voltage of about 1.2 V.2 Nowadays, LIBs power electric vehicles and store energy from renewable solar and wind sources.3–7 The demands on LIB performance and safety are higher for these applications than in portable electronic devices. Conventional LIBs with organic carbonate electrolytes suffer from several issues including leakage, sensitivity to moisture electrochemical instability, and flammability.2,8,9 These issues raise significant safety concerns, especially in electric vehicles.
Solid-state (ceramic) lithium-ion electrolytes have attracted ever-increasing attention as an alternative to conventional liquid electrolytes since these ceramic electrolytes can overcome all of the above safety issues while offering possibilities for developing other battery chemistries10 such as Li–O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and Li–S12–15 that exhibit higher gravimetric and volumetric energy densities than traditional LIBs. The theoretical energy densities are 2567 W h kg−1/2199 W h l−1 for Li–S16 and 3505–3582 W h kg−1/2234–3436 W h l−1 for Li–O2,16 while the practical energy densities of conventional LIBs17,18 only reach up to 250 W h kg−1 and 650 W h l−1.
11 and Li–S12–15 that exhibit higher gravimetric and volumetric energy densities than traditional LIBs. The theoretical energy densities are 2567 W h kg−1/2199 W h l−1 for Li–S16 and 3505–3582 W h kg−1/2234–3436 W h l−1 for Li–O2,16 while the practical energy densities of conventional LIBs17,18 only reach up to 250 W h kg−1 and 650 W h l−1.
The solid-state electrolyte used in the present study has a garnet-type structure that is stable when in contact with the Li metal anode. Elemental Li exhibits one of the highest specific capacities (3860 mA h g−1) and the lowest reduction potential (−3.05 V vs. SHE), allowing for high-energy-density solid-state lithium batteries. Aside from chemical stability against the Li metal, garnet-type solid electrolytes possess high ionic conductivities,19–21 10−4 to 10−3 S cm−1 at room temperature, and a wide electrochemical stability window, up to 6 V vs. Li+/Li.19 The key challenge in utilizing the garnet material, like other solid-state electrolytes, is the high interfacial impedance between the solid-state electrolyte and the electrodes.22 Specifically, a significant limitation of garnet solid electrolytes is their poor adhesion with the Li metal due to their rigid nature, resulting in high interfacial area specific resistance (ASR) in the garnet–Li metal interface and uneven current distribution on the surface which hamper the utilization of Li-based garnets in all-solid-state Li batteries.23 For commercialization, there should be a scalable and cost-effective technique to reduce the ASR.
Melting the elemental Li anode on the garnet offers limited interfacial resistance reduction.24,25 In recent years, several groups have studied various approaches to induce intimate interfacial contact between the garnet and Li metal.16–25 These studies can be grouped into two general methods.26 The first approach involves removal of impurities that form naturally on the surface of Li–garnets upon exposure to air during processing and sintering. Li2CO3 forms at the surface of the garnet in air and is reported as the source of high interfacial resistance with Li electrodes.27 Cheng et al.27 removed surface impurities by polishing, and achieved a moderate interfacial ASR of 109 Ω cm2 at 25 °C. A similar polishing route and heating of the surface in Ar was performed by Sharafi et al.28 which resulted in an impressive room temperature interfacial ASR reduction to 2 Ω cm.2 The polishing approach is arguably a more straightforward route to reduce interfacial ASR. However, this approach still require heating up to 500 °C in Ar and is limited to flat surfaces. Moreover, polishing will not be feasible in 3D designs that are currently being pursued for increased active material loading in solid-state batteries.29 The second approach involves the deposition of a transition layer that aids in uniform and intimate contact with the Li metal. Various materials and deposition techniques are reported, including poly(vinylidene fluoride-hexafluoropropylene) (PVDF-HFP)30 with a liquid electrolyte and polyethylene oxide (PEO)29 by solution casting, Au by sputtering,31 Si by plasma enhanced chemical vapor deposition,32 Ge by electron beam thermal evaporation,33 and ZnO34 and Al2O323 by atomic layer deposition (ALD). Solution casting by PVDF–HFP or PEO is promising due to its scalability but the reduction in interfacial ASR is not significant (e.g., 214 Ω cm2 for PVDF-HFP).30 The most effective technique to date in reducing the interfacial ASR is Al2O3 deposition by ALD, which reduced the interfacial ASR from 1710 to 1 Ω cm2 at room temperature.23 Although this reduction is unprecedented and represents a critical milestone towards the path to commercialization of solid-state batteries, the ALD technique is currently relatively expensive and complex in operation,35 making it unfavourable for large-scale application.
