
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mo-doped TiO2 photoanodes using [Ti4Mo2O8(OEt)10]2 bimetallic oxo cages as a single source precursor†
Miriam
Regue
 ab,
Katherine
Armstrong
c,
Dominic
Walsh
b,
Emma
Richards
c,
Andrew L.
Johnson
ad and
Salvador
Eslava
*ab
ab,
Katherine
Armstrong
c,
Dominic
Walsh
b,
Emma
Richards
c,
Andrew L.
Johnson
ad and
Salvador
Eslava
*ab
aCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: S.Eslava@bath.ac.uk
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 19th September 2018
Abstract
Photoelectrochemical solar water splitting is a promising and sustainable technology for producing solar fuels such as clean hydrogen from water. A widely studied photoanode semiconductor for this application is TiO2, but it suffers from a large band gap (3.2 eV) and fast recombination of electrons and holes. Herein, we present a novel, facile and rapid strategy to develop Mo-doped TiO2 (Mo:TiO2) mixed anatase–rutile photoanodes using [Ti4Mo2O8(OEt)10]2 bimetallic oxo cages as a single source precursor. These cages dissolved in tetrahydrofuran deposit by spray pyrolysis at 150 °C forming films with hierarchical porosity on the micrometer and nanometer scale. XPS, EDXS and UV-Vis spectroscopy reveal Mo atoms evaporate during annealing in air at temperatures 650–800 °C, contributing to the formation of nanostructures and porosity. XPS depth profiling, XRD, EDXS, Raman, and electron paramagnetic resonance indicate that the remaining Mo atoms are well spread and incorporated in the TiO2 lattice, at interstitial or substitutional sites of the rutile or anatase phases depending on the annealing temperature. Photocurrent measurements show that Mo:TiO2 photoanodes optimized at 700 °C outperform a TiO2 photoanode prepared in a similar manner by a factor of two at 1.23 VRHE. Finally, UV-Vis spectroscopy, conduction and valence band calculations, and incident-to-photon efficiency measurements show these Mo:TiO2 photoanodes possess a narrower band gap than TiO2 and higher efficiency in the visible light range (5% at 400 nm). These outcomes open a new avenue in the exploitation of titanium oxo cages and advance the development of photoelectrodes for water splitting and energy applications.
1. Introduction
The abundant solar energy, 1.3 × 105 TW per year reaching the Earth's surface, can be utilized to produce hydrogen fuel by splitting water, offering an excellent and sustainable alternative to fossil fuels. Among solar absorber candidates in photoelectrochemical (PEC) solar water splitting devices, TiO2 is one of the most promising ones due to its chemical and thermal stability, long durability, excellent optical and electronic properties, low cost and non-toxicity.1–3 However, TiO2-based PEC cells are still far from commercialization mainly due to both fast recombination of photogenerated electrons and holes in TiO2 and its large band gap (3.2 eV for the anatase phase) that results in a reduced use of the solar spectrum. One strategy to overcome its limitations is doping it with transition metals. The metal doping increases the solar light absorption of TiO2 and performance by incorporating additional energy levels within the band gap of the semiconductor.4,5 Nevertheless, this only seems to occur when there is an actual substitution of Ti atoms in the TiO2 lattice structure with the external metal (known as substitutional doping), that reduces the band gap of the material without compromising the surface of the photocatalyst.6 Otherwise, if metals are simply impregnated on the surface of the semiconductor they may result in electron–hole recombination and blocking of reaction sites.6,7Research on metal doping of TiO2 with Ta, Fe, Co, Mn, and Ni is abundant and has proved to be successful in both reducing the band gap and improving the photoelectrochemical performance (PEC).8–11 For instance, Yan et al. reported an enhancement in the PEC performance for Ta-doped TiO2 nanotube photoanodes in comparison to pristine TiO2.8 This enhanced PEC performance was attributed to a decrease in the band gap, lower charge transfer resistance and higher charge carrier density. Similarly, Zhao et al. successfully demonstrated that Fe-doped TiO2 results in a better PEC performance than pristine TiO2, attributed to a smaller band gap and thus a better light absorption.9 However, very few studies have been reported for Mo doping of TiO2 and those few were mainly focused on photodegradation of organic dyes, not PEC water splitting.12–16 In 2015 Zhang et al. reported Mo-doped TiO2 photoanodes prepared using two steps of Ti foil anodization and a third one of hydrothermal doping, obtaining an improved PEC performance over pure TiO2 photoanodes.17 The improvement was attributed to a decrease in the recombination of electrons and holes in the mixed phase of anatase and rutile and to better light absorption.
Heterometallic titanium oxo (HMTO) cages such as [TixOy(L)zMnXm], where L is a ligand, M a metal and X a halide, can be prepared with a wide range of metal compositions (e.g. with M = Co, Ni, and Mo), nuclearities, spatial arrangements, and functionalities via simple solvothermal methods and isolated by crystallization.18–23 Some of these cages, also called clusters, have successfully been used as precursors for photocatalysts or electrocatalysts, for example using graphene oxide as a sacrificial template or impregnating WO3 photoanodes.20,24 However, their wide adoption as single source precursors still requires finding facile and effective approaches that overcome their instability in water or humid environments.
In this publication, we report for the first time a facile and rapid synthesis of porous Mo-doped TiO2 photoanodes using a [Ti4Mo2O8(OEt)10]2 HMTO cage as a single source precursor. Stability of the cages during the deposition is ensured using anhydrous tetrahydrofuran as solvent and spray pyrolysis as deposition method. We reveal that upon deposition, the calcination at temperatures above 600 °C induces sublimation of Mo atoms adding porosity and nanostructured features to remaining TiO2, while the rest of Mo atoms effectively occupy substitutional or interstitial sites in the TiO2 lattice. The resulting Mo:TiO2 photoanode optimized at 700 °C outperforms by a factor of two a pure TiO2 photoanode prepared in a similar manner in PEC solar water splitting. The results herein presented therefore reveal new strategies to develop efficient photoelectrodes for water splitting applications using Mo as both a sacrificial agent and dopant and opens an avenue to exploit HMTO cages as single source precursors using spray pyrolysis.
2. Experimental
Materials
Titanium(IV) ethoxide [Ti(OEt)4], anhydrous molybdenum(V) chloride (MoCl5, 99.99%), anhydrous tetrahydrofuran (≥99.9%, inhibitor free), anhydrous ethanol (<0.0003% water), titanium isopropoxide (TTIP, 97%), acetylacetone (AcAc), cobalt nitrate (Co(NO3)2), iron chloride hexahydrate (FeCl3·6H2O), sodium acetate (NaOAc) and tetrabutylammonium hexafluorophosphate (TBAPF6) were provided by Sigma Aldrich. Cobalt(II) chloride (CoCl2, anhydrous 97%) was provided by Alfa Aesar. Aluminoborosilicate glass (ABS) coated with fluorine-doped tin oxide (FTO) transparent conductive layer (8 Ω sq−1) was provided by Solaronix, SA. These FTO-ABS substrates were cleaned ultrasonically in a 2% aqueous Hellmanex III solution followed by dipping in deionized water, acetone and isopropyl alcohol, ultrasonicating in each step for 3 min. Finally, the substrates were rinsed with deionized water and dried with compressed air.Synthesis of [Ti4Mo2O8(OEt)10]2
[Ti4Mo2O8(OEt)10]2 oxo cages (Fig. 1) were synthesized using a solvothermal process as described in a previous article by Eslava et al.25 Briefly, Ti(OEt4) (14 mL, 66.8 mmol), MoCl5 (1.128 g, 4.12 mmol) and anhydrous ethanol (14 mL) were carefully mixed in a 45 mL Teflon-lined stainless steel autoclave and heated to 150 °C for 24 hours. The autoclave was left overnight to cool down to room temperature and mm size brown-red crystals were obtained (88% yield). Elemental analysis (%) calculated for C40H100Mo4O36Ti8: C 25.7, H 5.3; found: C 25.6, H 5.3. Due to the sensitive nature of these reactants, all manipulations were carried out in an air-free atmosphere involving the use of a glove box and Schlenk line when necessary.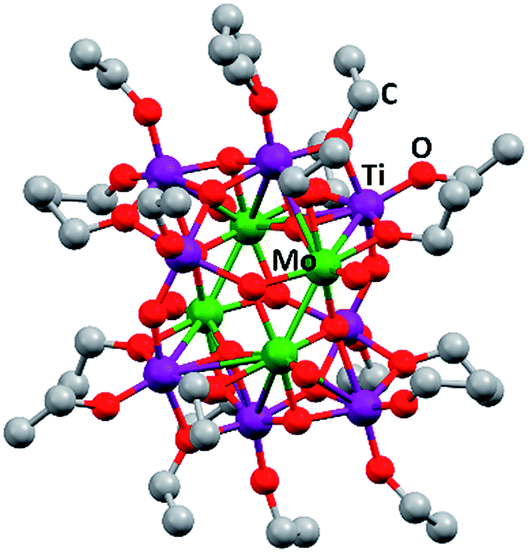 | ||
| Fig. 1 Ball and stick drawing of [Ti4Mo2O8(OEt)10]2 represented from CCDC no. 812604.25 Mo: green; Ti: purple; O: red; C: grey. | ||
Preparation of Mo:TiO2 and TiO2 films
Mo-doped TiO2 (abbreviated as Mo:TiO2) and pure TiO2 films to be studied as photoanodes were prepared using a manual spray-pyrolysis system (Clarke CAB3P) on FTO-ABS. The solutions employed for the spray pyrolysis deposition were prepared in an argon atmosphere using a Schlenk line. Nevertheless, the actual spray pyrolysis deposition process was conducted in air, but vessels were kept closed when possible. The precursor solution for the Mo:TiO2 photoanodes was prepared by dissolving [Ti4Mo2O8(OEt)10]2 (0.96 g) in anhydrous tetrahydrofuran (20 mL). The precursor solution for the preparation of pure TiO2 was carried out following an established method.26 Briefly, a 0.2 M solution of TTIP was prepared by diluting TTIP and AcAc in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 volumetric ratio and topping up with absolute ethanol in order to obtain 0.2 M solution of TTIP.
