
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced alkaline stability in a hafnium-substituted NaSICON ion conductor†
Leo J.
Small
 ,
Jill S.
Wheeler
,
Jon F.
Ihlefeld‡
,
Paul G.
Clem
and
Erik D.
Spoerke
*
,
Jill S.
Wheeler
,
Jon F.
Ihlefeld‡
,
Paul G.
Clem
and
Erik D.
Spoerke
*
Sandia National Laboratories, PO Box 5800, Albuquerque, NM MS 1411, USA 87185. E-mail: edspoer@sandia.gov
First published on 14th May 2018
Abstract
We present here a multi-length scale integration of compositionally tailored NaSICON-based Na+ conductors to create a high Na+ conductivity system resistant to chemical attack in strongly alkaline aqueous environments. Using the Pourbaix Atlas as a generalized guide to chemical stability, we identify NaHf2P3O12 (NHP) as a candidate NaSICON material for enhanced chemical stability at pH > 12, and demonstrate the stability of NHP powders under accelerated aging conditions of 80 °C and pH = 13–15 for a variety of alkali metal cations. To compensate for the relatively low ionic conductivity of NHP, we develop a new low temperature (775 °C) alkoxide-based solution deposition chemistry to apply dense NHP thin films onto both platinized silicon wafers and bulk, high Na+ conductivity Na3Zr2Si2PO12 (NZSP) pellets. These NHP films display Na+ conductivities of 1.35 × 10−5 S cm−1 at 200 °C and an activation energy of 0.53 eV, similar to literature reports for bulk NHP pellets. Under aggressive conditions of 10 M KOH at 80 °C, NHP thin films successfully served as an alkaline-resistant barrier, extending the lifetime of NZSP pellets from 4.26 to 36.0 h. This integration of compositionally distinct Na+ conductors across disparate length scales (nm, mm) and processing techniques (chemically-derived, traditional powder) represents a promising new avenue by which Na+ conducting systems may be utilized in alkaline environments previously thought incompatible with ceramic Na+ conductors.
Introduction
The Na Super Ionic CONductor (NaSICON) is a ceramic ion conductor with a nominal composition of Na1+x Zr2P3−xSixO12 known to selectively transport Na+ at high rates (>10−3 S cm−1 at room temperature) through atomic channels formed between zirconia octahedra and phosphate or silica tetrahedra within its crystal structure.1–4 As a class of materials, NaSICONs are regarded for their compositional flexibility, accommodating numerous variations of chemical composition and crystallographic structure. This versatility has made NaSICONs attractive, selective ion conductors that can be engineered to selectively promote or inhibit ion transport based on ion size or charge, making this a particularly versatile electrochemically active ceramic material system.5–8 Although NaSICON in its over 110 varied forms9 has found widespread application in energy storage,10–16 chemical separations,17,18 sensors,19–21 and other technologies, many NaSICON compositions suffer from significant chemical instabilities.22,23The most widely used, highest Na+ conductivity NaSICON, Na3Zr2Si2PO12 (NZSP), is known to be susceptible to degradation in strongly acidic or basic aqueous media.24 In the present work, we focus explicitly on the alkaline instability of NaSICON. In strongly alkaline environments (pH > 12) both Si–O and/or Zr–O bonds are susceptible to attack,25,26 limiting the potential utility of these ion conductors in aggressive alkaline media. The ability to extend NaSICON's operating pH range would enable ionic separations in caustic sodas, rechargeable alkaline batteries, and significantly expand the design space for redox flow batteries or separations. Using the Pourbaix Atlas as a general guide, it is seen that Si–O and Zr–O bonds are generally unstable at pH > 12.25,26 To simplify our initial studies, we consider first NaSICON containing no Si: NaZr2P3O12. According to the Pourbaix Atlas, then, we expect Hf–O bonds would be considerably more stable than Zr–O bonds at high pH.25 Based on this expectation, we present here a study a modified, Hf-substituted NaSICON, NaHf2P3O12 (NHP), as an alkaline stable variation of the NaSICON. While Hf-containing NaSICONs have previously been synthesized and shown to conduct Na+, their chemical stability is relatively unexplored.27–30 Here we explicitly compare the alkaline aqueous stability of NHP to its Zr-based analogue, NaZr2P3O12 (NZP), interrogating the material with highly alkaline NaOH, LiOH, KOH, and CsOH aqueous media. We explore this material in two forms, both as a bulk ceramic powder and as a novel ion-conducting ceramic thin film, capable of serving as an ion-conducting barrier to protect NaSICON compositions with more vulnerable chemical compositions.
Experimental
Powder synthesis
To synthesize the NHP powders, P2O5 (1.067 g, 7.517 mM, 99.99%, Sigma-Aldrich) was saturated with hexanes (anhydrous, 99.9%, Sigma-Aldrich) in a dry N2-purged glove box and then NaH2PO4·H2O (1.038 g, 7.522 mM, 99.0%, Sigma-Aldrich) and HfO2 (3.166 g, 15.04 mM, 99.99%, Alfa-Aesar) were added. This mixture was thoroughly ground with a mortar and pestle, placed in an alumina crucible, and ramped at 5 °C min−1 to 1050 °C in air, where it was held for 12 h before cooling to room temperature at 5 °C min−1.NZP powders were synthesized in the same manner as NHP powders, except an equimolar quantity of ZrO2 (99%, Sigma-Aldrich) was substituted for HfO2.
Thin film synthesis
NHP thin films were prepared via chemical solution deposition, adapting procedures recently reported for the growth of NZP thin films.31,39 It should be noted that the present chemistry is distinct from methods used by other groups, including 2-methoxyethanol-based routes for thin films,32 or oxalic acid-based sol–gel routes for NaSICON powders.33In a dry, argon-filled glovebox, hafnium(IV) n-butoxide (1.695 g, 3.60 mmol, 99%, Sigma-Aldrich), 1-butanol (9.56 mL, 104 mmol, 99.8%, Sigma-Aldrich), dibutyl phosphate (1.135 g, 5.400 mmol, >97.0%, Sigma-Aldrich), and sodium ethoxide (0.123 g, 1.81 mmol, >95%, Fluka) were sequentially added to a 20 mL glass vial, ensuring thorough mixing between additions. This NHP precursor solution was magnetically stirred overnight at room temperature. These solutions were further diluted with 1-butanol to obtain NHP concentrations of 0.15 or 0.3 M.
In air, platinized silicon wafers37 (100 nm Pt//40 nm ZnO//400 nm SiO2//Si (001)) were coated with 0.15 M NHP precursor solution filtered through a 0.2 μm syringe filter, spun at 3000 rpm for 30 s, placed on a 300 °C hotplate for 5 minutes, and loaded into an air-filled tube furnace preheated to 775 °C for 10 minutes. Samples were subsequently removed and allowed to cool to room temperature. This process was repeated for 3 layers total.