Herein, we report a scalable, novel deposition method that enables the formation of uniformly distributed, nanoporous ZnO. This method involves a Zn(NO3)2 solution mixed with a copolymer surfactant (Pluronic P123) deposited onto the surface of the garnet electrolyte pellet, followed by heating. The triblock copolymer surfactant facilitates dispersion of Zn ions that results in a uniform distribution of the ZnO particles after heating. The nano/microporous network of ZnO with a thickness of 6 μm reacts very readily with Li, reducing the garnet–Li metal interfacial ASR from 70 to 10 Ω cm2 at room temperature. This solution-coating method is easy to implement and feasible for mass-production with negligible cost, and can also be adopted to different solid-state battery systems.
A garnet-type cubic electrolyte with the nominal composition Li6.5La2.9Ba0.1Zr1.4Ta0.6O12 (LLBZT) was derived from the chemically stable garnet-type Li7La3Zr2O1236 by simultaneous substitution of Ba2+ for La3+ and Ta5+ for Zr4+. Ta-doping increases the Li-ion vacancy concentration and reduces the degree of local ordering which aids in increasing the Li-ion conductivity, and like Zr(IV), Ta(V) is not readily reduced when in contact with lithium.21 In addition to lowering the sintering temperature, Ba2+ has a larger ionic radius than La3+ which increases the lattice constant and also aids in improving Li-ion conductivity.37
The experimental details on the synthesis and characterization of the LLBZT electrolyte are shown in the ESI.† The powder X-ray diffraction (XRD) pattern of LLBZT (Fig. S1 in the ESI†) matches that of cubic garnet-phase Li5La3Nb2O12 (powder diffraction file (PDF) number 45-0109), indicating that LLBZT is a pure cubic garnet phase. Microstructural analysis of the fracture cross-section of the pellet by scanning electron microscopy (SEM) reveals a dense morphology with a mixture of inter-granular and trans-granular fractures, as shown in Fig. S2 in the ESI.† The Li-ion conductivity of LLBZT was found to be 2.4 × 10−4 S cm−1 at 25 °C with an activation energy of 0.30 eV at 25–150 °C, obtained from electrochemical impedance spectroscopy (EIS) with Li-ion blocking Au electrodes (Fig. S3 in the ESI†). The LLBZT top layer after final polishing with 1500 grit size SiC paper shows distinctive scratch marks from polishing that cover a wide area of the surface with only a few spots left unpolished (Fig. S4 in the ESI†). Energy dispersive X-ray spectroscopy (EDX) was performed on the uncoated sample, and no traces of impurities from the SiC polishing paper were observed.