:2 volumetric ratio and topping up with absolute ethanol in order to obtain 0.2 M solution of TTIP.
The spray pyrolysis deposition of previous precursor solutions to prepare Mo:TiO2 and TiO2 photoanode films was conducted as follows. In a first stage, FTO-ABS was pre-heated on a hot plate at 150 °C. Secondly, the as-prepared precursor solutions were sprayed on top of the pre-heated FTO-ABS, at a constant distance of ca. 5 cm from the surface of the FTO-ABS to the spray pyrolysis nozzle. Three deposition layers were performed per sample. Finally, the as-prepared Mo:TiO2 and TiO2 films were annealed at different temperatures between 450 and 800 °C for 2 h in air at a ramp rate of 10 °C min−1. This range of temperatures was chosen to ensure full conversion of the precursor to the metal oxide and to evaluate the effect that different annealing conditions might have on the photoanode performance. The resultant photoanodes were denoted as Mo:TiO2-### and TiO2-###, where ### is the corresponding annealing temperature (°C).
Cobalt phosphate (Co-Pi) loading on Mo:TiO2-700 photoanodes was carried out by photo-electrodeposition.27,28 The electrolyte consisted of 0.05 mM of cobalt nitrate in 0.1 M potassium phosphate buffer (pH = 7) and the applied potential 1 VRHE was kept for 20 s under simulated sunlight (AM 1.5G, 100 mW cm−2) from a filtered 300W Xenon lamp source. Illumination was directed towards the back of the FTO-ABS working electrode. CoFeOx was deposited on Mo:TiO2-700 by electrodeposition.29 The electrolyte consisted of 10 mM FeCl3·6H2O, 16 mM CoCl2 and 0.1 M NaOAc dissolved in deionized water. The pH of the solution was 4.90. The deposition was carried out by positively sweeping the voltage from 1.1 to 1.4 VAg/AgCl three times. In both depositions, a Compactstat. potentiostat (Ivium Technologies) was used and an electrochemical cell consisting of a Pt counter electrode, a silver chloride (Ag/AgCl/3.5 M KCl) reference electrode, and a Mo:TiO2-700 photoanode as working electrode.
Characterization
13C NMR spectroscopy was conducted at room temperature using a 500 MHz Agilent Propulse spectrophotometer. Samples were dissolved in dried deuterated benzene (C6d6). Elemental analysis was performed on a Carlo Erba Flash 2000 Elemental Analyser. Field emission scanning electron microscopy images (FE-SEM) were acquired using a JEOL FESEM6301F and energy dispersive X-ray spectroscopy (EDXS) was carried out in a SEM 6480LV equipped with a high sensitivity Oxford INCA X-Act SDD X-ray detector. X-Ray diffraction (XRD) patterns were collected in the 2 theta range 10–80° with a Bruker AXS D8 Advance using Cu Kα (0.154 nm) radiation with a total integration time of 960 s. Raman spectroscopy was carried out on a Renishaw inVia system using a 532 nm diode-pumped solid state laser (DPSS) manufactured by Cobolt. The laser beam was focused onto the sample using a 50× long distance objective. X-Ray photoelectron spectroscopy (XPS) depth profiling was performed using a ESCALAB 250 Xi instrument manufactured by Thermo Fisher Scientific. Measurements were carried out using a monochromated Al Kα X-ray source with an energy of 1486.68 eV. The X-ray spot size was of 900 μm and the pass energy for the high resolution scans was of 50 eV. The depth profile for the sample was obtained by etching the surface of the sample with an Ar+ ion gun (2000 eV, high current) for different times (0, 60, 180 and 420 s). C 1s XPS spectra was used as an internal charge correction. Samples studied via electron paramagnetic resonance (EPR) spectroscopy were evacuated at 393 K for over 12 h to reduce the influence of physisorbed water. Samples were maintained under static vacuum (10−5 mbar) for the duration of the experiments. For EPR analysis, powder samples were prepared by drying and calcining in air at 650, 700 and 800 °C in a porcelain dish a solution of 0.96 g of [Ti4Mo2O8(OEt)10]2 in 20 mL of anhydrous tetrahydrofuran. High resolution transmission electron microscopy (HRTEM) images were obtained using a JEOL JEM-2100Plus microscope. Ultraviolet–visible (UV-Vis) spectra were collected in a Cary 100 diffuse reflectance UV-Vis spectrophotometer.(Photo)Electrochemical measurements
(Photo)electrochemical performance of photoanodes was evaluated using a CompactStat. potentiostat (Ivium Technologies). Photocurrents were measured under simulated sunlight (AM 1.5G, 100 mW cm−2) from a filtered 300W Xenon lamp source (Lot Quantum Design) or under UV illumination (365 nm, 3.6 mW cm−2) from a ModuLight IM3412 LED light (Ivium Technologies). PEC cells consisted of three electrodes with Pt as the counter electrode, silver chloride (Ag/AgCl/3.5 M KCl) as the reference electrode and as-prepared photoanodes as the working electrodes.Electrochemically active surface area (ECSA) of photoanodes was investigated using cyclic voltammetry (CV), scanning from 0 to 0.17 VAg/AgCl at scan rates between 10 and 250 mV s−1, in 1 M KOH solution (pH = 13.7). ECSA is proportional to the double layer capacitance (Cdl), which is estimated from the slope of the plot Δj vs. scan rate and dividing by two.30 Δj is equal to (ja–jc), where ja and jc are the anodic and cathodic current densities, respectively, in this case taken at 0.1 VAg/AgCl in the CV scans.31
Conduction and valence band (CB & VB) positions were measured from CV curves recorded in acetonitrile containing 0.1 M of tetrabutylammonium hexafluorophosphate (TBAPF6) at a scan rate of 50 mV s−1 and using the following formula:32–34
| CB (or VB) (eV) = −4.8−(E − E1/2) | (1) |
Photoelectrochemical performances of photoanodes were carried out in a 1 M KOH (pH = 13.7) electrolyte solution. Illumination was directed towards the back of the FTO-ABS working electrode and a mask was placed on top of the photoelectrode to define the illuminated area. Photocurrent–time curves were performed at an applied bias of 1.23 V vs. the reversible hydrogen electrode (VRHE). Photocurrent–potential curves were recorded at a scan rate of 20 mV s−1. The measured Ag/AgCl potentials (EAg/AgCl) were converted to RHE potentials 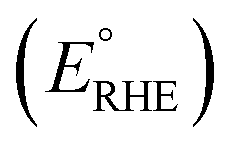 following the Nernst equation:
following the Nernst equation:
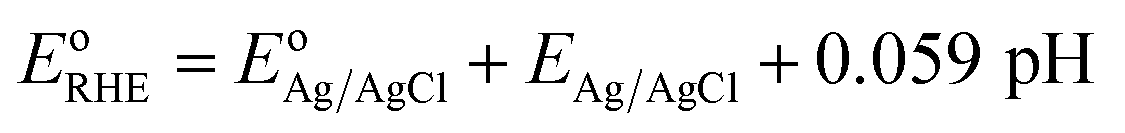 | (2) |
 is 0.205 V at 25 °C (3.5M KCl). Photoelectrochemical impedance spectroscopy (PEIS) was carried out under simulated sunlight (AM 1.5G, 100 mW cm−2) at a direct current (DC) potential of 1.23 VRHE and an alternating current (AC) potential frequency range of 100000–0.01 Hz with an amplitude of 5 mV. Incident photon-to-current efficiency (IPCE) measurements were calculated using the same Xe light source and a triple grating Czerny–Turner monochromator. The intensity of monochromatic light was measured at the working electrode position with SEL033/U photodetector (International Light Technologies). The following equation was used to calculate the IPCE values:36
is 0.205 V at 25 °C (3.5M KCl). Photoelectrochemical impedance spectroscopy (PEIS) was carried out under simulated sunlight (AM 1.5G, 100 mW cm−2) at a direct current (DC) potential of 1.23 VRHE and an alternating current (AC) potential frequency range of 100000–0.01 Hz with an amplitude of 5 mV. Incident photon-to-current efficiency (IPCE) measurements were calculated using the same Xe light source and a triple grating Czerny–Turner monochromator. The intensity of monochromatic light was measured at the working electrode position with SEL033/U photodetector (International Light Technologies). The following equation was used to calculate the IPCE values:36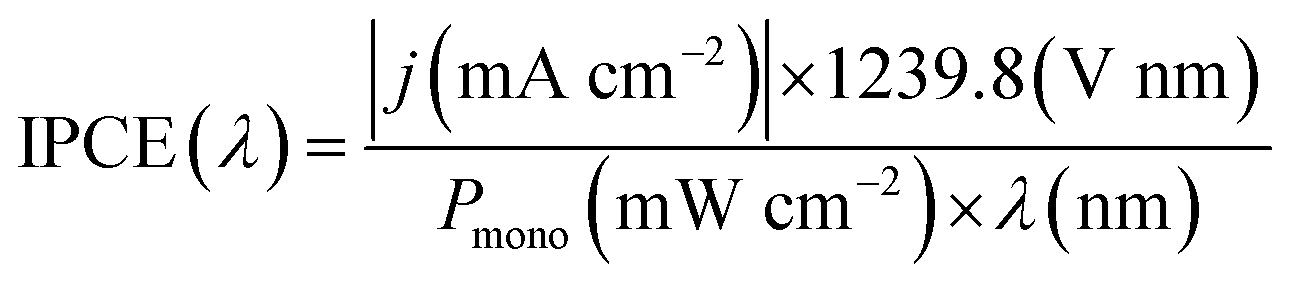 | (3) |
3. Results and discussion
The synthesis of [Ti4Mo2O8(OEt)10]2 cages was successfully performed using a solvothermal synthesis. The 13C NMR spectra of the product showed the ten characteristic sharp resonances of the cage at δ 75.24, 75.12, 73.80, 73.49, 73.38, 73.30, 72.65, 71.67, 71.22 and 70.06 ppm belonging to the different –OCH2– environments in the structure, in agreement with literature (Fig. S1, ESI†).25 In addition, CHN elemental analysis further confirmed the successful formation of [Ti4Mo2O8(OEt)10]2 (wt% calculated for C40H100Mo4O36Ti8: C 25.7, H 5.3; found: C 25.6, H 5.3). Fig. 1 shows a ball and stick model of the cage, which highlights a heterometallic oxo core with four MoV atoms in a rectangle and eight TiIV atoms. Mo and Ti atoms bridge together by a combination of μ3-, μ4-O and μ2-OEt (CCDC no. 812604).Spray pyrolysis was used to deposit solutions of [Ti4Mo2O8(OEt)10]2 cages or titanium isopropoxide-acetylacetone (TTIP-AcAc), respectively. The as-deposited films were then annealed in air at temperatures between 450 and 800 °C for 2 hours and allowed to cool to room temperature. The morphology of the as-prepared films was evaluated using SEM. Fig. 2 shows SEM images at different magnifications of Mo:TiO2 prepared at different annealing temperatures (650, 700 and 800 °C), along with a film prior to calcination for comparison (i.e. a film of [Ti4Mo2O8(OEt)10]2 cages deposited by spray pyrolysis at 150 °C). At lower magnification (Fig. 2, 1st row), all Mo:TiO2 films, even the un-annealed sample, exhibit almost the same morphology, showing aggregated islands evenly distributed on top of FTO-ABS support. At slightly higher magnification (Fig. 2, 2nd row), a large amount of micrometer cavities on Mo:TiO2 films are observed. These cavities contribute to an increase of surface area, which in turn must result in more active sites for the PEC oxygen evolution reaction. At even higher magnification (Fig. 2, 3rd row), it can be observed that well-defined nanostructures form as the annealing temperature increases. A highly smooth and fine surface is present before calcination (Fig. 2i), but after calcination, grain-rice-shaped nanostructures appear creating nanometer-size porosity and extra surface area (Fig. 2j–l).
