
Eu(III)-coordination polymer sub-micron fibers: material for selective and sensitive detection of Cu2+ ions via competition between photoinduced electron transfer and energy transfer†
Chanchal
Hazra
a,
Sajjad
Ullah
 ab,
Laís G.
Caetano
a and
Sidney J. L.
Ribeiro
*a
ab,
Laís G.
Caetano
a and
Sidney J. L.
Ribeiro
*a
aInstitute of Chemistry, São Paulo State University, UNESP, 14800-060, Araraquara, SP, Brazil. E-mail: sidney@iq.unesp.br; Tel: +55-1633019631
bInstitute of Chemical Sciences, University of Peshawar, Peshawar, 25120, KP, Pakistan
First published on 1st December 2017
Abstract
In this article, we report a unique strategy based on the competition between photoinduced electron transfer (PET) and intramolecular energy transfer (ET) processes occurring among the components of guanine-based europium(III) coordination polymer sub-micron fibers (CPMFs) for the selective and sensitive detection of Cu2+. The novel CPMFs were synthesized via a hydrothermal method using guanine, europium ions (Eu3+) and 3,5-dinitrosalicylic acid (DNSA) as an auxiliary linking molecule that can sensitize the luminescence of Eu3+ ions via intramolecular ET. The photoluminescence (PL) emission signal from the CPMFs is found to be weak due to the existence of PET from guanine to DNSA. The PET process from guanine to DNSA prevents the intramolecular ET from DNSA to Eu3+ ions, leading to the quenching of the luminescence from the CPMFs. However, 11 times enhancement of the PL emission signal from the CPMFs is observed in the presence of Cu2+ ions (12 μM) which is attributed to the suppression of the PET process (or enhancement of ET) as a result of coordination of Cu2+ with guanine molecules. This forms the basis of Cu2+ ion detection and the prepared CPMFs exhibited excellent selectivity and good sensitivity toward Cu2+ ions up to the 1.42 μM detection limit. The study was also extended to real tap water samples, where spiking with Cu2+ ions led to a 10 times enhancement of the PL emission intensity from the CPMFs, though hardly any change was noted for the unspiked tap water sample. We envision that our CPMFs are potential candidates for application in ultrasensitive time-resolved fluorometric assays owing to their long luminescence lifetime, good dispersion and stability in solution.
Introduction
Coordination polymers are a special category of hybrid materials built from metal ions and organic bridging ligands. The proper choice of metal ions and organic ligands can lead to the formation of coordination polymers with different shapes, sizes and physicochemical properties against several organic or inorganic materials.1–3 Coordination polymers have received great attention due to their flexible components and their potential for application in gas storage, catalysis, chemical sensing and nonlinear optics, among others.4–7 The use of different metal ions and organic ligands with different structural motifs provides numerous possible combinations. Coordination polymers are thus of great constructional flexibility for obtaining tailored functional materials. Several potential functions and applications of functional coordination polymer materials require them to be constructed from initial building blocks that are environmentally friendly and biologically compatible.8,9 To achieve this specific goal, natural biomolecules are extensively used because they are non-toxic and water soluble and have several binding sites for various metal ions and even excellent self-assembly properties.10–12 The blending of these biomolecules could provide some interesting properties to coordination polymer (nano/micro) particles, which cannot be retrieved using simple organic molecules as building blocks and hence coordination polymers are promising candidates for a wide range of applications. There are few reports available on the use of biomolecules as building blocks to constitute coordination polymer materials. Among them, nucleobases like adenine and guananine are of great interest.13–15 Some important characteristics of nucleobases, for example, lone pair of electrons on the nitrogen atom, rich metal binding sites and H-bonding ability render them ideal building blocks for constructing metal organic functional coordination polymers.16 Moreover, their multiple Lewis basic sites (amino group and pyrimidine nitrogen atoms) may allow PET and, when linked to a fluorophore, could be exploited to obtain a suitable PET sensor system. A few literature reports suggest that the PET mechanism of fluorescence sensing could be applied in the design of fluorescent sensors based on a fluorescence off/on strategy. For example, Ghosh and coworkers constructed a sensor connecting adenine and anthracene molecules