Fig. 1 summarizes the Surfactant-Processed Interlayer for Ceramic Electrolyte (SPICE) coating of ZnO, the resulting microstructures, and the effectiveness of the SPICE deposition method to improve garnet–Li wetting. We have chosen a zinc nitrate solution that will eventually be converted into ZnO to demonstrate the SPICE procedure since it has already been shown that a layer of this oxide deposited by ALD improves garnet–Li wetting.34 As long as the target material can be prepared in solution, the SPICE deposition method can be adopted to develop the ceramic coating. A key component in preparing the solution is a surfactant that aids in spreading the solution on the ceramic surface and dispersing the cations to form a uniformly distributed layer. The Pluronic P123 surfactant has been shown as an excellent soft templating agent to prepare nano-sized mesoporous structures for LIB electrodes.38,39
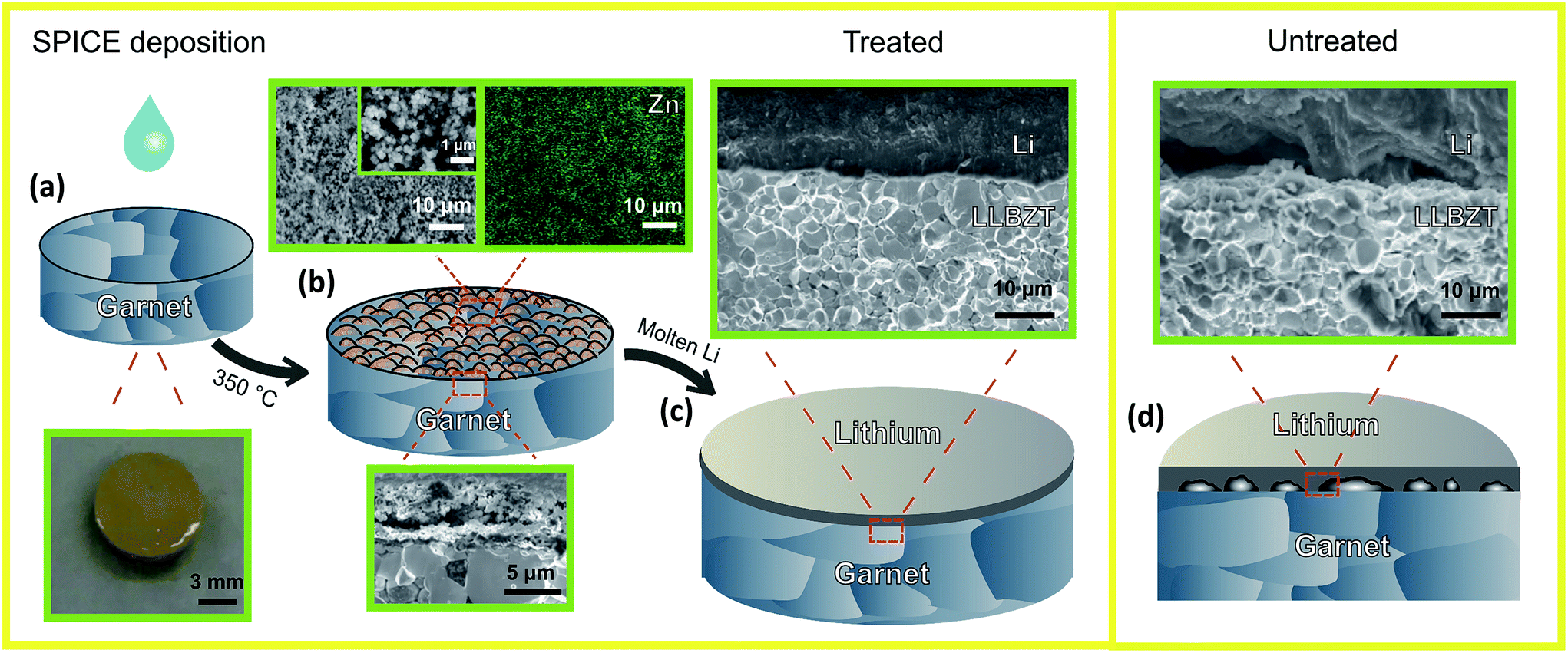 | ||
| Fig. 1 Schematic of the surfactant-processed interlayer for ceramic electrolyte (SPICE) deposition method with a ZnO interlayer: (a) known volume of the solution is dispensed onto the surface of the garnet. A digital photograph of the pellet after the solution was deposited is shown below the pellet schematic. (b) Heating of the solution to decompose Zn(NO3)2 to ZnO at 350 °C in air. SEM images of the top surface (general area and magnified image), including the EDX mapping of Zn, and cross-section of the layer are shown. (c) Li foil is heated on top of the SPICE-treated garnet at 250 °C for 1 h inside an Ar-filled glove box. The SEM image shows the resulting interface after heating and after electrochemical tests and (d) untreated pellet after similar Li heat treatment at 250 °C. The magnified SEM image shows the interface between the untreated garnet and lithium after electrochemical tests. | ||
0.3 M Zn(NO3)2 in isopropanol and 10 wt% P123 is coated onto a polished LLBZT pellet (Fig. 1a). After air-drying, the pellet is placed in a preheated oven at 350 °C for 20 min, and the resulting surface microstructure is schematically shown in Fig. 1b (further experimental details are described in the ESI†). Microstructural analysis of the garnet surface layer reveals a 6 μm thick micro-/nano-porous distribution of ZnO particles about 200 nm in diameter (Fig. 1b, magnified surface SEM image). EDX mapping confirms that the surface is mainly composed of uniformly distributed