 | ||
| Fig. 2 SEM images of Mo:TiO2 at different magnifications. (a, e and i) Film after spray-pyrolysis deposition at 150 °C, (b, f and j) Mo:TiO2-650, (c, g and k) Mo:TiO2-700 and (d, h and l) Mo:TiO2-800. | ||
SEM images of the pure TiO2 film at different magnifications are shown in Fig. S2 (ESI†). Unlike Mo:TiO2 films, TiO2-650 (prepared with TTIP-AcAc) does not show either the presence of cavities nor nanostructures. This comparison therefore reveals the benefits of using [Ti4Mo2O8(OEt)10]2 cages as a precursor. Their decomposition and transformation to Mo-doped TiO2 during the spray pyrolysis and posterior calcination leads to the formation of cavities, nanostructures and porosity. Cavities must result from the drying step upon spray pyrolysis at 150 °C. More cavities may form during the calcination and oxidation of the cages' carbon content up to 400–500 °C. Above this temperature, the grain-rice-shaped nanostructures and its associated porosity must result from the sintering of TiO2 and especially from the very likely sublimation of Mo atoms. Previous reports have shown that MoO3 sublimes above 700 °C.37 The sublimation of Mo atoms was confirmed by atomic quantification from XPS data (Fig. 3a). The amount of Mo in the films decreased with temperature. For example, at the top surface (XPS-etching time 0 s) it went from 10.6 at% for Mo:TiO2-650 to 4.9 and 4.3 at% for Mo:TiO2-700 and Mo:TiO2-800, respectively, indicating Mo sublimes within the temperatures of study. Accordingly, the percentage of carbon (C) decreases with temperature and that of Ti increases. Carbon is present from solvents and cage ethoxides and deposition of volatile organic compounds during storage. The Mo sublimation was further confirmed by an experiment of simply heating MoCl5 up to 700 °C in air, which showed its complete sublimation. Therefore, Mo atoms work as pore formers, sacrificial agents that increase the porosity in the films and allow their nanostructuring.
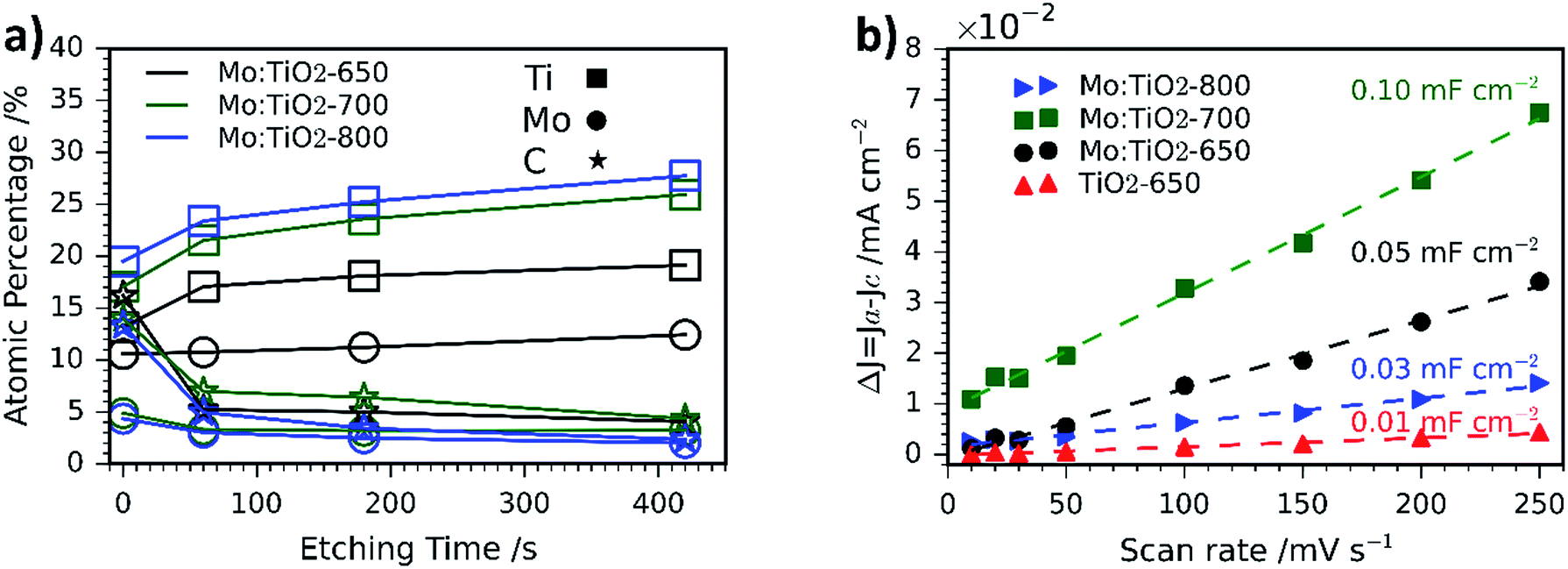 | ||
| Fig. 3 (a) Atomic percentage (at%) distribution of Ti, Mo and C obtained via XPS depth profiling. (b) Capacitive current Δj versus scan rate curves of Mo:TiO2 and TiO2-650. | ||
The XPS depth profiling indicated that Mo is homogeneously distributed at different depths, but some gradient is formed at highest temperatures of 700 and 800 °C, with more C and Mo present at the surface (Fig. 3a). This may result from the gasification and sublimation taking place and accumulation at the top during the process.
ECSA measurements are shown in Fig. 3b and the corresponding CV curves in Fig. S3 (ESI†). In such measurements, the slope of the current density vs. scan rate can be related to the double layer capacitance, which is directly proportional to the ECSA.31 Based on the obtained results, Mo:TiO2-700 possesses the highest surface area (Cdl = 0.10 mF cm−2), whereas TiO2-650 is the sample with the smallest surface area (Cdl = 0.01 mF cm−2). These results agree well with SEM images, where grain-rice-shaped nanostructures and porosity are observed for Mo:TiO2-700 (Fig. 2k) and very flat films for TiO2-650 (Fig. S2, ESI†). Among all Mo:TiO2 photoanodes, Mo:TiO2-800 is the sample with the smallest surface area, due to the largest grains formed at highest temperature (Fig. 2l).
SEM-EDXS analysis was also carried out to evaluate the distribution of Ti, O and Mo atoms at the surface of the films. Fig. S4 (ESI†) reveals a uniform distribution of Ti and Mo atoms at the surface of Mo:TiO2-650 and Mo:TiO2-700. The amount of Mo atoms at the surface of Mo:TiO2-800 is considerably lower supporting the hypothesis that Mo species sublime during the calcination.