A similar process was used to coat NZSP pellets. Here 0.3 M NHP precursor solution was filtered through a 0.2 μm syringe filter, spun at 3000 rpm for 30 s, and loaded into an air-filled furnace which was ramped at 10 °C min−1 from room temperature to 775 °C, held for 10 minutes, and cooled back to room temperature at 10 °C min−1. This process was repeated for three or six layers. Later, a revised thermal treatment was developed where 0.15 M NHP precursor solution was filtered through a 0.2 μm syringe filter, spun at 3000 rpm for 30 s, placed on a 300 °C hot plate for 5 minutes, and then cooled to room temperature. This process was repeated for three layers. After the third layer, the NZSP pellets were transferred directly from the 300 °C hotplate to a furnace preheated to 300 °C. The furnace was then ramped at 10 °C min−1 to 775 °C, held for 10 minutes, and cooled to room temperature at 10 °C min−1. This three-layer coating and annealing process was performed twice to create 6 layers of NHP on the NZSP pellet (“6 L revised NHP”). All NZSP pellets were 25 mm in diameter, 1.0 mm thick, obtained from Ceramatec Corporation (Salt Lake City, UT, USA), and characterized in depth elsewhere.14,15
Chemical stability of ceramic powders
Bulk powder stability was evaluated by immersing 0.250 g of NHP or NZP powder in 10 mL of electrolyte (e.g., 10 M KOH), sealed in a Teflon-lined autoclave, and heated to 80 °C for 72 h. After removing the autoclave from the oven and cooling to room temperature, the solution was transferred to a 25 mL centrifuge tube and spun at 2450 × g for 10 minutes. The supernatant was removed, and 10 mL fresh 18.2 MΩ cm deionized water used to resuspend and wash the powder before centrifuging again. This washing process was repeated until the pH of the supernatant registered 7, as judged by pH paper. The solids were separated and dried overnight under 20 mTorr vacuum. The dried solid was interrogated by X-ray diffraction (XRD).Electrochemical measurements
90 nm thick platinum top electrodes approximately 0.003 cm2 were sputtered through a shadow mask onto the NHP thin films; actual areas were measured with optical microscopy and image analysis. NHP thin film ionic conductivity of NHP films was evaluated using impedance spectroscopy with an HP 4192 LCR meter over a range of 1 MHz to 112 Hz with a 50 mV (rms) oscillator in a metal–electrolyte–metal geometry. The top and bottom platinum electrodes serve as Na-ion blocking electrodes. Fits to the complex plane impedance data were performed with Bio-Logic EC-Lab software. A circuit element of a constant phase element in parallel with a resistor was used to model the ionic conductivity of the NHP electrolyte and a constant phase element in series to model the blocking electrodes. An additional series resistor was used to account for resistances of the metal electrodes, which proved to be negligible. Temperature dependent measurements were performed within a Delta Design 9023 environmental chamber between 25 and 200 °C.NZSP pellets with and without NHP coatings on one side were evaluated via electrochemical impedance spectroscopy (EIS), and open circuit potential (OCP) measurements in electrolyte heated to 80 °C. EIS measurements occurred at 0 V DC with respect to the open circuit potential (OCP) with a 5 mV RMS AC amplitude over 1 MHz to −100 mHz using a Solartron Modulab system.
For conductance measurements in aqueous NaOH, an NZSP pellet was sealed in a NW-10 vacuum flange in a water-jacketed U-shaped glass cell (Adams and Chittenden, Berkeley, CA, USA). 5 mL of electrolyte was placed on each side of the cell. A four electrode cell was constructed: on one side a large area platinum mesh served as the current source, while a freshly polished 1.6 mm diameter platinum disc electrode (MF-2013 BASi, Inc.), placed near the NZSP pellet, measured the potential of the electrolyte. On the other side of the NZSP pellet, another platinum disc electrode measured the electrolyte potential and a platinum mesh served as the current sink. To evaluate the chemical stability of the NZSP pellets, a three electrode electrochemical cell was fashioned using the NZSP pellet as the working electrode, a large area platinum mesh as the counter electrode, and a freshly polished 1.6 mm platinum disc as the pseudo reference electrode (standard reference electrodes were avoided, as their porous glass frits etch rapidly in hot, concentrated KOH). A gold wire was electrically contacted to an NZSP pellet using silver paint (Ted Pella Leitsilber 200), added to the side not coated with NHP, and the paint was dried at 80 °C for 1 h in air. Upon cooling, a standard two part epoxy (Extra fast setting, Royal Adhesives & Sealants, Wilmington, CA) was used to mechanically stabilize the silver–gold connection. A viton gasket sealed the NHP-coated side of the pellet against a glass NW-10 vacuum flange in a water-jacketed U-shaped glass cell (Adams and Chittenden, Berkeley, CA, USA). The dry assembled cell was then preheated to 80 °C using a circulating bath to heat the cell jacket, and the electrolyte was separately heated to 80 °C on a hotplate. After all components reached 80 °C, 5 mL hot electrolyte was added to the electrochemical cell (on the side of the pellet coated with NHP) and the measurements commenced.