Zn. XRD and thermogravimetric analysis (Fig. S5 and S6,† respectively) of the solution components, Zn(NO3)2 and P123, show that Zn(NO3)2 transforms into ZnO in the temperature range of 225–350 °C, and P123 decomposes around 200–250 °C. A similar solution without P123 was also studied, and dark areas seen in the SEM images are most likely due to Li2CO3 (as they are carbon and oxygen-rich based on EDX analysis) (Fig. S7†). The formation of the dark spots in the layer without the added surfactant is consistent with the idea that large areas of the LLBZT are exposed during the heat treatment at 350 °C due to the non-uniform distribution of ZnO when it was transformed from Zn(NO3)2. It appears that the uniformly coated ZnO limited the formation of Li2CO3, but further investigation is required to confirm this hypothesis.
The electrochemical performance and the characteristics of Li plating and stripping of symmetric cells with untreated and SPICE-treated LLBZT electrolytes that are sandwiched between two Li metal electrodes, i.e., Li|LLBZT|Li and Li|ZnO|LLBZT|ZnO|Li, respectively, were investigated. The LLBZT pellets used were ∼0.9 mm thick, and the area of Li-foil attached to the pellet was 0.28 cm2. The attached Li was heated to 250 °C for 1 h in an Ar-filled glove box. The process to attach the lithium was similar to the ZnO-treated (ALD) garnet by Wang et al.34 Based on the studies of Li et al., the lithiation of ZnO follows a two-step chemical reaction:40
| ZnO + 2Li+ + 2e− → Li2O + Zn | (1) |
| Zn + xLi+ + xe− → LixZn | (2) |
In the ALD-deposited ZnO study by Wang et al.,34 they observed through XRD that the most lithium-rich alloy phase, LiZn, was formed after melting lithium on top of a ZnO pellet. We performed a similar procedure by placing ZnO powder on the top of the Li metal and heating to 250 °C for 1 h. After cooling, the resulting mixture was examined by XRD which confirmed that the primary phase formed was a LiZn alloy (see Fig. S8†). As reported by Liang et al.,41 LiZn, like other lithium alloys such as LiIn and Li3Bi, exhibit a high bulk chemical diffusion coefficient for lithium. The resulting garnet–Li metal interface is schematically shown in Fig. 1c, as confirmed by the actual image taken by SEM after the electrochemical tests. Excellent wetting of Li onto the garnet pellet is seen. In contrast, the untreated sample shows poor interfacial contact between Li and LLBZT (Fig. 1d).