Fig. 4a and b show the XRD patterns of the resulting Mo:TiO2 and TiO2 films on FTO-ABS. Mo:TiO2-650 and Mo:TiO2-700 exhibit diffraction peaks corresponding to both anatase TiO2 and rutile TiO2. The diffraction peaks at 2θ 25.3 and 48.0° are indexed to the diffraction planes (1 0 1) and (2 0 0) of anatase TiO2, respectively (ICDD-JCPDS no. 75-1537). The diffraction peaks at 2θ 27.4, 36.1, 39.2, 44.0, 54.3, 69.0 and 69.8° correspond to (1 1 0), (1 0 1), (2 0 0), (2 1 0), (2 1 1), (3 0 1) and (1 1 2) diffraction planes, resp., of rutile TiO2 (ICDD-JCPDS no. 88-1173). The diffraction of the rutile phase increases and that of anatase decreases with annealing temperature (Fig. 4a), indicating that the conversion of anatase to rutile was promoted at highest temperatures.38 Actually, Mo:TiO2-800 only shows diffraction peaks indexed to rutile TiO2, due to the high calcination temperature employed. No diffraction peaks corresponding to any Mo phase such as MoO3 are observed in any of the samples, which suggests that Mo6+/5+ could be incorporated into the lattice of TiO2. MoO2 was unlikely to be formed since phase transformation from tetragonal MoO2 to orthorhombic MoO3 occurs at temperatures above 350 °C.39
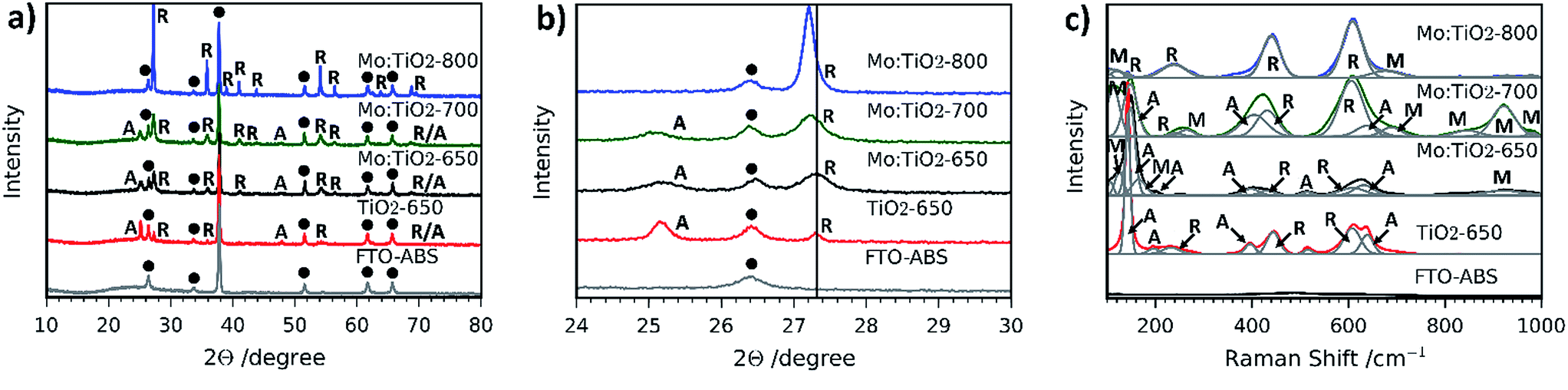 | ||
| Fig. 4 (a) XRD patterns and (b) XRD diffraction profiles on (101) plane of anatase TiO2 and (1 1 0) plane of rutile TiO2 of Mo:TiO2, TiO2-650 and FTO-ABS substrate. A: anatase, R: rutile and black dot: FTO. (c) Raman spectra from 100 to 1000 cm−1 of Mo:TiO2, TiO2-650 and FTO-ABS substrate. A: anatase, R: rutile, M: Mo. | ||
In Fig. 4b an expansion of the (1 0 1) and (1 1 0) diffraction peaks of anatase and rutile, respectively, of the different Mo:TiO2 and pure TiO2 films is shown. A shift of the anatase and rutile diffraction peaks towards lower angles for Mo:TiO2-700 and Mo:TiO2-800 in comparison to pure TiO2 is observed, which indicates incorporation of Mo6+ or Mo5+ atoms in the TiO2 lattice structure upon exposure to those temperatures.13 The reported ionic radius for Mo6+ and Mo5+ are 0.0620 and 0.0610 nm, respectively, whereas the Ti4+ ionic radius is 0.0605 nm.13,40,41 The similarity in the ionic radius of these ions facilitates the aliovalent substitution of Ti4+ atoms for Mo6+/5+ in the TiO2 lattice, and due to the slightly larger size of Mo6+/5+ in comparison to Ti4+ a shift in the diffraction pattern towards lower angles is observed. However, Mo:TiO2-650 shows a smaller shift towards lower angles, suggesting that at lower temperatures the majority of Mo6+/5+ atoms are not occupying Ti4+ sites in the TiO2 lattice structure. Instead, Mo6+/5+ must be distributed on the surface of TiO2 or occupying interstitial sites within the TiO2 lattice, without distorting the crystal structure of either anatase or rutile TiO2. In fact, this may also suggest that Mo6+ atoms could be present in the form of MoO3 that could be either highly dispersed on the TiO2 surface or of amorphous structure, and therefore not detectable by XRD analysis.
Raman spectroscopy was also carried out to further verify the presence of rutile TiO2, anatase TiO2 and the substitution of Mo for Ti atoms in the lattice structure of TiO2. Fig. 4c shows the Raman spectra of all films. The presence of anatase-TiO2 and rutile-TiO2 is confirmed by the sharp peaks observed in all Raman spectra, with the exception of Mo:TiO2-800 for which only rutile TiO2 is observed, in agreement with the XRD patterns (Fig. 4a and b). The sharp bands at ca. 145, 395, 515 and 635 cm−1 correspond to Raman active modes of anatase TiO2 and bands at 230, 445 and 610 cm−1 to Raman active modes of rutile TiO2.42,43 Interestingly, some bands ascribed to the presence of Mo are also observed. For instance, in the region between 870–970 cm−1 there are bands belonging to hydrated terminal Mo–O and Mo–O–Mo vibrations.44,45 The presence of crystalline MoO3 can be discarded since its characteristic main bands at 996, 820 and 666 cm−1 are not observed, which is in accordance with XRD analysis. Moreover, the weak bands observed in the range 100–200 cm−1 are attributed to bending modes of Mo–O–Mo.46 This verifies the incorporation of Mo atoms into the lattice of TiO2, suggesting the presence of Ti–O–Ti, Mo–O–Mo and Mo–O–Ti bonds in Mo:TiO2.47 The Mo Raman vibrations are most evident for Mo:TiO2-700 and drastically decrease for Mo:TiO2-800, indicating that at 700 °C the Mo incorporation into the oxide crystalline structure is at its maximum. This temperature dependence is attributed to evaporation of the Mo species, which as previously shown, sublime at relatively low temperatures. Along the same lines, the Mo Raman vibrations for Mo:TiO2-650 are relatively weak, which further confirms that the vast majority of Mo atoms are not incorporated in the TiO2 lattice structure, instead they are impregnated on the surface of TiO2 or distributed in interstitial sites of the TiO2 structure, which agrees well with XRD patterns.
The composition and chemical state of Mo:TiO2 films were further characterized by XPS. Fig. 5 shows the XPS high resolution spectra at the surface of Mo:TiO2 samples. Ti 2p resolution spectra show the two characteristic peaks of Ti4+ in TiO2 at 458 and 464 eV in the Mo:TiO2 films.42,48 The O 1s spectra for Mo:TiO2 films are shown in Fig. 5b. The peak at lower binding energies mainly corresponds to crystal lattice oxygen O–Ti4+, whereas the smaller peaks at higher binding energies correspond to hydroxyl groups or adsorbed water on the surface i.e. Ti–OH or Mo–OH.42,48Fig. 5c shows the high-resolution XPS surface spectra of Mo 3d. Surface Mo 3d spectra of all Mo:TiO2 films show the characteristic peaks of Mo6+ situated at binding energies at 232 and 235 eV.49–51 An additional doublet appears at lower binding energies for all Mo:TiO2 films, attributed to the presence of some Mo5+ centers in the films.52 XPS O 1s and Ti 2p spectra of TiO2-650 are shown in Fig. S5 (ESI†). The binding energies for Ti 2p and O1s are in agreement with Mo:TiO2 samples.
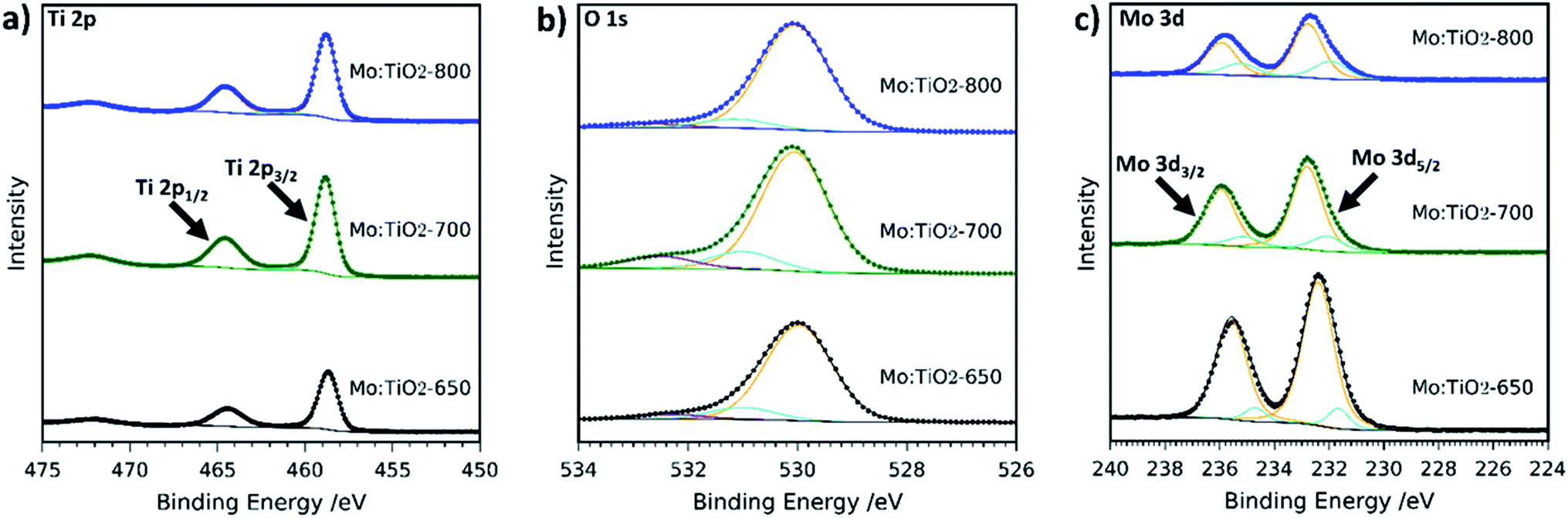 | ||
| Fig. 5 XPS spectra of (a) Ti 2p, (b) O 1s and (c) Mo 3d of Mo:TiO2. Scattered points correspond to raw data acquired in the measurements and solid lines to the fitted values. | ||
Electron paramagnetic resonance (EPR) spectroscopy was undertaken (at 120 K) to investigate the nature of the Mo doping within the series of TiO2 lattice structures. EPR spectroscopy only detects the presence of paramagnetic species, thus the Mo5+ species are observed whereas there is no detection of Mo6+ centers. The EPR data (Fig. 6) indicates the presence of Mo5+ in all the films, in agreement with the XPS analysis. Variation in g values for the different samples demonstrated the existence of multiple Mo5+ species. As an example, the EPR spectrum of Mo:TiO2-650 indicated the presence of MoO3+ species on the surface, characterized by an axial g-tensor (g⊥ = 1.932 and g∥ = 1.886) and corresponding weak hyperfine satellite lines originating from coupling of the unpaired electron to the two nuclear spin active isotopes of molybdenum (95,97Mo, both with spin I = 5/2 and total natural abundance of 25.5%; A⊥ = 112 MHz and unresolvable A∥). Additional contributions from bulk Mo5+ (g1 = 1.944, g2 = 1.944, g3 = 1.839; A1 = 198, A2 = 75 and A3 = 93 MHz) and a small contribution from Mo5+ in substitutional anatase lattice positions (g1 = 1.917, g2 = 1.828, g3 = 1.828) was also detected in the Mo:TiO2-650 sample. In contrast, the EPR spectra obtained for both the Mo:TiO2-700 and Mo:TiO2-800 samples confirmed the existence of substitutional and interstitial doping in the TiO2 rutile lattice. The variation in Mo sites displayed via EPR for Mo:TiO2-650 and both Mo:TiO2-700 and Mo:TiO2-800 confirms that substitutional doping mainly occurs in rutile TiO2.