Material characterization
NaSICON phase chemistry and crystallinity were evaluated using X-ray diffraction (XRD) with a Bruker D2 Phaser system set in the traditional Bragg–Brentano geometry with Cu Kα radiation. NHP thin film microstructure was evaluated in plan-view and cross section using a Zeiss Supra 55VP scanning electron microscope (SEM) at 3 kV and 5 mm working distance in an in-lens imaging mode.Results and discussion
Chemical stability of NHP and NZP powders
The as-synthesized NHP and NZP powders were characterized by XRD, and the resulting data are plotted in Fig. 1A and B, respectively, along with reference powder diffraction files (PDF) for NHP (04-002-2743)30 and HfO2 (04-001-7440),40 or NZP (01-071-0959)34 and ZrO2 (04-002-8305).41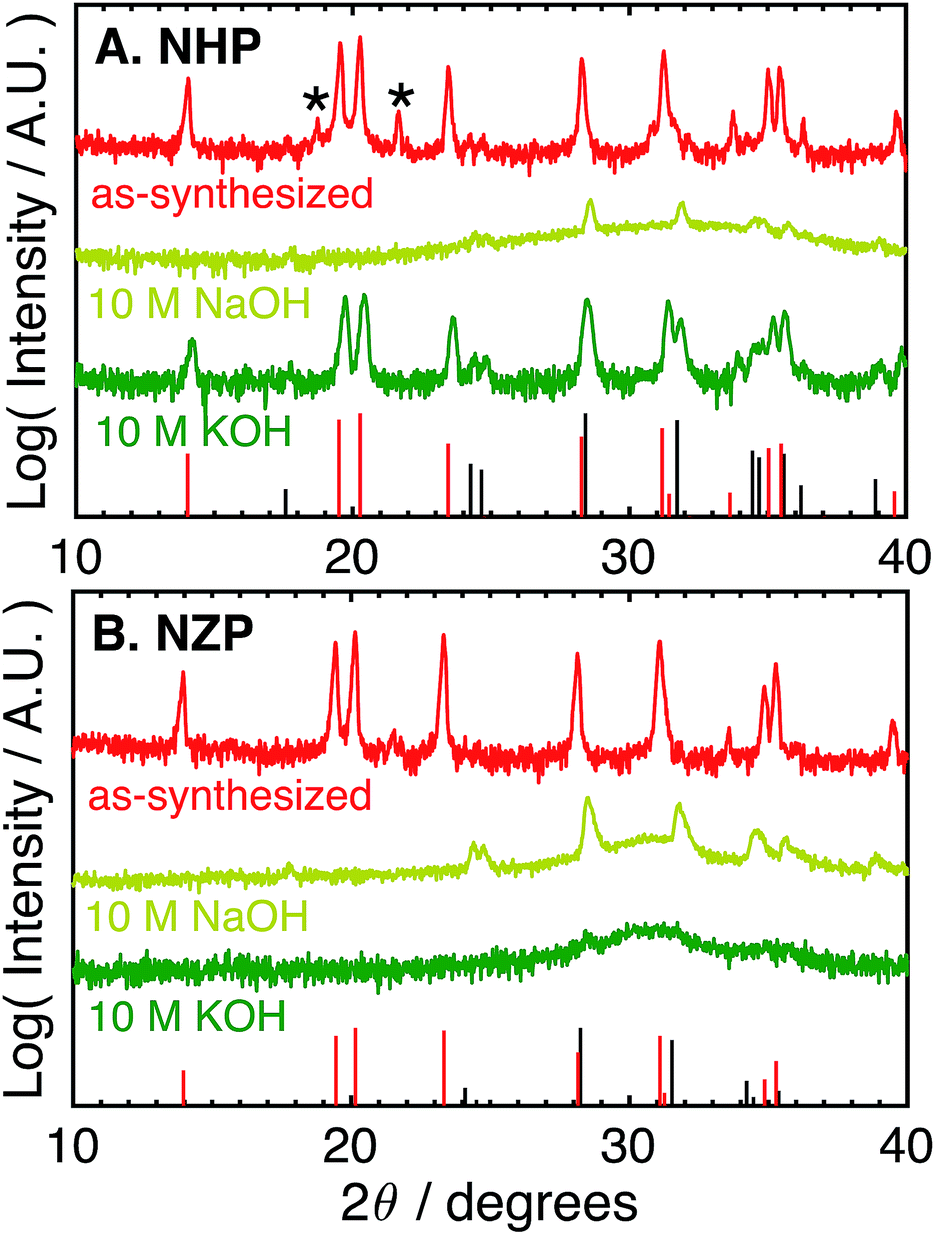 | ||
| Fig. 1 Log-scale X-ray (Cu Kα) diffraction patterns of (A) NHP and (B) NZP powders as-synthesized and after aging for 72 h at 80 °C in different aqueous 10 M NaOH or KOH. Asterisks (*) denote a slight HfP2O7 impurity. Red bars at the bottom of each plot denote PDFs for (A) NHP (04-002-2743) and (B) NZP (01-071-0959). Black bars denote PDFs for (A) HfO2 (04-001-7440) and (B) ZrO2 (04-002-8305). | ||
For the NHP powder, a slight HfP2O7 impurity, labeled by an asterisk in Fig. 1A, is observed. Minor peaks between 24–25° indicate a very small amount of poorly crystalline HfO2 is also present. The NZP powder produced diffraction primarily from the NaSICON phase in Fig. 1B, with the slight suggestion of a peak near 25° to indicate a very minor ZrO2 secondary phase. The chemical stability of these powders was then evaluated by immersion in aqueous alkaline base at 80 °C for 72 hours and recharacterizing washed powders by XRD to determine if the NZP and NHP phases persisted through the alkaline exposure. Initial tests explored stability in particularly aggressive 10 M NaOH and 10 M KOH. As seen in the diffraction patterns in Fig. 1A and B, both the NZP and NHP powders were completely degraded in the 10 M NaOH solution, producing fine-grained ZrO2 and HfO2 powders, respectively. In addition to evident metal oxide peaks (e.g., peaks near 25°, 28°, 32° and 35° 2θ), there is a broad feature in the spectra centered around 30–32° that suggests a fine-grained (nanoscale) oxide. The formation of these oxides phases is believed to result from precipitation of dissolved zirconium and hafnium species as the alkaline solutions cooled and the pH of the solutions was reduced during washing.26 In the 10 M KOH, although the NZP phase was dissolved and reprecipitated as ZrO2, the crystalline NHP phase was predominantly preserved, though a small amount of HfO2 is still visible, perhaps even more pronounced, in the washed powder. Interestingly, the initial HfP2O7 contaminant phase was effectively removed during this process. Potentially increased HfO2 phase in the exposed NHP powder may be have been reprecipitation from a small amount of either dissolved NHP or dissolved contaminant HfP2O7 phase.
The difference in the apparent stability of the NHP powders as a function of alkali cation size motivated an additional study of powder stability in less aggressive 1 M alkaline solutions comprising alkali salts with variable cation size. These results are summarized in Table 1 and the associated diffraction patterns are shown in Fig. 2A and B. Looking first at the NZP data in Fig. 2B, it is clear that every alkaline salt led to significant decomposition of the NZP and reprecipitation of ZrO2. There appears to be a trend of decreasing NZP crystallinity as the alkali cation increases from Li+ to K+ to Cs+. This trend is attributed to the increasing activity coefficients and resulting pHs of these solutions (LiOH < KOH < CsOH).42,43 Similar trends in dissolution rates have been observed for Si etching.43 Interestingly, the most degradation occurred in the NaOH sample, where the behavior would be expected to fall between LiOH and KOH. This increased degradation in NaOH is likely due to specific interactions between Na+ in the alkaline media and the highly mobile Na+ ions as the active species in the NaSICON ceramic structure.