via a spacer and they observed a decrease in the emission signal from antharacene while coordination between the analytes and adenine occurred.17 The same group have shown the use of adenine-based fluorescent PET sensors for the selective detection of dicarboxylic acids and iodide ions.18,19 Most of the fluorescent PET sensors were demonstrated by using conventional fluorescent molecules although very few examples of nano/micro materials were reported. Recently, Chen et al. have shown the potential application of adenine/terbium(III)/dipcolinic acid coordination polymer nanoparticles as fluorescence sensors for the detection of Hg2+, which is based on the suppression of the PET process from adenine to dipcolinic acid.20In the past few years, many luminescent coordination polymers have been reported and lanthanide-based coordination polymers continuously draw particular attention due to their unique physicochemical properties. Lanthanide ions (Ln3+) have a larger coordination number, versatile coordination geometry and great affinity to ligands containing oxygen or nitrogen atoms. These special characteristics of lanthanides are most essential for the construction of functional coordination polymers.21,22 In addition, they possess narrow transitions, large Stokes shift and longer luminescence lifetime (μs to ms)23–25 which make them useful potential candidates for use in the construction of tunable luminescent sensors as well as probes for chemical entities.26–30 In fact, some lanthanide coordination polymers have been successfully used as fluorescent sensors for the detection of metal ions, anions, and biomolecules, for example.31–34
Among the metal ions, Cu2+ is an essential trace element which plays a pivotal role in the biological processes occurring in living organisms.35,36 Cu2+ ions, along with proteins and metalloenzymes, may facilitate many essential metabolic functions like electron transport and oxygen transportation, for instance.37,38 However, excess accumulation of Cu2+ ions in neuronal cytoplasm may cause some serious health problems like Alzheimer's, Parkinson's and Wilson's diseases.39,40 Moreover, Cu2+ is one of the most abundant pollutants, leading again to various infectious diseases in animals. From this point of view, it is important to develop simple and selective sensors for Cu2+ ions. There are fewer reports available on the detection of Cu2+ ions.41,42 Various analytical methods, for example, atomic absorption spectroscopy (AAS), inductively coupled plasma mass spectroscopy (ICP-MS), and voltammetry have so far been used for the selective detection of Cu2+ ions.43–45 Although these methods are quantitative, they are monotonous, time consuming and expensive. In addition, quantum dots (e.g. ZnS, CdSe) and nano/micro materials (functionalized Ag, Au NPs) and Ln3+-doped down/upconverting nanoparticles as fluorescence probes have recently been tested for the selective detection of Cu2+ ions.46–48
In this research article, we report the synthesis of novel guanine-based europium(III) coordination polymer sub-micron fibers (CPMFs) via a hydrothermal method using guanine, europium ions (Eu3+) and DNSA for the selective and sensitive detection of Cu2+ ions. To the best of our knowledge, there is no report available on Cu2+ detection using a lanthanide (Ln3+) coordination polymer via competition between PET and ET. More importantly, we provide a detailed study of the relationship/competition between the PET and ET processes occurring in CPMFs and show how the presence of Cu2+ ions favors the ET process to dominate, leading to an enhancement in the Eu3+-based PL emission signal. This, in turn, provides a simple basis for the selective detection of Cu2+ ions using spectroscopic techniques. In the absence of Cu2+ ions, the PL emission from the CPMFs was found to be weak due to PET from guanine to DNSA that suppressed the intramolecular ET from DNSA to Eu3+ ions, leading to the quenching of the luminescence from the CPMFs. Upon the addition of Cu2+ ions, however, the PL emission signal from the CPMFs enhanced because of the breakdown in the PET mechanism by the coordination of Cu2+ with guanine. The CPMFs show good selectivity and high sensitivity up to the 1.42 μM detection limit. The study was also extended to real tap water samples and the detection limit value is found to be 1.15 μM. The unique strategy based on the competition between PET and intramolecular ET proposed here is general and may be extended to other sensing systems based on Ln3+ ions and suitable electron donor/acceptor ligands. We believe that our CPMFs are suitable candidates for potential applications in ultrasensitive time-resolved fluorometric assays in the solution phase.