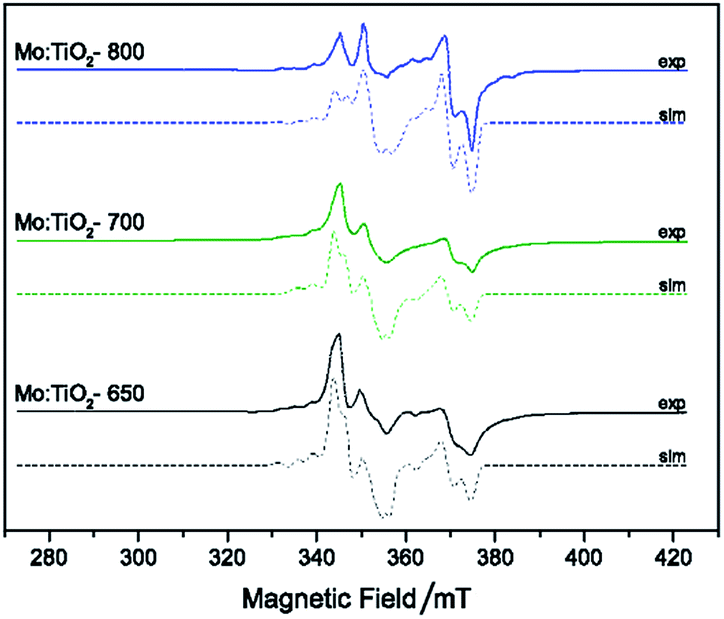 | ||
| Fig. 6 CW X-band EPR spectra T (T = 120 K) of degassed samples at 393 K of Mo:TiO2, experimental (solid line) and simulation (dashed-line). | ||
The particle size and crystallinity of Mo:TiO2 were evaluated using TEM and SAED-TEM. Fig. 7 shows the corresponding SAED diffraction patterns, bright-field TEM and HRTEM images of Mo:TiO2. To perform these analyses, a few milligrams of film deposited on FTO-ABS was scratched and dispersed in ethanol followed by TEM grid loading. The SAED-diffraction patterns (Fig. 7, 1st column) indicate a high polycrystalline character of these Mo:TiO2 and TiO2 particles. The corresponding diffraction pattern along with peak identification is shown in the inset of these figures. These data agree well with XRD where diffraction peaks corresponding to TiO2 anatase and rutile are shown in Mo:TiO2-650, Mo:TiO2-700 and pure TiO2, whereas only TiO2 rutile is observed in Mo:TiO2-800. Particle size distributions are shown in Fig. S6 (ESI†). In all Mo:TiO2 samples, the particles possess a well-defined particle shape in comparison to pure TiO2. Nevertheless, a large variability of particle sizes is observed in Mo:TiO2-650 suggesting an insufficient annealing temperature for the formation of uniform particles. Mo:TiO2-800 has the highest particle size due to aggregation and sintering of particles at high calcination temperatures.53,54
 | ||
| Fig. 7 SAED-TEM diffraction patterns, TEM and HRTEM images: (a–c) Mo:TiO2-800, (d–f) Mo:TiO2-700, (g–i) Mo:TiO2-650, (j–l) TiO2-650, (m) TiO2-650, (n) Mo:TiO2-650, (o) Mo:TiO2-700 and (p) Mo:TiO2-800. For clarity, images (b), (i) and (p) do not have scales consistent with the rest. | ||
HRTEM images are shown in Fig. 7m–p. These images also reveal the highly crystalline character of Mo:TiO2 and pure TiO2. The measured lattice spacing for the TiO2-650 sample agrees well with XRD analysis, since lattice spacings corresponding to the (101) diffraction plane of TiO2 anatase and (101) of TiO2 rutile are observed. In line with this, lattice spacings for Mo:TiO2 also agree well with XRD and Raman analysis. More precisely, Mo:TiO2-650 and Mo:TiO2-700 show diffraction planes corresponding to both TiO2 rutile and anatase crystalline phases, whereas only rutile TiO2 particles are observed in Mo:TiO2-800.
UV-Vis spectroscopy Tauc plots of Mo:TiO2 and pure TiO2 films are depicted in Fig. 8a and the corresponding absorption spectra are shown in Fig. S7 (ESI†). All Mo:TiO2 samples exhibit lower band-gap energy values ranging from 2.6 to 2.7 eV compared to TiO2-650, ca. 3.1 eV, being in accordance with literature reports.7 The overall band-gap reduction for all Mo:TiO2 is attributed to the incorporation of Mo6+/5+ centers in the TiO2 lattice structures, contributing to the formation of a shallow donor energy level below the CB of TiO2.47,55 Two distinguished slopes are observed for Mo:TiO2-800. The lower energy one is related to the band gap, while the higher one we assign it to an Urbach tail caused by sample disorder (i.e. Ti–O, Ti–O–Mo bond breaking due to anatase transformation to rutile and Mo sublimation).56
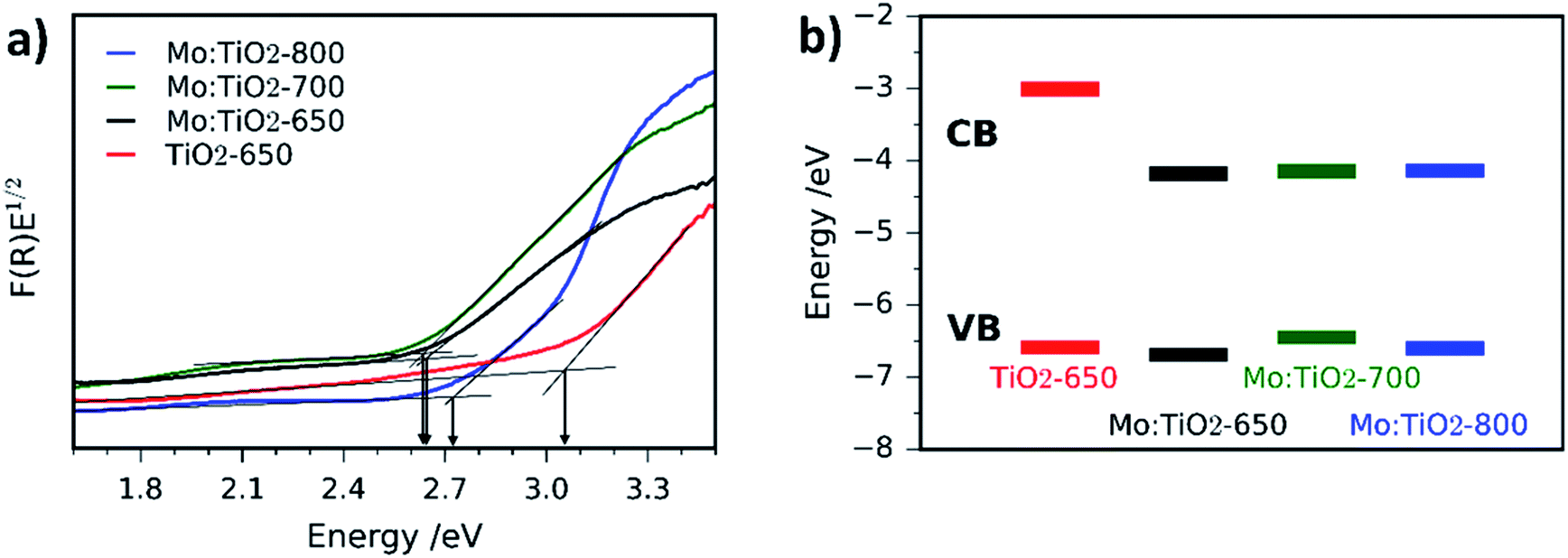 | ||
| Fig. 8 Tauc plots of Mo:TiO2-650, Mo:TiO2-700, Mo:TiO2-800 and pure TiO2-650 photoanodes measured via diffuse reflectance UV-Vis spectroscopy. (b) Schematic diagram of CB and VB energy levels obtained by CV of Mo:TiO2 and TiO2 photoanodes. | ||
The CB and VB position of TiO2-650 and Mo:TiO2 photoanodes were determined using CV curves (Fig. S8, ESI†). Detailed information of the electrochemical characteristics of TiO2 and Mo:TiO2 are shown in Table S1 (ESI†). A schematic diagram of the relative position of CB and VB energy levels for TiO2-650 and Mo:TiO2 samples is shown in Fig. 8b. Interestingly, in all Mo:TiO2 samples the CB offset is narrowed down from approximately −3.01 eV for TiO2-650 to ca. −4.2 eV for Mo:TiO2. This further confirms that the incorporation of Mo5+/6+ atoms at the TiO2 lattice structure reduces the overall band-gap of TiO2. The electrochemical band gap values obtained from CV curves are also shown in Table S1 (ESI†). The difference between the electrochemical band gap and optical band gap of TiO2 and Mo:TiO2 photoanodes is of 0.1–0.5 eV, which falls within the range of error.57
The photocurrent density (at 1.23 VRHE) as a function of annealing temperature for front illumination (via the electrolyte–film interface) and back illumination (via the ABS) was evaluated for Mo:TiO2 and pure TiO2 films and the results are shown in Fig. 9a. Photocurrent performances are higher when films are illuminated from the back, which indicates the porous photoanodes have a sufficient thickness. Fig. 9a also indicates that the optimal annealing temperature for pure TiO2 is 650 °C (and for Mo:TiO2 is 700 °C). In view of these results, all the following photoelectrochemical experiments were carried out directing the light towards the back of the photoelectrode (via the ABS) and using TiO2-650 as a benchmark against Mo:TiO2 films.