Typical impedance plots of the SPICE-treated and untreated samples before Li plating and stripping are shown in Fig. 2. The thickness of the pellets is similar (∼0.9 mm). The spectra are normalized to the geometric area of the Li electrode to highlight the difference of the interfacial area specific resistance (ASR) of the garnet–Li interface in the SPICE-treated and untreated samples. The shape of the impedance spectra of the SPICE-treated and untreated samples with Li electrodes is similar, and both exhibit one high-frequency arc and one low-frequency arc that appears like a straight line. We have characterized at least three SPICE-treated and untreated samples, and this low frequency feature was consistently observed. We attribute the high frequency arc to the total electrolyte impedance (bulk resistance, Rb + grain-boundary resistance, Rgb), as indicated in Fig. 2a, for the untreated sample. This small arc at low-frequency is attributed to the interfacial resistance, Ri. This description of the electrolyte and interface contributions follows the analysis performed by Sharafi et al.28
 | ||
| Fig. 2 Area normalized impedance spectra of (a) untreated LLBZT and (b) SPICE-treated LLBZT symmetric cells with Li electrodes at 25 °C under open circuit potential in Ar. From the origin up to the real impedance part that separates the high-frequency and low-frequency arcs in the impedance spectra was considered as the total resistance (bulk (Rb) + grain boundary (Rgb)) contribution from the electrolyte pellet. The interfacial area specific resistance (ASR) can be estimated by taking the width of the low-frequency arc and dividing it by two to account for the garnet–Li surface on either side of the LLBZT pellet. The inset in (b) shows the magnified part of the low-frequency arc observed before and after the electrochemical measurements in the SPICE-treated sample. | ||
As seen in Fig. 2b, the SPICE-treated sample shows lower overall area-normalized impedance than the untreated LLBZT. This difference in the total electrolyte resistance contribution can be examined by comparing the results of the SPICE-treated and untreated samples to those of a reference sample in a Au blocking experiment (Fig. S3†). The thickness- and area-normalized impedance of the LLBZT electrolyte (bulk and grain contribution) in the Au Li-blocking experiment is ∼4.2 kΩ cm, while the total electrolyte resistances in the SPICE-treated and untreated Li|LLBZT|Li cells are 3.8 and 13 kΩ cm, respectively. The impedance of the SPICE-treated LLBZT electrolyte is closer to the expected area- and thickness-normalized electrolyte resistance of Au electrodes. Considering that the dimensions of the pellet used in the SPICE-treated sample is the same as that of the untreated pellet, the higher electrolyte resistance from the untreated sample is most likely due to the poor interfacial contact between the LLBZT and Li. We believe that only a few points of contact were achieved in the untreated sample, creating a much lower actual surface area than the SPICE-treated sample (Fig. 1d). More striking in Fig. 2b is the lower interfacial ASR, Ri, of the SPICE-treated sample compared to the untreated sample. After dividing it by two to account for the two sides of the pellet, the interfacial ASR of the untreated sample is estimated to be 70 Ω cm2, while that of the SPICE-treated sample is 10 Ω cm2 at room temperature. This interfacial ASR of the SPICE-treated pellet is about 2 times lower than that of ZnO-treated LLCZNO via ALD from the study of Wang et al.34 On comparing the specific resistance values of the SPICE-treated and untreated samples, it is interesting to note that the decrease in interfacial ASR (70 vs. 10 Ω cm2; 7× decrease) is greater than the decrease in the electrolyte normalized resistance (13 vs. 3.8 kΩ cm; 3.4× decrease). This observation implies that, not only did the contact area increase, but a process/processes that enhance(s) the transfer of Li-ions in the SPICE-treated sample may also be present. This mechanism is unclear at the moment, but presents an intriguing case for further studies.