Experimental
Materials
Eu2O3 (99.99%, Sigma-Aldrich), Gd2O3 (99.99%, Sigma-Aldrich), HNO3 (65%, Qhemis), guanine (98%, Aldrich), HCl (fuming 37%, Sigma-Aldrich), NaOH (Synth, Brazil), 3,5-dinitrosalicylic acid (DNSA, 98%, Merck), CaCl2·2H2O, Cd(Ac)2·4H2O, CoCl2·6H2O, Fe(NO3)2·9H2O, FeCl2·4H2O, KNO3, MnCl2·4H2O, NaNO3, NiCl2·6H2O, Pb(NO3)2, ZnCl2, CuSO4·5H2O, AgNO3, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), methanol (CH3OH, Synth) and absolute ethanol (C2H5OH, Synth) were used for the synthesis and all the experiments in our work. All chemicals were used without further purification. Preparation of HEPES buffer and stock solutions of interference ions is described in the ESI†.Preparation of europium hydroxide
First, europium nitrate [Eu(NO3)3·6H2O] was obtained by mixing an appropriate amount of Eu2O3 with HNO3 and residual HNO3 was removed by evaporation. The pH of this salt was highly acidic. To increase the pH, water was added to the salt and evaporated to dryness. The process was repeated six times until the pH of the final solution was around 6. The concentration for the stock solution of europium nitrate was found to be 0.882 M as determined by titration with standard EDTA solution. Europium hydroxide was prepared by the addition of 0.16 M NaOH aqueous solution to a 40 mM Eu(NO3)3·6H2O aqueous solution in a 15 mL centrifuge tube. The white precipitate was collected by centrifugation at 8000 rpm for 10 min. To remove unreacted reagents, the precipitate was washed thrice with absolute ethanol and once with deionized water. Finally, the europium hydroxide precipitate was dried in air for 30 min.Synthesis of the Eu–DNSA complex
The Eu–DNSA complex was obtained with the dropwise addition of aqueous NaOH solution to a warm water mixture of freshly prepared europium hydroxide and DNSA. The molar ratio Eu3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNSA:Na+ was equal to 1:3:6. The mixture was vigorously stirred for 30 minute at room temperature and then transferred to a 100 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 160 °C temperature for 8 hours. Subsequently, the reaction mixture was allowed to cool to room temperature. The yellowish product was collected by centrifugation at 8500 rpm for 10 min and washed thrice with absolute ethanol and finally once with deionized water.
:DNSA:Na+ was equal to 1:3:6. The mixture was vigorously stirred for 30 minute at room temperature and then transferred to a 100 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 160 °C temperature for 8 hours. Subsequently, the reaction mixture was allowed to cool to room temperature. The yellowish product was collected by centrifugation at 8500 rpm for 10 min and washed thrice with absolute ethanol and finally once with deionized water.
Preparation of Eu–guanine–DNSA CPMFs
The Eu–guanine–DNSA CPMFs were prepared via a hydrothermal method. Typically, an aqueous NaOH solution of guanine (10 mL) was added to a warm water mixture (20 mL) of freshly prepared europium hydroxide and DNSA. The Eu3+:DNSA:guanine:Na+ molar ratio was equal to 1:3:3:6. The mixture was vigorously stirred for 30 min at room temperature and then transferred to a 100 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 160 °C temperature for 8 hours. Subsequently, the reaction mixture was allowed to cool to room temperature naturally. The faint yellowish precipitate was collected by centrifugation at 8500 rpm for 15 min. To remove unreacted reagents, the precipitate was washed thrice with absolute ethanol and finally once with deionized water. Finally, 10 mg of the product was dispersed in 10 mL methanol:water (3:1, v/v) mixture to form a Eu–guanine–DNSA CPMF suspension for use. As a control experiment, Eu–guanine CPMFs were synthesized by the same experimental steps and conditions followed in the synthesis of the Eu–DNSA complex except using guanine instead of DNSA.