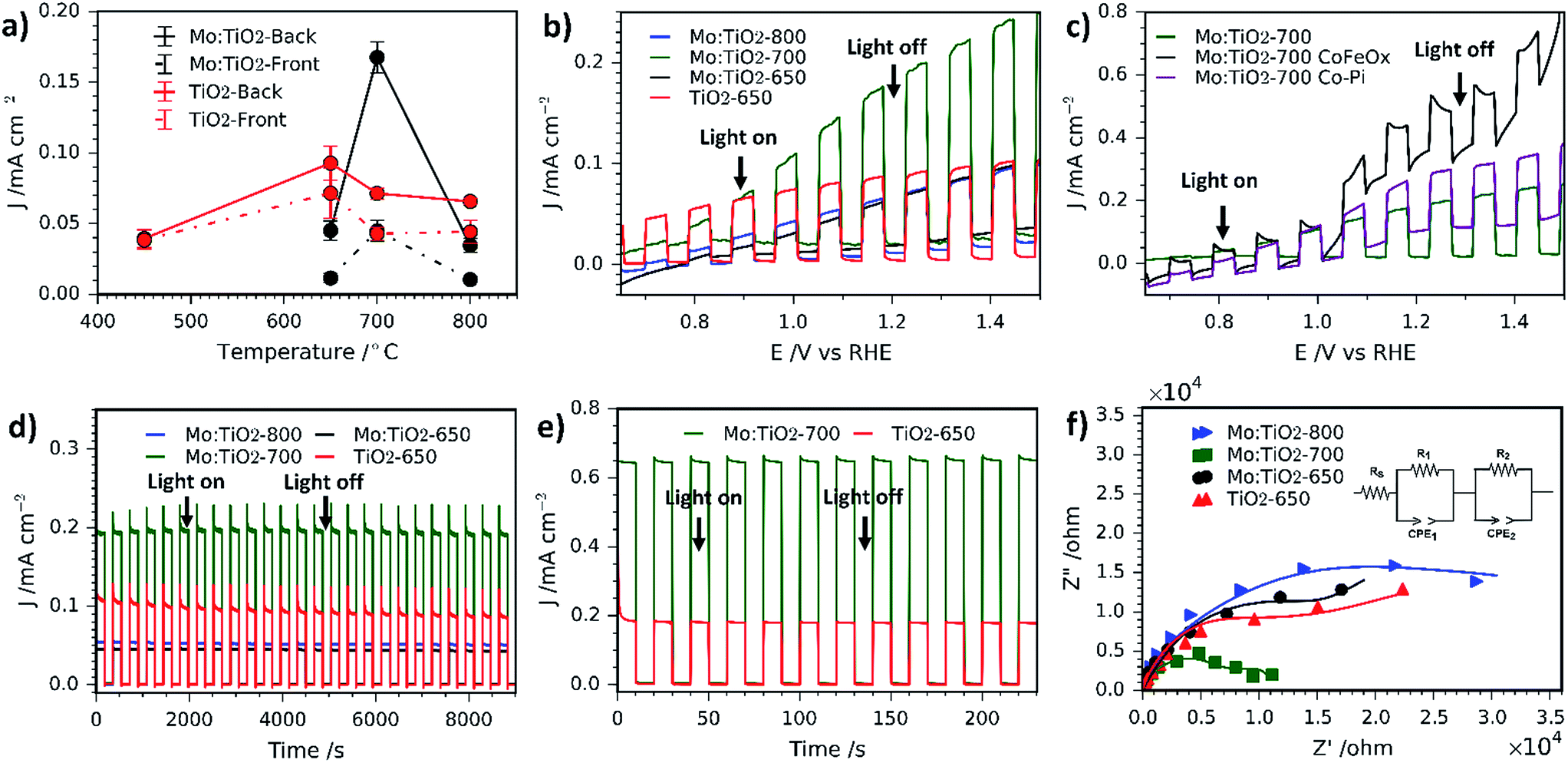 | ||
| Fig. 9 (a) Variation of photocurrent density as a function of annealing temperature for Mo:TiO2 and pure TiO2 (at 1.23 VRHE). Solid lines correspond to photoanodes where illumination was directed towards the back of the FTO-ABS and dashed-lines where illumination was directed towards the front of the working electrode. Error bars indicate standard error above and below the mean. (b) Photocurrent–potential curves of Mo:TiO2 and pure TiO2. (c) Photocurrent potential curves of Mo:TiO2-700, Mo:TiO2-700-CoFeOx and Mo:TiO2-700-Co-Pi. (d) Photocurrent–time curves for 9000 s of Mo:TiO2 and pure TiO2 at an applied bias of 1.23 VRHE. (e) Photocurrent–time curves for 250 s of Mo:TiO2 and TiO2-650 at an applied bias of 1.23 VRHE under UV illumination (365 nm, 3.6 mW cm−2). (f) Nyquist plots of Mo:TiO2 and pure TiO2 in 1 M KOH. DC of 1.23 VRHE; AC potential frequency range 105 to 0.01 Hz with an amplitude of 5 mV. The inset of the figure shows the equivalent circuit used to fit the data (solid lines). All electrochemical measurements were carried out in 1 M KOH aqueous electrolyte. All were irradiated with simulated sunlight (AM 1.5G, 100 mW cm−2) except (e). | ||
Photocurrent–potential (J–V) and photocurrent–time (J–t) curves are also shown in Fig. 9. J–V curves (Fig. 9b) under chopped simulated solar light indicate that Mo:TiO2-700 outperforms the rest by almost a factor of two, reaching about 0.20 mA cm−2 at 1.23 VRHE. This suggests that the annealing temperature has a significant effect. We assign this enhancement to a combination of (i) a smaller band gap, able to capture a higher fraction of the solar spectrum; (ii) higher surface area, as evidenced by FE-SEM and ESCA measurements (Fig. 2 and 3b); and (iii) to the presence of an anatase–rutile type-II heterojunction, that lowers the electron and hole recombination rate.43,58–60 The slightly lower photocurrent observed for Mo:TiO2 samples at lower applied bias in comparison to TiO2-650 might arise from the different charge distribution at the space-charge region of Mo:TiO2 that can affect the band-bending properties of the as-prepared photoanodes.36 In fact, Z-potential measurements of powdered suspensions of Mo:TiO2 particles revealed a highly negatively charged surface at a broad pH range (pH 1–14). This highly negative surface may lead to the formation of an accumulation layer when the Mo:TiO2 films are in contact with the electrolyte, requiring a larger applied bias to switch to a depletion layer and promote the migration of photocarriers.36
Two different co-catalysts, cobalt phosphate (Co-Pi) and CoFeOx have been evaluated in order to reduce the onset potential of Mo:TiO2-700. Previous reports have shown that both co-catalysts are excellent candidates for reducing the on-set potential owing to the decrease in electron–hole recombination at the electrode/electrolyte interface and reduction of surface charge recombination.27,29,61Fig. 9c shows the J–V curves for Mo:TiO2-700 without co-catalyst and with either Co-Pi or CoFeOx. Optimal deposition conditions were found to be 20 s of deposition time for CoPi loading and 3 cycles for CoFeOx (positively sweeping the voltage from 1.1 to 1.4 VAg/AgCl). Interestingly, both co-catalysts show a similar behaviour, where an enhancement in photocurrent is observed at low bias. At higher bias the driving force for the electron–hole separation comes from the higher applied bias itself rather than the co-cocatalyst, so main improvements with co-catalyst addition are only seen at low bias.27
Photocurrent–time curves at 1.23 VRHE also confirm that Mo:TiO2-700 exhibits a twofold increase in photocurrent performance and reveals better photostability in comparison to TiO2-650 (Fig. 9d). The photostability for Mo:TiO2 and TiO2-650 photoanodes was quantified as the percentage of the photocurrent performance at the end of the last illuminated cycle (J) against the photocurrent performance at the end of the first illumination cycle (J0), as previously reported by Paracchino et al.62 After 9,000 s of chopped light irradiation, Mo:TiO2-700 presented the best photostability with a J/J0 of 99.0%, followed by Mo:TiO2-650 (J/J0 = 95.7%), Mo:TiO2-800 (J/J0 = 93.5%) and finally TiO2-650 (J/J0 = 81.7%). Interestingly, all Mo:TiO2 photoanodes resulted in an enhancement in the photostability in comparison to pure TiO2, although only Mo:TiO2-700 showed an improvement in the photocurrent performance over TiO2-650. Unlike Mo:TiO2-700, Mo:TiO2-800 exhibits a lower photocurrent than TiO2 at an applied bias of 1.23 VRHE. This lower photocurrent performance is attributed to a combination of plausible reasons: first, Mo:TiO2-800 only exhibits rutile TiO2 in its composition due to the high annealing temperature employed for the preparation, as depicted in XRD and Raman experiments. Rutile TiO2 is known to be less active than the anatase one, despite having a narrower band gap.63 Second, unlike a rutile–anatase heterojunction where electrons and holes separate, the single phase rutile Mo:TiO2-800 must suffer from higher electron–hole recombination.43,58–60 Third, SEM images (Fig. 2) and ECSA measurements (Fig. 3b) also indicate a smaller surface area for Mo:TiO2-800 with larger grains compared to Mo:TiO2-700. Finally, many other factors such as particle size, aggregate shape and size may also influence the final photoelectrochemical performance of the Mo:TiO2-800 film. In fact, TEM images showed higher particle size and aggregation for the Mo:TiO2-800 photoanode, mainly due to the high annealing temperature employed.