Galvanostatic cycling experiments, where Li is plated and stripped on either side of the LLBZT pellet, were employed to investigate the Li-ion transport between Li and LLBZT at the interface in both SPICE-treated and untreated garnet samples. As shown in Fig. 3a, the Li plating and stripping at 25 °C is stable at 40 mV at a current density of 0.1 mA cm−2 for about 33 h and 100 cycles in the SPICE-treated sample. The untreated LLBZT showed significant voltage hysteresis in only five cycles (Fig. S9†). The total ASR (electrolyte + interface) of the untreated sample can also be estimated from the Li plating and stripping experiment. Assuming that the sample follows Ohm's law, the total ASR of the treated sample is 400 Ω cm2. Following subtraction of the expected electrolyte ASR from the Au/Au measurements, which is 381 Ω cm2, and dividing it by two to account for the two sides, the interfacial ASR is estimated to be ∼10 Ω cm2. Hence, both impedance spectroscopy and the DC method show similar ASR value, indicating that no additional electrode contribution is anticipated in the investigated impedance study. Higher current densities were also applied, and as shown in Fig. 3b and c, cycling at current densities of 0.2 and 0.5 mA cm−2 results in relatively stable voltage responses. The amount of lithium that is plated on either side of the pellet can be estimated using Faraday's law, assuming that no side reactions are present:
| m = (ItM/nF) | (3) |
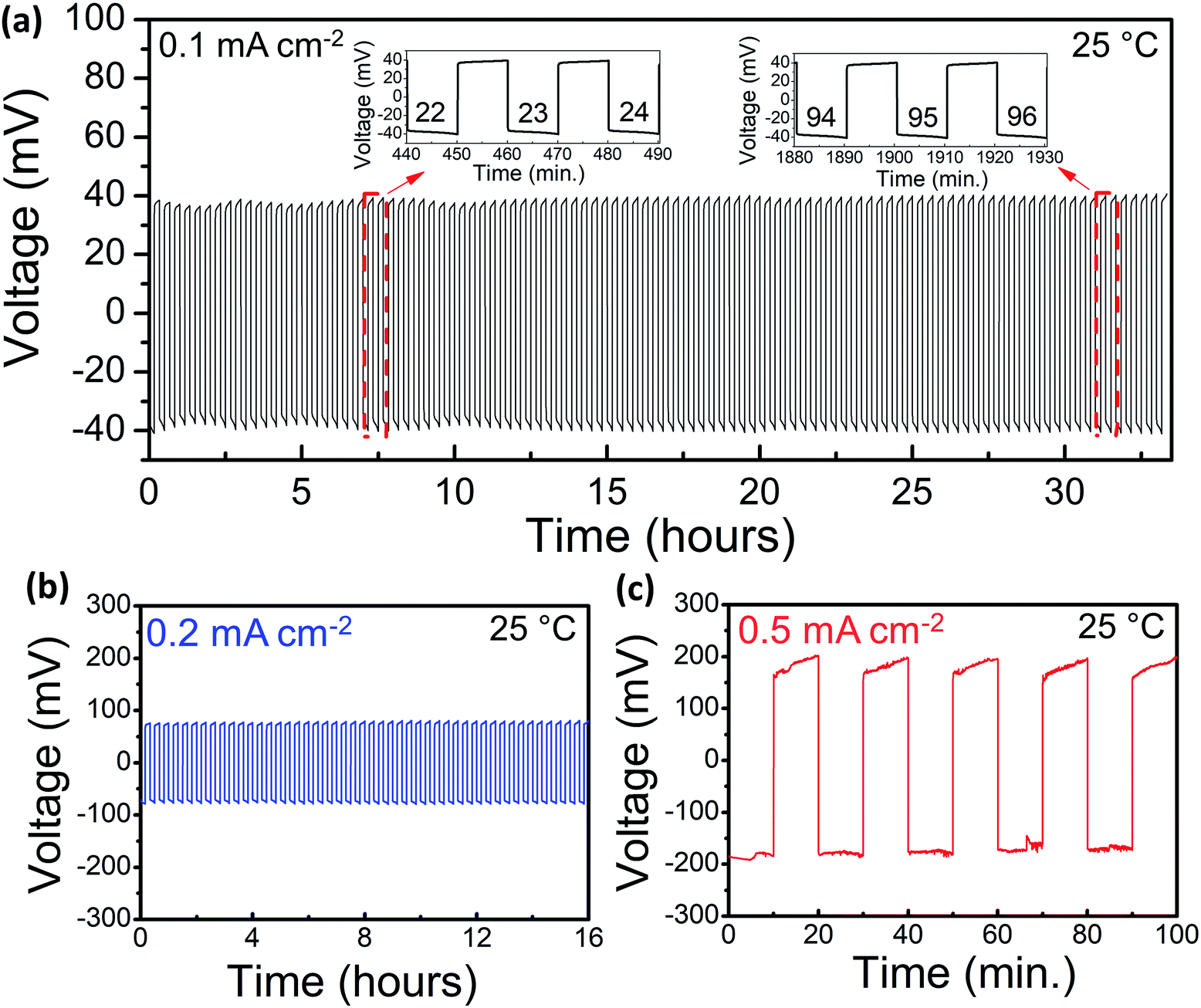 | ||
| Fig. 3 (a) Galvanostatic cycling of the SPICE-treated sample at 0.1 mA cm−2. Lithium plating or stripping is set to 10 min. Cycles 22–24 and 94–96 are magnified for clarity. Galvanostatic cycling of the SPICE-treated sample at (b) 0.2 and (c) 0.5 mA cm−2. | ||
The impedance spectrum of the sample was measured after the Li plating and stripping experiment (Fig. 2b). The stable cycling and low interfacial ASR provide evidence of the excellent interfacial contact between lithium and the coated garnet surface. After Li cycling, the samples were dismantled, and it was observed that most of the Li-ions were easily detached from the untreated sample, while the Li in the SPICE-treated sample could not be easily removed. The digital photographs of the pellets are shown in Fig. S10.