Measurement of the luminescence signal from Eu–guanine–DNSA CPMFs upon the addition of Cu2+ aqueous solution
For the detection of Cu2+, the Eu–guanine–DNSA CPMF suspension was diluted in a CH3OH:H2O (3:1, v/v) mixture. For the sensitivity experiments, 100 μL of Cu2+ aqueous solution with concentrations from 50 to 300 μM was added to 500 μL of Eu–guanine–DNSA CPMF suspension and the CH3OH:H2O (3:1, v/v) mixture was added until the total volume reached 2.5 mL. The minimum and maximum concentrations of Cu2+ ions are set to be 2 μM and 12 μM, respectively. After reacting for 10 min, the luminescence spectra were collected using an excitation wavelength of 355 nm. To examine the selectivity of Eu–guanine–DNSA CPMFs to Cu2+, 100 μL of 300 μM stock solutions of interfering ions were added to 500 μL of Eu–guanine–DNSA CPMF suspension. The total volume was made up to 2.5 mL by adding CH3OH:H2O (3:1, v/v) mixture. The reaction lasted for 10 min before measuring the luminescence intensities of these mixtures at 615 nm. To check the effect of pH on the luminescence intensity of the CPMFs, 500 μL (0.1 wt%) of an Eu–guanine–DNSA CPMF suspension were added to 2.0 mL CH3OH:H2O (3:1, v/v) mixture and buffered at different pH with HEPES at room temperature. The mixture was shaken well and reacted for 10 min before recording the luminescence intensities. The experimental procedure for the detection of Cu2+ ions in tap water samples is given in the ESI.†
Characterizations
The morphology of the CPMFs was characterized by Field Emission Scanning Electron Microscopy (FESEM). FESEM imaging was performed using a JEOL (JSM 6390 LV) instrument with samples first coated with a thin film of gold and palladium in order to avoid charging effects prior to loading into the chamber. The average width (±SD) and length (±SD) of the fibers were measured using Image Tool processing software where the ±SD values represent the width of size distribution and do not refer to any experimental error. The XRD patterns in the 2θ range of 10–60° were collected using a Rigaku-SmartLab diffractometer operating at 35 mA and 70 kV and employing CuKα radiation. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was performed with a Thermo Scientific XSERIES2 ICP-MS instrument. Fourier transform infrared (FTIR) spectra were collected with a PerkinElmer FT-IR spectrometer 1000 at a resolution of 2 cm−1 and averaged over four scans. The room temperature optical absorption spectrum of the sample was recorded on a Varian model Cary 5000 spectrophotometer. The samples were taken in a 3 mL quartz cuvette (path length, 1 cm). The photoluminescence (PL) spectra were measured on a Horiba Jobin Yvon Fluorolog-3 machine (USA), FL3-122 model, spectrometer equipped with a 450 W Xe continuous lamp. All the spectra were acquired in an identical fashion under the same experimental conditions. The PL emission measurements were carried out at an optical spectral resolution of 0.93 nm. The excitation and emission light were dispersed using a monochromator (double grating) with a resolution of 0.3 nm. The emitted photons were counted using a photomultiplier tube (PMT) detector; model R 928 P from Spex 180–850/860 nm. The PL lifetime measurements were performed with the Horiba Jobin Yvon Fluorolog-3 machine (USA), FL3-122 model, spectrometer equipped with a Xenon pulsed bulb with 0.15 Joules per flash. The temporal uncertainty value of the emission decay curve acquisition is ±0.001 ms (accuracy of initial delay).Results and discussion
The Eu–guanine–DNSA CPMFs were successfully synthesized using Eu3+, guanine and DNSA via a hydrothermal method. As a control, CPMFs consisting of only Eu3+ and guanine were also prepared under the same experimental conditions. The chemical structures of the guanine and DNSA molecules are shown in Fig. S1 (ESI†). The morphology of the Eu–guanine and Eu–guanine–DNSA CPMFs was examined by SEM. The SEM images of the Eu–guanine CPMFs indicate the formation of fiber-shaped morphology (Fig. 1) with an estimated average length (±SD) of 3 ± 1 μm and an average width of around 90 ± 30 nm. In the presence of DNSA, the average length of the Eu–guanine CPMFs decreased by half (1.5 ± 0.3 μm), though the average width increased to around 150 ± 50 nm. The shrinkage in the length occurs probably due to the interaction of DNSA with the Eu3+ ions on the surface of the CPMFs. The width distribution histograms of the Eu–guanine CPMFs and Eu–guanine–DNSA CPMFs are shown in Fig. S2 (ESI†). | ||
| Fig. 1 SEM images of (a) Eu–guanine CPMFs and (b) Eu–guanine–DNSA CPMFs. | ||
The qualitative chemical composition of the Eu–guanine–DNSA CPMFs and the incorporation of Eu3+ ions in the fibers were studied by energy-dispersive X-ray spectroscopy (EDS) (Fig. S3, ESI†). The EDS spectrum of the sample shows clear X-ray emission peaks of Eu, C, N and O, suggesting that Eu3+, guanine and DNSA participate in the formation of CPMFs. A quantitative measure of the Eu3+ loading of the fibers was obtained using ICP-MS analysis. Both Eu–guanine–DNSA and Eu–guanine CPMFs were found to contain 0.5 wt.% Eu3+ in the polymer matrix. The X-ray diffraction (XRD) patterns of both Eu–guanine and Eu–guanine–DNSA CPMFs lack the presence of any clear and defined diffraction peaks, indicating the amorphous nature of the fibers (Fig. S4, ESI†).