Unlike Mo:TiO2-800, Mo:TiO2-650 shows both anatase and rutile TiO2 crystalline phases, as demonstrated by XRD analysis and Raman spectroscopy and the measured band gap is considerably lower than pure TiO2. Therefore, the poorer photoresponse behaviour of Mo:TiO2-650 must be due to the type of Mo doping and to the film morphology. As shown in SEM-EDXS and XPS analyses, Mo:TiO2-650 possesses the highest amount of Mo amongst the Mo:TiO2 photoanodes and XRD analysis shows a small shift towards lower angles. This fact suggests that the clear majority of Mo6+/5+ atoms are dispersed around the surface or occupying interstitial sites of the TiO2 lattice structure, rather than occupying Ti4+ positions in the TiO2 lattice structure. The presence of larger quantities of Mo6+/5+ atoms on the surface of TiO2 can reduce the photocurrent performance by creating recombination sites and blocking reaction sites.6,7 Morphology and surface area can also play an important role in the photocurrent performance of photoanodes. As shown in the SEM images (Fig. 2j), very fine and poorly defined nanostructures are observed, which results in less surface area exposed for the water oxidation. This is further confirmed with ECSA measurements (Fig. 3b), where Mo:TiO2-650 shows a significantly smaller surface area in comparison to Mo:TiO2-700.
Photocurrent–time curves of Mo:TiO2-700 and TiO2-650 photoanodes at 1.23 VRHE were also recorded using a UV lamp (365 nm, 3.6 mW cm−2) and are shown in Fig. 9e. Under these conditions, Mo:TiO2-700 also outperforms the performance of pure TiO2, reaching a photocurrent value of ca. 0.65 mA cm−2 compared to ca. 0.2 mA cm−2, respectively.
In order to further understand the enhancement in photoresponse for the Mo:TiO2-700 photoanode, the charge transfer properties of photogenerated electrons and holes were studied using PEIS. Fig. 9f shows Nyquist plots of the as-prepared photoanodes recorded at a DC potential of 1.23 VRHE under illumination and AC potential frequency range of 100000–0.01 Hz. The inset of Fig. 9f shows the equivalent circuit used to fit the Nyquist plots. It consists of an ohmic resistance and two RC elements in series, where Rs corresponds to the resistance of the cell, R1 to the resistance of the electronic process in the bulk semiconductor along with Constant Phase Element 1 (CPE1), and R2 to the resistance of the interfacial charge transfer between the electrolyte and the photoanode along with CPE2.64 The corresponding fitted resistance values are listed in Table 1. As expected, the resistance values of the Mo:TiO2-700 photoanode are smaller than that of Mo:TiO2-650, Mo:TiO2-800 and TiO2-650, suggesting a better separation efficiency and faster transfer rate of photogenerated electrons and holes. This enhancement in the charge transfer properties of the Mo:TiO2-700 photoanode agrees well with the J–t curves (Fig. 9d), that showed better photostability and a twofold photocurrent increase. This improvement is attributed to the presence of oxygen vacancies, formed to balance charges after partial doping with Mo6+/5+. Oxygen vacancies are known to improve the electrical conductivity and charge transportation of TiO2.65,66 Since Mo:TiO2-700 shows an optimal substitutional doping of Mo atoms into TiO2, an enhancement in electron conductivity and charge transportation occurs, giving rise to higher photocurrents and stability.65,66
| Sample | R s (Ω) | R 1 (Ω) | R 2 (Ω) |
|---|---|---|---|
| Mo:TiO2-650 | 11.99 | 10098 |
64870 |
| Mo:TiO2-700 | 13.11 | 1512 | 9902 |
| Mo:TiO2-800 | 12.07 | 10098 |
36055 |
| TiO2-650 | 11.77 | 6931 | 34431 |
IPCE measurements for Mo:TiO2-700 and pure TiO2 photoanodes are shown in Fig. 10a. Pure TiO2 values slowly increase from 0% at 500 nm to 1.4% at 400 nm and reach a maximum of 35% at 320 nm. However, Mo:TiO2-700 IPCE values increase from 0% at 500 nm to 5% at 400 nm and reach a maximum of 40% at 330 nm. These IPCE results with monochromatic light confirm the superior performance of Mo:TiO2-700 over pure TiO2 on absorbing and utilizing visible light, and corroborate the photocurrent results and impedance analysis with polychromatic solar light.
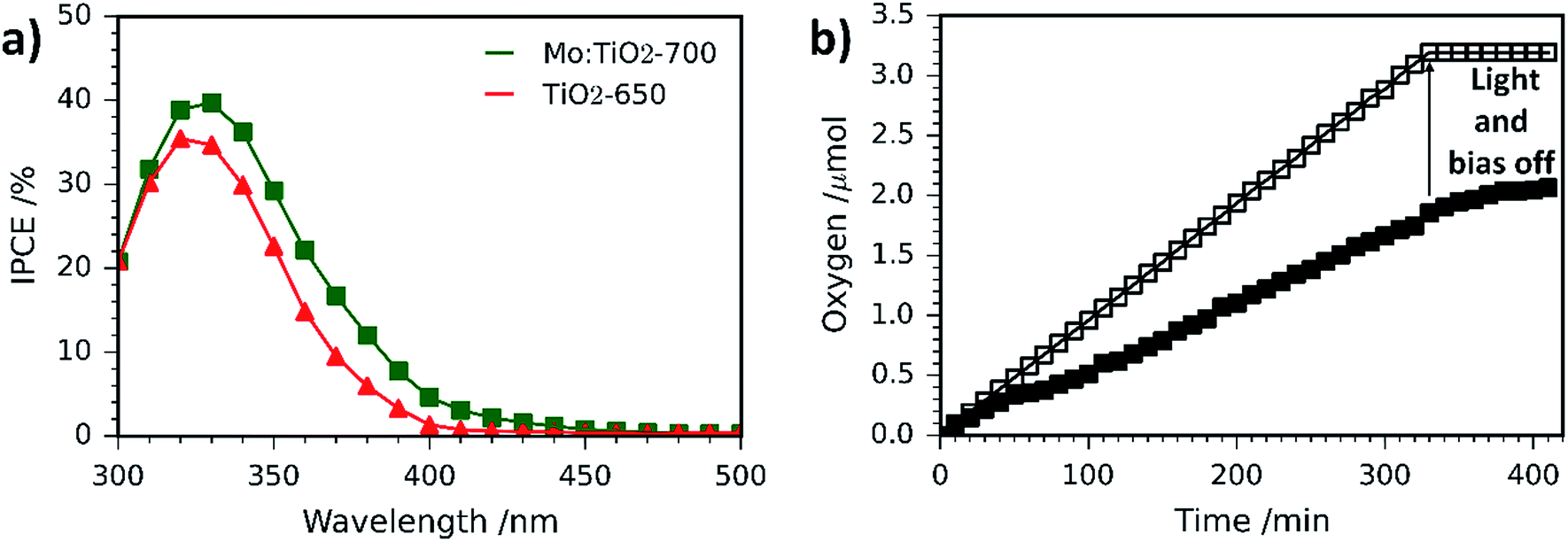 | ||
| Fig. 10 (a) IPCE spectra at 1.23 VRHE of Mo:TiO2-700 and pure TiO2. (b) Amount of O2 gas evolved at 1.23 VRHE under simulated sunlight (AM 1.5G, 100 mW cm−2). The amount of O2 quantified with a fluorescence probe is represented by solid markers, whereas the theoretical amount of O2 calculated assuming a 100% faradaic efficiency is shown with empty markers. | ||
O2 evolution and photocurrent measurements were performed on Mo:TiO2-700 at 1.23 VRHE under 1 sun illumination for 340 min (Fig. 10b). The amount of O2 in the headspace of the PEC cell increased linearly with time during irradiation. Using the photocurrent–time curve obtained (Fig. S9, ESI†), the theoretical amount of O2 expected for a water oxidation reaction with 100% faradaic efficiency was calculated and also represented in Fig. 10b. Comparison between values indicated that Mo:TiO2-700 photoanode has a faradaic efficiency of approx. 60% (details of calculations are shown in ESI†). Similar efficiency values have been obtained on bare photoanodes without oxygen evolution electrocatalysts.67
4. Conclusions
In this work a facile and rapid approach for the design of Mo:TiO2 photoanodes using a heterometallic oxo cage of the type [Ti4Mo2O8(OEt)10]2 as a single source precursor has been demonstrated. The performance of the resultant photoanodes is highly reliant on the annealing temperature employed owing to its effects on the crystallinity, morphology and doping of TiO2, being 700 °C the optimal annealing temperature. At this temperature, the Mo:TiO2 photoanode (Mo:TiO2-700) presents better photostability and a two-fold increase in photocurrent performance (0.20 mA cm−2 at 1.23 VRHE) in comparison to a TiO2 photoanode (0.10 mA cm−2 at 1.23 VRHE). This improvement both in the photocatalytic performance and stability is attributed to a combination of several factors that become optimized at 700 °C: first, Mo:TiO2-700 exhibits the presence of anatase TiO2 and in minor amount rutile TiO2, forming a heterostructure that is known to reduce the electron and hole recombination rate. Second, Mo:TiO2-700 has a smaller band gap than the obtained at different annealing temperatures or without Mo doping, which allows for a better use of the solar spectrum and higher efficiencies (IPCE: 5% at 400 nm). Third, in Mo:TiO2-700 there is preferred substitutional doping of Mo6+/5+ atoms for Ti4+ atoms in the TiO2 lattice structure, causing the presence of oxygen vacancies which improve the electrical conductivity and charge transportation of the film. Fourth, Mo:TiO2-700 shows a large amount of cavities, porosity and well-defined nanostructures, resulting in the photoanode with the highest surface area. This characteristic morphology is associated to the spray pyrolysis deposition of [Ti4Mo2O8(OEt)10]2 and to the partial sublimation of Mo species during the annealing process. On balance, these results clearly demonstrate a simple and effective methodology for preparing Mo-doped TiO2 photoanodes with tuned electronic band properties and morphology and reveal the crucial parameters that allow the exploitation of heterometallic oxo cages in thin films for energy applications. These results open up the possibility for exploring a wide range of different heterometallic oxo cages for the fabrication of metal oxides photoanodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge both EPSRC for funding the Centre for Doctoral Training in Sustainable Chemical Technologies at the University of Bath (EP/L016354/1), the microscopy and analysis (MAS) suite at the University of Bath and the Cavendish Laboratory from the University of Cambridge. SE would like to acknowledge the financial support from EPSRC (EP/P008097/1).References
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renew. Sustain. Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- A. Folli, J. Bloh, M. Strøm, T. Pilegaard Madsen, T. Hensriksen and D. E. Macphee, J. Phys. Chem. Lett., 2014, 5, 830–832 CrossRef CAS PubMed.