| Electrolyte | NHP | NZP |
|---|---|---|
| 1 M LiOH | NHP | NZP + ZrO2 |
| 1 M NaOH | NHP + HfO2 | ZrO2 |
| 1 M KOH | NHP | NZP + ZrO2 |
| 1 M CsOH | NHP | NZP + ZrO2 |
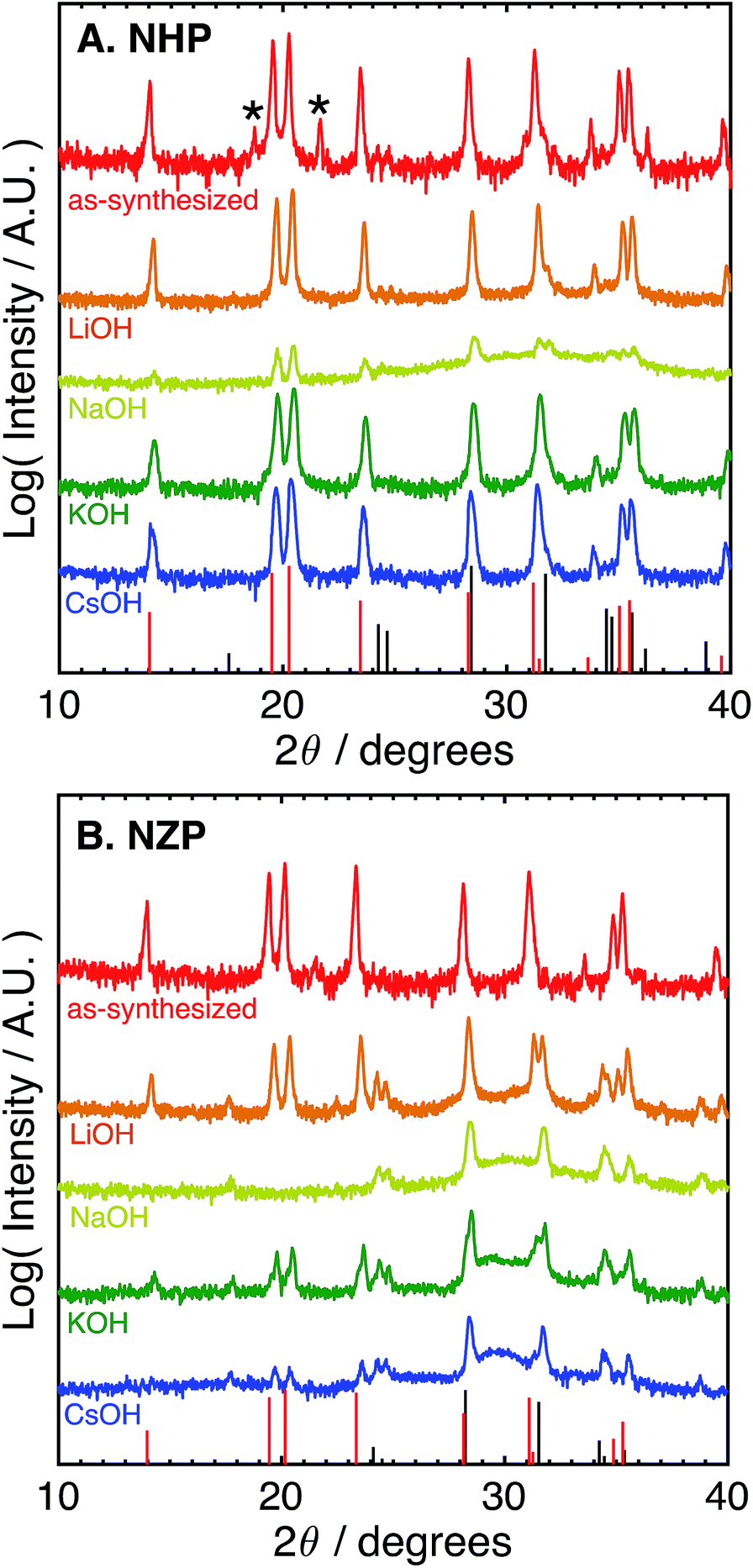 | ||
| Fig. 2 Log-scale X-ray (Cu Kα) diffraction patterns of (A) NHP and (B) NZP powders as-synthesized and after aging for 72 h at 80 °C in varied 1 M basic aqueous electrolytes. Asterisks (*) denote a slight HfP2O7 impurity. Red bars at the bottom of each plot denote PDFs for (A) NHP (04-002-2743) and (B) NZP (01-071-0959). Black bars denote PDFs for (A) HfO2 (04-001-7440) and (B) ZrO2 (04-002-8305). | ||
The NHP samples, in contrast, showed significantly improved stability in all salts as crystalline NHP peaks persist through all treatments. In fact, only the NaOH solution appears to produce any evidence of secondary HfO2 precipitate. This increased sensitivity is consistent with that observed in the NZP, where ion exchange with the active species in the NHP could again influence the ceramic stability. More generally, however, these results do clearly show excellent chemical stability of the NHP ceramic, relative to the NZP analogue. These results are summarized in Table 1.
These data suggest that although NHP is more stable than NZP in each alkaline exposure, the NHP does exhibit some significant, concentration-dependent solubility in NaOH. Fig. 3 compares the phases present in NHP and NZP after exposure to NaOH solutions at 80 °C for 0.5 M, 1 M, and 10 M. Again, in each case, the diffraction patterns shows the loss of the NZP peaks in each case (Fig. 3B), but the NHP peaks, highlighted in Fig. 3A, show increased retention of strong NHP peaks with decreasing NaOH concentration. In fact, the only significant evidence of NHP degradation in the 0.5 M experiment is the large, diffuse hump in the spectrum around 32°, a feature attributed to the formation of fine-grained (nanocrystalline) HfO2.
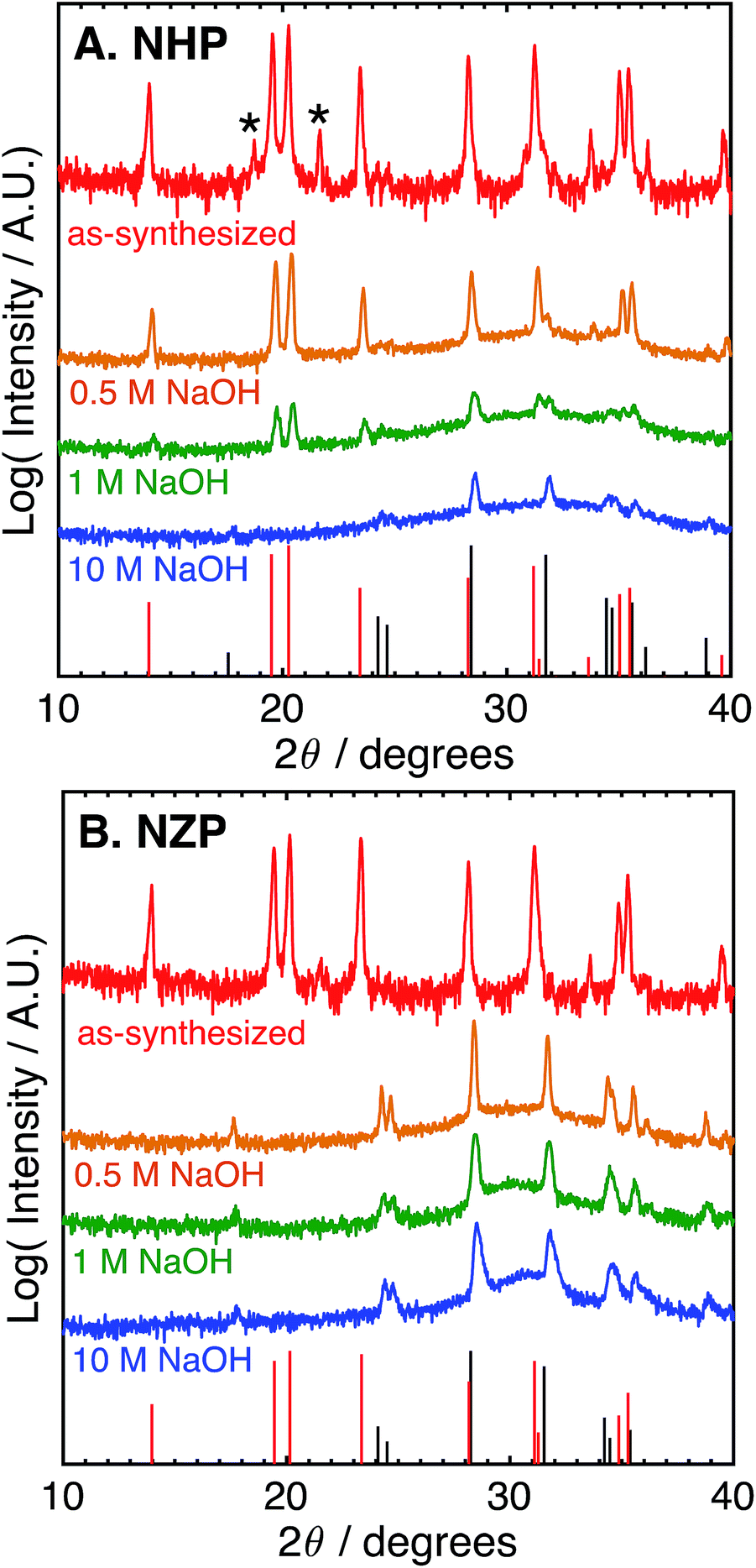 | ||
| Fig. 3 Log-scale X-ray (Cu Kα) diffraction patterns of (A) NHP and (B) NZP powders before (virgin) after aging for 72 h at 80 °C in 0.5, 1, or 10 M aqueous NaOH. Asterisks (*) denote a slight HfP2O7 impurity. Red bars at the bottom of each plot denote PDFs for (A) NHP (04-002-2743) and (B) NZP (01-071-0959). Black bars denote PDFs for (A) HfO2 (04-001-7440) and (B) ZrO2 (04-002-8305). | ||
These results imply significantly enhanced chemical stability of NHP vs. NZP in aggressive alkaline conditions, with most conditions being in excess of pH 14. We attribute this enhanced stability to the strength of the Hf–O bond versus that of the weaker Zr–O bond, and the influence of this increased stability on the overall NaSICON crystal structure.