† Post-test SEM analysis was also performed to examine the interface of the SPICE-treated and untreated samples, and the results are already shown in Fig. 1c and d. The poor interfacial contact between Li and the garnet is evident in Fig. 1d, with large areas that appear to be not connected at all. Fig. 1c confirms the excellent contact between Li and LLBZT in the SPICE-treated sample. Poor interfacial contact between Li and LLBZT in the untreated sample (Fig. 1d) is consistent with the high impedance in the EIS analysis (Fig. 2a) and the unstable Li cycling (Fig. S9†).
Conclusions
In summary, we demonstrated a new Surfactant-Processed Interlayer for Ceramic Electrolyte (SPICE) method to form a porous ZnO interlayer structure that improves the wetting of lithium onto the garnet pellet. The excellent contact of the SPICE-treated garnet and lithium, as confirmed by microstructural analysis, increased the contact area between Li and LLBZT and reduced the interfacial resistance from 70 Ω cm2 in the untreated sample to 10 Ω cm2. The SPICE-treated sample also showed excellent stability during lithium plating and stripping at current densities up to 0.5 mA cm−2. The flexibility of the SPICE method regarding the choice of precursor materials, solvents, and surfactants can provide other researchers the opportunity to advance material interface research, not only in solid-state lithium batteries but also in other energy and storage conversion devices such as solid oxide fuel cells and supercapacitors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (NSERC) have supported this work through the discovery grants of Prof. V. Thangadurai (Award number: RGPIN-2016-03853). We also thank the University of Calgary for the support.Notes and references
- K. M. Abraham, J. Phys. Chem. Lett., 2015, 6, 830–844 CrossRef PubMed.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- C. Yu, S. Ganapathy, E. R. H. v. Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1086 CrossRef PubMed.
- S. B. Schougaard, Science, 2016, 353, 543 CrossRef PubMed.
- J. M. Tarascon, Philos. Trans. R. Soc., A, 2010, 368, 3227 CrossRef PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2014, 7, 19 CrossRef PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef PubMed.