- R. M. Navarro, F. del Valle, J. A. Villoria de la Mano, M. C. Álvarez-Galván and J. L. G. Fierro, in Advances in Chemical Engineering, ed. B. S. R. Hugo I. de Lasa, Academic Press, 2009, vol. 36, pp. 111–143 Search PubMed.
- C. Sanchez, L. Rozes, F. Ribot, C. Laberty-Robert, D. Grosso, C. Sassoye, C. Boissiere and L. Nicole, C. R. Chim., 2010, 13, 3–39 CrossRef CAS.
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325, 1–14 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- Y. Yan, J. Lee and X. Cui, Vacuum, 2017, 138, 30–38 CrossRef CAS.
- W. Zhao, Y. Li, M. Zhang, J. Chen, L. Xie, Q. Shi and X. Zhu, Chem. Eng. J., 2016, 283, 105–113 CrossRef CAS.
- C. Wang, Z. Chen, H. Jin, C. Cao, J. Li and Z. Mi, J. Mater. Chem. A, 2014, 2, 17820–17827 RSC.
- Q. Liu, D. Ding, C. Ning and X. Wang, Int. J. Hydrogen Energy, 2015, 40, 2107–2114 CrossRef.
- S. Wang, L. N. Bai, H. M. Sun, Q. Jiang and J. S. Lian, Powder Technol., 2013, 244, 9–15 CrossRef CAS.
- O. Avilés-García, J. Espino-Valencia, R. Romero, J. L. Rico-Cerda, M. Arroyo-Albiter and R. Natividad, Fuel, 2017, 198, 31–41 CrossRef.
- Y. Yang, X. J. Li, J. T. Chen and L. Y. Wang, J. Photochem. Photobiol., A, 2004, 163, 517–522 CrossRef CAS.
- B. Miljević, J. M. van der Bergh, S. Vučetić, D. Lazar and J. Ranogajec, Ceram. Int., 2017, 43, 8214–8221 CrossRef.
- V. Štengl and S. Bakardjieva, J. Phys. Chem. C, 2010, 114, 19308–19317 CrossRef.
- T. Zhang, B. Yu, D. Wang and F. Zhou, J. Power Sources, 2015, 281, 411–416 CrossRef CAS.
- P. D. Matthews, T. C. King and D. S. Wright, Chem. Commun., 2014, 50, 12815–12823 RSC.
- Y. Lv, M. Yao, J. P. Holgado, T. Roth, A. Steiner, L. Gan, R. M. Lambert, D. S. Wright, E. Reisner, X. Liang, R. Li, Z. Xu and Q. Ren, RSC Adv., 2013, 3, 13659–13662 RSC.
- Y. H. Lai, T. C. King, D. S. Wright and E. Reisner, Chem.–Eur. J., 2013, 19, 12943–12947 CrossRef CAS PubMed.
- J.-L. Hou, W. Luo, Y.-Y. Wu, H.-C. Su, G.-L. Zhang, Q.-Y. Zhu and J. Dai, Dalton Trans., 2015, 44, 19829–19835 RSC.
- Y.-H. Lai, C.-Y. Lin, Y. Lv, T. C. King, A. Steiner, N. M. Muresan, L. Gan, D. S. Wright, E. Reisner, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Chem. Commun., 2013, 49, 4331–4333 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1076 CrossRef CAS PubMed.
- S. Eslava, A. Reynal, V. G. Rocha, S. Barg and E. Saiz, J. Mater. Chem. A, 2016, 4, 7200–7206 RSC.
- S. Eslava, B. P. R. Goodwill, M. McPartlin and D. S. Wright, Inorg. Chem., 2011, 50, 5655–5662 CrossRef CAS PubMed.
- M. Ibadurrohman and K. Hellgardt, Int. J. Hydrogen Energy, 2014, 39, 18204–18215 CrossRef CAS.
- G. Ai, R. Mo, H. Li and J. Zhong, Nanoscale, 2015, 7, 6722–6728 RSC.
- D. K. Zhong, M. Cornuz, K. Sivula, M. Grätzel and D. R. Gamelin, Energy Environ. Sci., 2011, 4, 1759–1764 RSC.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- B. K. Kang, G. S. Han, J. H. Baek, D. G. Lee, Y. H. Song, S. Bin Kwon, I. S. Cho, H. S. Jung and D. H. Yoon, Adv. Mater. Interfaces, 2017, 4, 1700323 CrossRef.
- Y. Duan, S. Zhou, Z. Chen, J. Luo, M. Zhang, F. Wang, T. Xu and C. Wang, Catal. Sci. Technol., 2018, 8, 1395–1403 RSC.
- X. Jin, W. Sun, C. Chen, T. Wei, Y. Cheng, P. Li and Q. Li, RSC Adv., 2014, 4, 46008–46015 RSC.
- Y. Li, H. Zhong, R. Li, Y. Zhou, C. Yang and Y. Li, Adv. Funct. Mater., 2006, 16, 1705–1716 CrossRef CAS.
- Z. Chen, Q. Li, C. Chen, J. Du, J. Tong, X. Jin, Y. Li, Y. Yuan, Y. Qin, T. Wei and W. Sun, Phys. Chem. Chem. Phys., 2014, 16, 24499–24508 RSC.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- Z. Chen, H. N. Dinh and E. Miller, Photoelectrochemical water splitting: standards, experimental methods, and protocols, Springer-Verlag, New York, 2013 Search PubMed.
- S. Choopun, P. Mangkorntong, P. Subjareon, N. Mangkorntong, H. Tabata and T. Kawai, Jpn. J. Appl. Phys., 2004, 43, L91–L93 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- M. Lu, C. Shao, K. Wang, N. Lu, X. Zhang, P. Zhang, M. Zhang, X. Li and Y. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 9004–9012 CrossRef CAS PubMed.
- R. C. Pullar, S. J. Penn, X. Wang, I. M. Reaney and N. Mcn, J. Eur. Ceram. Soc., 2009, 29, 419–424 CrossRef CAS.
- W. M. Haynes, D. Lide and T. Bruno, CRC handbook of chemistry and physics: A ready-reference book of chemical and physical data, CRC Press, Taylor & Francis Group, 97th edn, 2016 Search PubMed.
- J. Yan, G. Wu, N. Guan, L. Li, Z. Li and X. Cao, Phys. Chem. Chem. Phys., 2013, 15, 10978–10988 RSC.
- M.-C. Wu, P.-H. Lee and D.-L. Lee, Int. J. Hydrogen Energy, 2015, 40, 4558–4566 CrossRef CAS.
- X. Du, L. Dong, C. Li, Y. Liang and Y. Chen, Langmuir, 1999, 15, 1693–1697 CrossRef CAS.
- M. Dieterle and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 822–826 RSC.
- N. Illyaskutty, S. Sreedhar, G. Sanal Kumar, H. Kohler, M. Schwotzer, C. Natzeck and V. P. M. Pillai, Nanoscale, 2014, 6, 13882–13894 RSC.
- L. G. Devi and B. N. Murthy, Catal. Lett., 2008, 125, 320–330 CrossRef CAS.
- R. Sanjinés, H. Tang, H. Berger, F. Gozzo, G. Margaritondo and F. Lévy, J. Appl. Phys., 1994, 75, 2945–2951 CrossRef.
- J. G. Choi and L. T. Thompson, Appl. Surf. Sci., 1996, 93, 143–149 CrossRef CAS.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef CAS PubMed.
- X. Xiao, T. Ding, L. Yuan, Y. Shen, Q. Zhong, X. Zhang, Y. Cao, B. Hu, T. Zhai, L. Gong, J. Chen, Y. Tong, J. Zhou and Z. L. Wang, Adv. Energy Mater., 2012, 2, 1328–1332 CrossRef CAS.
- W. Li, C. Ni, H. Lin, C. P. Huang and S. I. Shah, J. Appl. Phys., 2004, 96, 6663–6668 CrossRef CAS.
- Y. F. Chen, C. Y. Lee, M. Y. Yeng and H. T. Chiu, J. Cryst. Growth, 2003, 247, 363–370 CrossRef CAS.
- Y. Gai, J. Li, S. S. Li, J. B. Xia and S. H. Wei, Phys. Rev. Lett., 2009, 102, 23–26 CrossRef PubMed.
- B. Choudhury and A. Choudhury, Phys. E, 2014, 56, 364–371 CrossRef CAS.
- K. Mahesh, S. Karpagam and F. Goubard, eXPRESS Polym. Lett., 2018, 12, 238–255 CrossRef CAS.
- Z. He, Q. Cai, H. Fang, G. Situ, J. Qiu, S. Song and J. Chen, J. Environ. Sci., 2013, 25, 2460–2468 CrossRef CAS.
- Z. Rui, S. Wu, C. Peng and H. Ji, Chem. Eng. J., 2014, 243, 254–264 CrossRef CAS.
- Y. Gao, H. Wang, J. Wu, R. Zhao, Y. Lu and B. Xin, Appl. Surf. Sci., 2014, 294, 36–41 CrossRef CAS.
- J. Zhang, R. García-Rodríguez, P. Cameron and S. Eslava, Energy Environ. Sci., 2018 10.1039/c8ee01346b.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 4043 CrossRef PubMed.
- R. R. Devarapalli, J. Debgupta, V. K. Pillai and M. V. Shelke, Sci. Rep., 2014, 4, 1–8 Search PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- D. C. Cronzmeyer, Phys. Rev., 1959, 113, 1222–1226 CrossRef.
- J. A. Seabold and K.-S. Choi, Chem. Mater., 2011, 23, 1105–1112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All data created during this research are openly available from the University of Bath data archive at https://doi.org/10.15125/BATH-00437. See DOI: 10.1039/c8se00372f |
| This journal is © The Royal Society of Chemistry 2018 |