NHP thin Films
Although these data suggest that NHP would be a more chemically robust material in strongly alkaline environments, its direct applicability could be limited in intermediate to low-temperature applications as reports in the literature show significantly lower Na+ conductivity at 200 °C for bulk NHP (0.1 mS cm−1) as compared to bulk NZSP (140 mS cm−1).15,30 To compensate for this decreased conductivity, a thin film approach was developed to create NHP structures with Na+ conductances similar to those of bulk NZSP pellets; by reducing the NHP thickness the overall conductance of Na+ in an NHP film can be equivalent to that of the more conductive bulk NZSP. In this way, we proposed that a thin coating of NHP, applied to a more conductive bulk ceramic (e.g., silicon and zirconium containing NZSP) could afford increased chemical stability to a vulnerable NZSP separator without sacrificing overall ion transport efficacy through the system. XRD analyses of NHP films spin-coated onto on platinized silicon wafer or NZSP pellets confirm nearly phase pure NHP, possibly including a trace amount of HfP2O7 when prepared on Pt/Si (Fig. 4). Fig. 5 shows both plan views and cross-section views of the coatings on Pt/Si and NZSP wafers. In plan view, the films present a relatively dense microstructure with grains on the order of 100 nm. For NHP films on platinized silicon, NHP grains and a minor, smaller grained phase, are observed, while for NHP films on NZSP, delineation of grains is less obvious. In cross section, films on both substrates display a dense microstructure with multiple grains spanning the film thickness. Clear delineation is seen between the NHP film and the underlying NZSP pellet.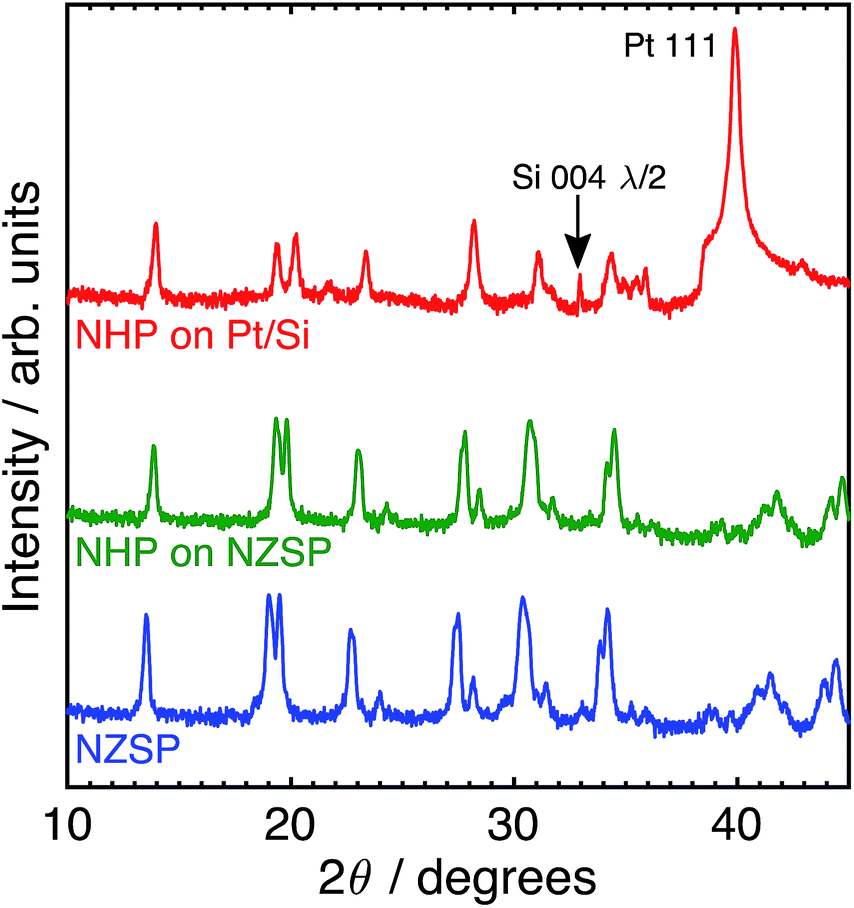 | ||
| Fig. 4 Log-scale X-ray (Cu Kα) diffraction patterns of (top) NHP on platinized silicon (Pt/Si), (middle) NHP on an NZSP pellet, and (bottom) an as-received NZSP pellet. | ||
 | ||
| Fig. 5 (A, B) Plan-view and (C, D) cross-sectional SEM micrographs of NHP thin films on (A, C) platinized silicon and (B, D) an NZSP pellet. | ||
Fig. S1 (ESI†) shows a cross-sectional electron micrograph of the NHP on NZSP, including elemental mapping to illustrate the distinction between the NHP and the NZSP materials. An NHP film thickness of 283 ± 6 nm was recorded for NHP (3 layers, 0.15 M precursor solution) on platinized silicon, while 610 ± 80 nm was recorded for of NHP (3 layers, 0.3 M precursor solution) on NZSP. The larger variation in NHP film thickness on the NZSP substrate is attributed to the increased surface roughness inherent to the polished NZSP pellet. Fig. S2 in the ESI† shows the rough texture of these NZSP pellets as well as cross-sections of the different NHP coating thicknesses on the NZSP pellets. Table S1† provides thickness measurements for each of these thin film preparations as well.