The coordination of guanine and DNSA with Eu3+ ions in Eu–guanine–DNSA CPMFs is verified using FTIR analysis (shown in Fig. S5, ESI†). From the FTIR spectra, it was observed that the peak at 2680 cm−1 (from pure guanine) disappeared in the case of Eu–guanine–DNSA CPMFs. This result suggests the probable involvement of N9 from guanine in binding to Eu3+ and N1 from guanine to Eu3+ and DNSA, respectively. The shoulder peak at 2990 cm−1 arising due to N–H stretching of amides (from pure guanine) was absent in the Eu–guanine–DNSA CPMFs, indicating that the N1 position of guanine was engaged in binding to Eu3+ and DNSA, respectively. The absence of the peak at 1259 cm−1 (N–H bending) from the free guanine molecule further indicates the involvement of the N1 or N9 positions in binding to Eu3+ ions in Eu–guanine–DNSA CPMFs. The peak at 1672 cm−1 in the case of pure guanine due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration shifted to 1600 cm−1 in the Eu–guanine–DNSA CPMFs, assigned to the probable binding of carbonyl oxygen to Eu3+. Moreover, from the spectra it is quite clear that the peak at 3099 cm−1 due to NH2 stretching (from pure guanine) disappeared in the Eu–guanine–DNSA CPMFs, indicating the interaction between the –NH2 group from guanine and DNSA. In addition, a new broad peak at 3686 cm−1 was observed due to O–H stretching vibrations, which might have arisen due to some trapped water molecules. The broadness of the peak additionally indicates the existence of H-bonding between guanine and DNSA. The symmetric stretching vibration at 1336 cm−1 of the –NO2 group from the free DNSA molecule shifted to a lower wavenumber (1294 cm−1) in the case of Eu–guanine–DNSA CPMFs, suggesting the coordination of the –NO2 group to the –NH2 group of guanine. The peak at 1681 cm−1 in the case of pure DNSA due to CO stretching vibration shifted to 1593 cm−1 in the Eu–guanine–DNSA CPMFs, assigned to the possible involvement of the carboxylic group with Eu3+. Moreover, in the presence of Cu2+ ions, the wavenumber of the –NO2 symmetric stretching vibration of DNSA (in Eu–guanine–DNSA CPMFs) shifted slightly from 1294 cm−1 to 1269 cm−1, reflecting the interaction of the –NO2 group of DNSA with the Cu2+ ion. Meanwhile, upon Cu2+ addition, there is hardly any presence of –NH2 stretching vibration observed which suggests the coordination of the Cu2+ ion with the N atom (from –NH2 group) of guanine.