- G. G. Eshetu, S. Grugeon, S. Laruelle, S. Boyanov, A. Lecocq, J.-P. Bertrand and G. Marlair, Phys. Chem. Chem. Phys., 2013, 15, 9145–9155 RSC.
- A. Manthiram, Y. Xingwen and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef PubMed.
- L. F. Nazar, M. Cuisinier and Q. Pang, MRS Bull., 2014, 39, 436–442 CrossRef.
- T. Cleaver, P. Kovacik, M. Marinescu, T. Zhang and G. Offer, J. Electrochem. Soc., 2018, 165, A6029–A6033 CrossRef.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19 CrossRef PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef PubMed.
- V. Thangadurai, D. Pinzaru, S. Narayanan and A. K. Baral, J. Phys. Chem. Lett., 2015, 6, 292–299 CrossRef PubMed.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- Y. Li, J.-T. Han, C.-A. Wang, H. Xie and J. B. Goodenough, J. Mater. Chem., 2012, 22, 15357–15361 RSC.
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599–4604 CrossRef PubMed.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2016, 16, 572 CrossRef PubMed.
- Y. Jin and P. J. McGinn, Electrochim. Acta, 2013, 89, 407–412 CrossRef.
- K. Hofstetter, A. J. Samson and V. Thangadurai, Solid State Ionics, 2018, 318, 71–81 CrossRef.
- K. Hofstetter, A. J. Samson, S. Narayanan and V. Thangadurai, J. Power Sources, 2018, 390, 297–312 CrossRef.
- L. Cheng, E. J. Crumlin, W. Chen, R. Qiao, H. Hou, S. Franz Lux, V. Zorba, R. Russo, R. Kostecki, Z. Liu, K. Persson, W. Yang, J. Cabana, T. Richardson, G. Chen and M. Doeff, Phys. Chem. Chem. Phys., 2014, 16, 18294–18300 RSC.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- B. Liu, Y. Gong, K. Fu, X. Han, Y. Yao, G. Pastel, C. Yang, H. Xie, E. D. Wachsman and L. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 18809–18815 CrossRef PubMed.
- C.-L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef PubMed.
- W. Luo, Y. Gong, Y. Zhu, K. K. Fu, J. Dai, S. D. Lacey, C. Wang, B. Liu, X. Han, Y. Mo, E. D. Wachsman and L. Hu, J. Am. Chem. Soc., 2016, 138, 12258–12262 CrossRef PubMed.
- W. Luo, Y. Gong, Y. Zhu, Y. Li, Y. Yao, Y. Zhang, K. Fu, G. Pastel, C.-F. Lin, Y. Mo, E. D. Wachsman and L. Hu, Adv. Mater., 2017, 29, 1606042 CrossRef PubMed.
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 565–571 CrossRef PubMed.
- X. Wang and G. Yushin, Energy Environ. Sci., 2015, 8, 1889–1904 RSC.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef PubMed.
- W. G. Zeier, S. Zhou, B. Lopez-Bermudez, K. Page and B. C. Melot, ACS Appl. Mater. Interfaces, 2014, 6, 10900–10907 CrossRef PubMed.
- C. Jiang, M. Ichihara, I. Honma and H. Zhou, Electrochim. Acta, 2007, 52, 6470–6475 CrossRef.
- Q. Wu, W. Li, Y. Cheng and Z. Jiang, Mater. Chem. Phys., 2005, 91, 463–467 CrossRef.
- H. Li, X. Huang and L. Chen, Solid State Ionics, 1999, 123, 189–197 CrossRef.
- X. Liang, Q. Pang, I. R. Kochetkov, M. S. Sempere, H. Huang, X. Sun and L. F. Nazar, Nat. Energy, 2017, 2, 17119 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00234g |
| This journal is © The Royal Society of Chemistry 2018 |