The bright, fine-grained phase observed in Fig. 5A may be a HfO2 or HfP2O7 secondary phase. Although HfO2 is not clearly seen as a contaminant phase in the thin film XRD of Fig. 4, it was observed in the bulk powder patterns in Fig. 2a, and prior research on NZP films identified similar grain morphologies and attributed those to ZrO2 secondary phases.31 The small crystal size, coupled with the low volume fraction of these secondary phase makes identification by XRD difficult. That these films could be grown as apparently dense, relatively phase pure materials at only 775 °C (relative, for example to the >1000 °C required to synthesize bulk NaSICON materials) means that they could be applied to a variety of substrates without the need for high temperatures that might drive deleterious secondary reactions in the substrate material.
The Na+ conductivity of NHP films on platinized silicon wafers was measured over 25–200 °C, using a sputtered platinum top electrode. A typical Nyquist plot for a room temperature measurement is presented in Fig. 6A, displaying a high frequency arc and low frequency tail. The high frequency arc is attributed to Na+ conduction through the NHP film, while the low frequency tail is characteristic of the capacitance at the ionically blocking platinum electrodes.31,36 From fitting these data, the Na+ conductivity was determined to be 1.35 × 10−5 S cm−1 at 200 °C, on par with previous reports for bulk NHP.30 The temperature dependence of Na+ conduction, plotted in Fig. 6B, displayed an Arrhenius behaviour above 25 °C. The observed activation energy of 0.53 eV is consistent with previous reports of bulk NHP,30 thin film NZP,31 and single crystal NZP,35 implying a similar Na+ conduction mechanism for both NHP and NZP. From these XRD and ionic conductivity results, it is concluded that crystalline thin film NHP was successfully produced with Na+ conductivity values and activation energies similar to those of bulk NHP.
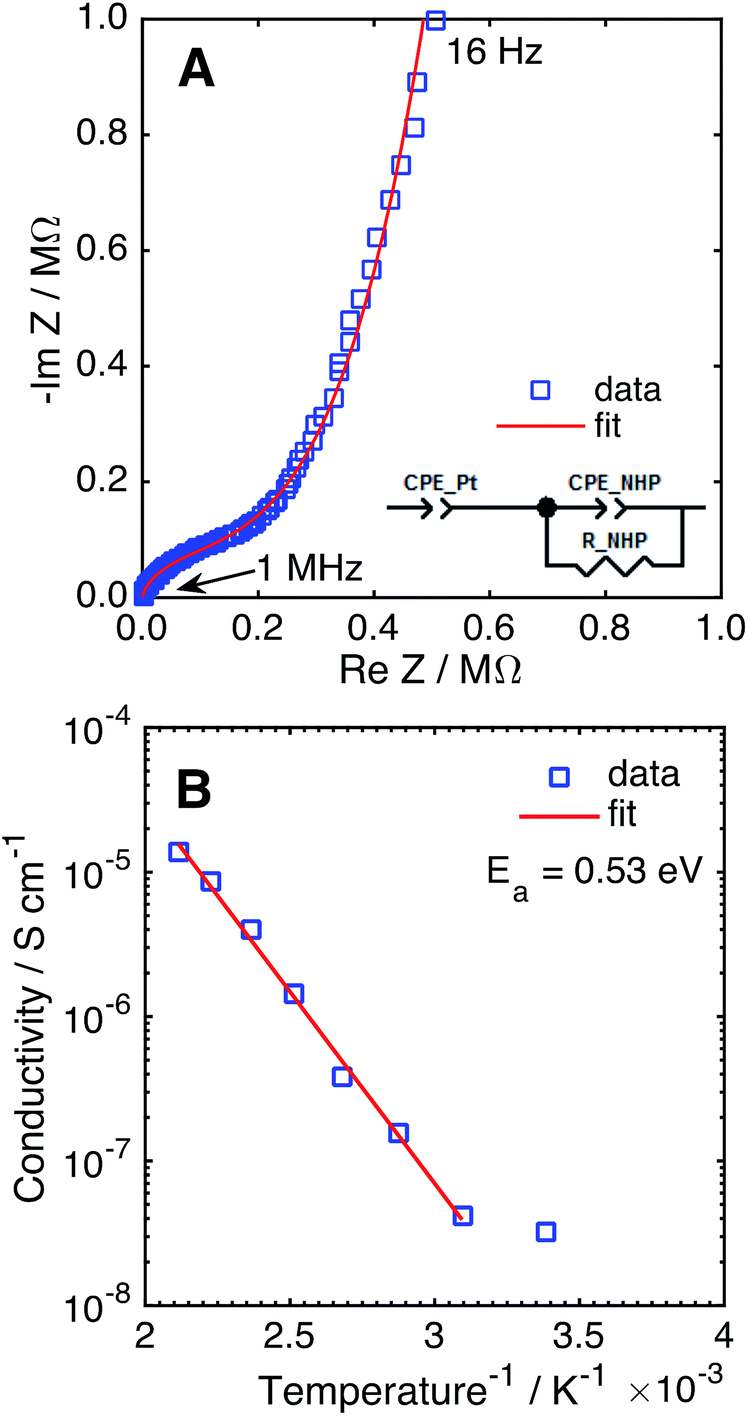 | ||
| Fig. 6 (A) Nyquist plot of NHP thin film in a Pt–NHP–Pt structure at 25 °C. (B) Na+ conductivity of a NHP thin film as a function of temperature. | ||
The Na+ conductance of NZSP pellets coated with NHP was evaluated at 80 °C in an electrochemical cell with 0.5 M NaOH. The cell conductance decreased from 30.1 to 26.8 mS cm−2 upon adding an uncoated NZSP pellet. The corresponding NZSP conductivity of 24.7 mS cm−1 agrees reasonably well with previous reports of Na-conductivity in NZSP using blocking electrodes in air.14,15 Addition of the NHP coating to the NZSP only decreased the conductance from 26.8 to 26.1 mS cm−2, suggesting application of this NHP coating would only minimally impact electrochemical performance. This conductance predicts a NHP conductivity (6.56 × 10−5 S cm−1 at 80 °C) greater than that calculated from platinum–NHP–platinum structures measured in Fig. 6. This higher conductivity may be due to pinholes in the film allowing intrusion of high conductivity aqueous NaOH. If pores are idealized as straight cylinders in the NHP film, porosity of 0.033% would account for this difference in conductivity values, assuming a NaOH conductivity of 0.2 S cm−1. This porosity level is reasonable provided that the coatings were not applied in a cleanroom environment and the rough pellet surface will result in thickness variations that could affect defect concentrations during film growth.