O stretching vibration shifted to 1600 cm−1 in the Eu–guanine–DNSA CPMFs, assigned to the probable binding of carbonyl oxygen to Eu3+. Moreover, from the spectra it is quite clear that the peak at 3099 cm−1 due to NH2 stretching (from pure guanine) disappeared in the Eu–guanine–DNSA CPMFs, indicating the interaction between the –NH2 group from guanine and DNSA. In addition, a new broad peak at 3686 cm−1 was observed due to O–H stretching vibrations, which might have arisen due to some trapped water molecules. The broadness of the peak additionally indicates the existence of H-bonding between guanine and DNSA. The symmetric stretching vibration at 1336 cm−1 of the –NO2 group from the free DNSA molecule shifted to a lower wavenumber (1294 cm−1) in the case of Eu–guanine–DNSA CPMFs, suggesting the coordination of the –NO2 group to the –NH2 group of guanine. The peak at 1681 cm−1 in the case of pure DNSA due to CO stretching vibration shifted to 1593 cm−1 in the Eu–guanine–DNSA CPMFs, assigned to the possible involvement of the carboxylic group with Eu3+. Moreover, in the presence of Cu2+ ions, the wavenumber of the –NO2 symmetric stretching vibration of DNSA (in Eu–guanine–DNSA CPMFs) shifted slightly from 1294 cm−1 to 1269 cm−1, reflecting the interaction of the –NO2 group of DNSA with the Cu2+ ion. Meanwhile, upon Cu2+ addition, there is hardly any presence of –NH2 stretching vibration observed which suggests the coordination of the Cu2+ ion with the N atom (from –NH2 group) of guanine.
The photoluminescence (PL) emission and excitation spectra of the 0.1 wt% Eu–guanine–DNSA CPMFs are shown in Fig. 2. The PL excitation spectrum (inset) obtained by monitoring the Eu3+ emission at 615 nm shows a broad band centered at 355 nm corresponding to the absorption from the DNSA molecule. To get more insight into the broad nature of the band and check the possibility of the presence of an embedded charge transfer band, we prepared the isostructural Gd–guanine–DNSA sample following the same procedure used to synthesize the Eu–guanine–DNSA CPMFs. The UV-Visible spectra of these two isostructural samples were obtained in methanol–water solvent mixtures of varying polarity (methanol:water = 3:1, 1:1, 1:3). From Fig. S6 (ESI†), it is observed that the absorption maxima of both the isostructural samples slightly shift to higher wavelength with increase in the polarity of the solvent mixture. However, the shift is not large enough to be attributed a prominent charge transfer band. We thus conclude that the broad excitation spectrum is primarily attributed to the absorption of DNSA molecules although the possibility of a small charge transfer contribution cannot be completely overlooked.
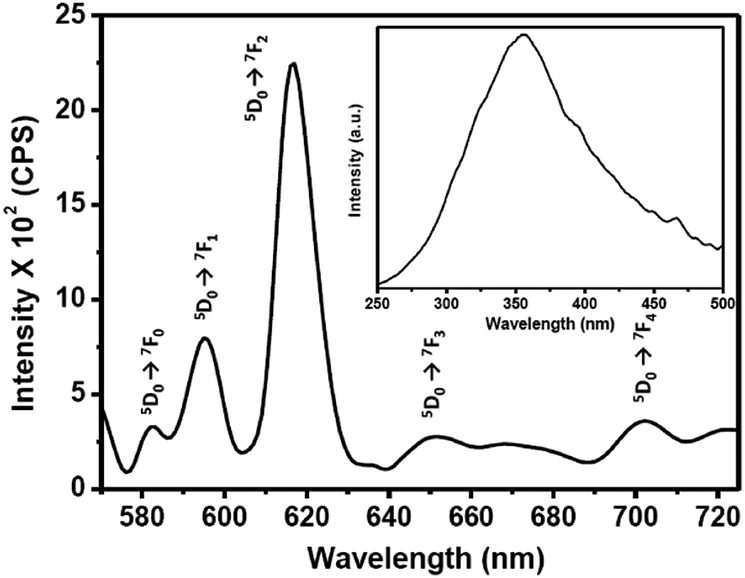 | ||
| Fig. 2 PL emission spectrum of 0.1 wt% Eu–guanine–DNSA CPMFs in the CH3OH:H2O (3:1, v/v) mixture (λex = 355 nm). The inset shows the PL excitation spectrum for 0.1 wt% Eu–guanine–DNSA CPMFs in the CH3OH:H2O (3:1, v/v) mixture (λemi = 615 nm). | ||