NHP-protected NZSP pellets
The ability of the thin film NHP coatings to chemically protect bulk NZSP pellets was evaluated in 10 M KOH at 80°C. These conditions represent the most extreme electrolyte previously used to test powders, wherein the NHP powders showed preferential stability relative to the Zr-analogue. The backside of the NZSP pellets were coated in silver paint and the NHP-coated front face was exposed to solution. The open circuit potential (OCP) between the silver back electrode and a platinum electrode in solution was measured over time, and the data are plotted in Fig. 7A. As the KOH etched through the NZSP pellet, the open circuit potential remained relatively constant, though some small variation in OCP was expected from the platinum pseudo reference electrode. Once the KOH reached the silver back electrode, however, the OCP immediately shifted >0.5 V more negative, indicating oxidation of the silver back electrode. This behavior is directly analogous to the corrosion potential of an engineering metal with or without a passivating oxide coating.26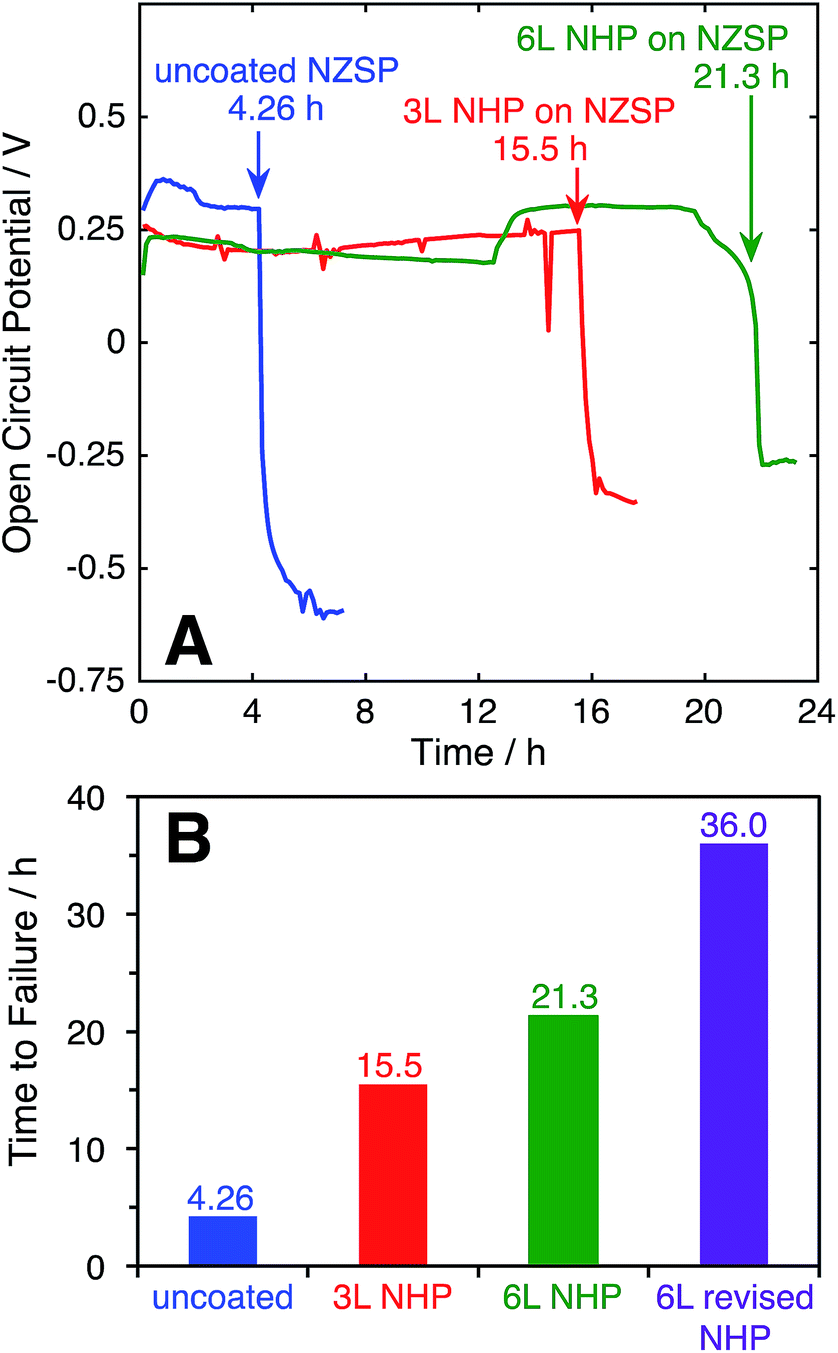 | ||
| Fig. 7 (A) Open circuit potential as a function of time for uncoated and NHP-coated NZSP pellets with a silver back electrode immersed in 10 M KOH at 80 °C. (B) Comparison of lifetimes of NZSP pellets with various NHP coatings subjected to 10 M KOH at 80 °C. | ||
From these experiments it is evident that the application of 3 layers (“3 L”) NHP coating increased the time to failure for an NZSP pellet from 4.26 (uncoated) to 15.5 h, while 6 layers (“6 L”) extended the time to failure to 21.3 h. The fact that doubling the NHP film thickness did not double the increase in time to failure suggests that the KOH is not uniformly etching the NHP, and could be related to defects in the coating.
For uncoated pellets, relatively uniform etching of NZSP was visually evident as the process produced a thick white paste, slumped against the pellet. XRD of this washed and dried paste appeared similar to that of NZP in KOH in Fig. 1B. Upon removal from the electrochemical cell, the uncoated pellets crumbled, having effectively and uniformly disintegrated. In contrast, large pieces of the NHP-coated pellets could be successfully removed, with chemical attack concentrated in select locations. This different behavior suggests that pin-hole defects or microcracks in the NHP films allowed solution to more directly access the vulnerable NZSP and led to premature failure. In an effort to decrease the film stresses during crystallization and minimize the resulting cracks or other defects, 6 layers of NHP precursor solution at half concentration (“6 L revised NHP”) were spin-coated onto NZSP pellets, pyrolyzed at 300 °C, and then crystallized at 775 °C after every third layer, as opposed to previous samples which were crystallized after every layer and lacked an intermediate pyrolysis step.38 This approach significantly reduced the frequency of film or pellet cracking, and the thickness of films produced using this method were 555 ± 136 nm. This approach yielded the best NHP film, lasting 36 hours to failure. Once again, the failed pellet came apart in pieces, indicating that the etching may have been mediated by defects in the film. Still, the NHP coating, only a few hundred nm thick, was able to increase the stability of the ceramic separator system by an order of magnitude. It is worth noting that at 555 nm thick, these films were comparable in overall thickness to the 3 L films reported above (610 nm thick), but yielded better protective properties. This result would suggest that film quality, more than overall film thickness is important in serving as a barrier to alkaline degradation. Collectively, these data suggest that the application of this newly developed NHP thin film barrier approach may have potential to dramatically expand the utility of alkaline-vulnerable Na+ conductors.
Conclusions
Guided by general rules for metal-oxide bond stability, we have designed, synthesized, and evaluated a Hf-substituted NaSICON variant with increased alkaline stability, relative to its zirconium-based analogue. Relative to the traditional NZP phase, in “accelerated” high temperature (80°C) exposure experiments, NHP powders showed dramatically increased stability against LiOH, NaOH, KOH, and CsOH, particularly at concentrations of 1 M or less. Greatest instability of the NHP coating occurred in NaOH, suggesting that ion-interactions with mobile Na+ ions in the ceramic may contribute to the increased solubility. The promise of this material was then explored as a newly developed thin film coating, demonstrated by sol–gel growth at reduced temperatures of 775°C on Pt/Si substrates and NZSP ceramic pellets. Integration of NHP thin films onto NZSP pellets was found to only nominally decrease the system Na+ conductance, while extending the time to failure in 10 M KOH at 80 °C from 4.26 to 36.0 h. Ultimately, failure of NHP-coated NZSP pellets is attributed to pinhole defects in the protective NHP films. This integration of compositionally distinct Na+ conductors across disparate length scales (nm, mm) and processing techniques (sol–gel, traditional powder) represents a promising new avenue by which Na+ conducting systems may be developed for previously inaccessible applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Ms B. B. McKenzie for performing SEM. This work was generously supported by program manager Dr Imre Gyuk through the U.S. Department of Energy, Office of Electricity Delivery and Energy Reliability. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525.References
- N. Anantharamulu, K. K. Rao, G. Rambabu, B. V. Kumar, V. Radha and M. Vithal, J. Mater. Sci., 2011, 46, 2821–2837 CrossRef.
- J. Goodenough, H. Y.-P. Hong and J. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef.
- H. Y.-P. Hong, Mater. Res. Bull., 1976, 11, 173–182 CrossRef.
- J. Fergus, Solid State Ionics, 2012, 227, 102–112 CrossRef.
- A. G. Jolley, D. D. Taylor, N. J. Schreiber and E. D. Wachsman, J. Am. Ceram. Soc., 2015, 98, 2902–2907 CrossRef.
- R. O. Fuentes, F. Figueiredo, F. M. B. Marques and J. I. Franco, Solid State Ionics, 2001, 140, 173–179 CrossRef.
- N. S. Bell, C. Edney, J. S. Wheeler, D. Ingersoll and E. D. Spoerke, J. Am. Ceram. Soc., 2014, 97, 3744–3748 CrossRef.
- A. G. Jolley, G. Cohn, G. T. Hitz and E. D. Wachsman, Ionics, 2015, 21, 3031–3038 CrossRef.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 Search PubMed.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2016, 3, 52–57 CrossRef PubMed.
- E. Ventosa, D. Buchholz, S. Klink, C. Flox, L. G. Chagas, C. Vaalma, W. Schuhmann, S. Passerini and J. R. Morante, Chem. Commun., 2015, 51, 7298–7301 RSC.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 Search PubMed.
- J. Kim, S. H. Jo, S. Bhavaraju, A. Eccleston and S. O. Kang, J. Electroanal. Chem., 2015, 759, 201–206 CrossRef.
- L. J. Small, A. Eccleston, J. Lamb, A. Read, M. Robins, T. Meaders, D. Ingersoll, P. G. Clem, S. Bhavaraju and E. D. Spoerke, J. Power Sources, 2017, 360, 569–574 CrossRef.
- E. Allcorn, G. Nagasubramanian, H. D. Pratt, E. Spoerke and D. Ingersoll, J. Power Sources, 2018, 378, 353–361 CrossRef.
- S. Balagopal, T. Landro, S. Zecevic, D. Sutija, S. Elangovan and A. Khandkar, Sep. Purif. Technol., 1999, 15, 231–237 CrossRef.
- D. Kurath, K. Brooks, G. Hollenberg, D. Sutija, T. Landro and S. Balagopal, Sep. Purif. Technol., 1997, 11, 185–198 CrossRef.
- H. Yagi and T. Saiki, Sens. Actuators, B, 1991, 5, 135–138 CrossRef.
- T. Kida, K. Shimanoe, N. Miura and N. Yamazoe, Sens. Actuators, B, 2001, 75, 179–187 CrossRef.
- H. Aono, Y. Sadaoka, L. Montanaro, E. D. Bartolomeo and E. Traversa, J. Am. Ceram. Soc., 2002, 85, 585–589 CrossRef.
- R. Fuentes, F. Figueiredo, F. Marques and J. Franco, Solid State Ionics, 2001, 139, 309–314 CrossRef.
- J.-I. Jung, D. Kim, H. Kim, Y. N. Jo, J. S. Park and Y. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 304–310 Search PubMed.
- P. Komorowski, S. Argyropoulos, J. Canaday, A. Kuriakose, T. Wheat, A. Ahmad and J. Gulens, Solid State Ionics, 1992, 50, 253–258 CrossRef.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, 2nd edn, 1974 Search PubMed.
- R. Revie, Uhlig's Corrosion Handbook, John Wiley and Sons, 3rd edn, 2011 Search PubMed.
- A. Essoumhi, C. Favotto, M. Mansori, K. Ouzaouit and P. Satre, Solid State Sci., 2007, 9, 240–246 CrossRef.
- E. Vogel, R. Cava and E. Rietman, Solid State Ionics, 1984, 14, 1–6 CrossRef.
- W. Wang, Z. Zhang, X. Ou and J. Zhao, Solid State Ionics, 1985, 28, 442–445 Search PubMed.
- H. Aono and E. Sugimoto, J. Am. Ceram. Soc., 1996, 79, 2786–2788 CrossRef.
- W. Meier, C. Apblett, D. Ingersoll, A. McDaniel and J. F. Ihlefeld, J. Electrochem. Soc., 2014, 161, A364–A367 CrossRef.
- A. Ahmad, C. Glasgow and T. Wheat, Solid State Ionics, 1995, 76, 143–154 CrossRef.
- S. Zhang, B. Quan, Z. Zhao, B. Zhao, Y. He and W. Chen, Mater. Lett., 2004, 58, 226–229 CrossRef.
- L. Hagman and P. Kierkegaard, Acta Chem. Scand., 1968, 22, 1822–1832 CrossRef.
- A. Ivanov-Schitz and A. Bykov, Solid State Ionics, 1997, 100, 153–155 CrossRef.
- E. Barsoukov and J. R. Macdonald, Impedance Spectroscopy, John Wiley & Sons, New York, 2nd edn, 2005 Search PubMed.
- C. T. Shelton, P. G. Kotula, G. L. Brennecka, P. G. Lam, K. E. Meyer, J.-P. Maria, B. J. Gibbons and J. F. Ihlefeld, Adv. Funct. Mater., 2012, 22, 2295–2302 CrossRef.
- G. L. Brennecka and B. A. Tuttle, J. Mater. Res., 2007, 22, 2868–2874 CrossRef.
- J. F. Ihlefeld, E. Gurniak, B. H. Jones, D. R. Wheeler, M. A. Rodriguez and A. H. McDaniel, J. Am. Ceram. Soc., 2016, 99, 2729–2736 CrossRef.
- F. M. Spiridonov and L. N. Komissarova, Russ. J. Inorg. Chem., 1970, 15, 445 Search PubMed.
- J. Stocker and R. Collongues, C. R. Hebd. Seances Acad. Sci., 1957, 244, 83 Search PubMed.
- G. S. Maku, Theoretical Principles of Inorganic Chemistry, McGraw-Hill, 1980 Search PubMed.
- O. J. Glembocki, E. D. Palik, G. R. de Guel and D. L. Kendal, J. Electrochem. Soc., 1991, 138, 1055–1063 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Thin film scanning electron microscopy images, an energy dispersive X-ray spectrum and map, and a tabulation of thin film thicknesses. See DOI: 10.1039/c7ta09924j |
| ‡ Present address: Dept. of Materials Science and Dept. of Electrical and Computer Engineering, University of Virginia, Charlottesville, VA, USA 22903. |
| This journal is © The Royal Society of Chemistry 2018 |