Upon excitation of the DNSA molecule (λex = 355 nm), the Eu–guanine–DNSA CPMFs show PL emission at 615 nm along with relatively weaker emissions near 580, 592, 654 and 700 nm, which are assigned to the 5D0 → 7F2, 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F3 and 5D0 → 7F4 transitions of Eu3+, respectively. The positions of the emission peaks from the Eu–guanine–DNSA CPMFs are the same as those of the Eu–DNSA complex, which is fluorescent in the CH3OH:H2O (3:1, v/v) mixture. Interesting to note is that the excitation of the Eu–guanine–DNSA CPMFs shifts to a higher wavelength (355 nm) compared to the absorption of the Eu–DNSA complex (335 nm) (Fig. S7, ESI†). This might be due to the crystal field effect after guanine coordination with Eu3+ ions and DNSA molecules to form CPMFs. In addition, it was observed that upon 355 nm excitation, the Eu–DNSA complex shows PL emission signals at 580, 592, 615, 654 and 700 nm, respectively, which are well comparable with that obtained using 335 nm excitation (Fig. S8, ESI†). As shown in Fig. S9 (ESI†), there is hardly any PL emission observed from the Eu–guanine CPMFs, which is due to the lower absorption coefficient of the Eu3+ ion. These results clearly indicate an intramolecular energy transfer from DNSA to Eu3+ in the Eu–guanine–DNSA CPMFs.
To investigate the detection ability of the Eu–guanine–DNSA CPMFs toward Cu2+ ions, an aqueous solution of Cu2+ was added to the CPMF dispersion. The PL emission spectra shown in Fig. 3 clearly indicate that upon the addition of Cu2+ solution, the Eu3+ emission intensity from the CPMFs increases dramatically. The luminescence behavior of the Eu–guanine–DNSA CPMFs was also confirmed by naked eye observation. In the presence of Cu2+ ions, the precipitate obtained after the centrifugation showed bright orange-red emission, whereas only a weak emission was observed in the absence of Cu2+ ions (Fig. S10, ESI†). Furthermore, to understand the effect of anions on the sensing of Cu2+ ions, copper salts with different anions such as copper chloride, sulfate and nitrate were used. From Fig. S11 (ESI†) it is clear that a similar enhancement of the Eu3+ luminescence was noticed in the presence of different counter anions, thus ruling out any affect of anions on the luminescence enhancement. To verify the selectivity, Eu–guanine–DNSA CPMFs were exposed to the distinct aqueous solution of other metal ions Ca2+, Cd2+, Co2+, Fe3+, Fe2+, K+, Mn2+, Na+, Ni2+, Pb2+, Zn2+, Cu2+, Hg2+, and Ag+, for example. The PL emission spectra shown in Fig. 3 hardly indicate any change in the emission intensity upon the addition of these metal ions. The bar diagram in the inset shows that the selective enhancement of Eu3+ luminescence intensity occurs only with the addition of Cu2+. The high selectivity of Eu–guanine–DNSA CPMFs toward Cu2+ is verified by measuring the Eu3+ luminescence from the Eu–guanine–DNSA CPMFs containing both Cu2+ and other metal ions. The aqueous solution of each foreign metal ion was first added to the CPMF dispersion followed by the addition of the same concentration of Cu2+. Fig. 4 shows that the Eu3+ emission intensity from the CPMFs is selectively enhanced only upon the addition of Cu2+. Upon the addition of Cu2+ ions, the enhancement rate in the luminescence signal from the CPMFs was demonstrated by measuring the PL emission intensity at different time intervals. The Eu3+ emission intensity from the Eu–guanine–DNSA CPMFs increases with time and hardly any change is observed after 10 minutes. This result clearly reveals a rapid response of our sensing system toward Cu2+ ions. The change in the emission spectra of the Eu–guanine–DNSA CPMFs (in the absence and the presence of Cu2+) as a function of time is shown in Fig. S12 (ESI†).
 | ||
| Fig. 3 PL Emission spectra of Eu–guanine–DNSA CPMFs after mixing with different metal ions including Cu2+ ions. The inset shows a bar diagram indicating the selective enhancement of the luminescence intensity of Eu3+ ions upon Cu2+ addition. | ||
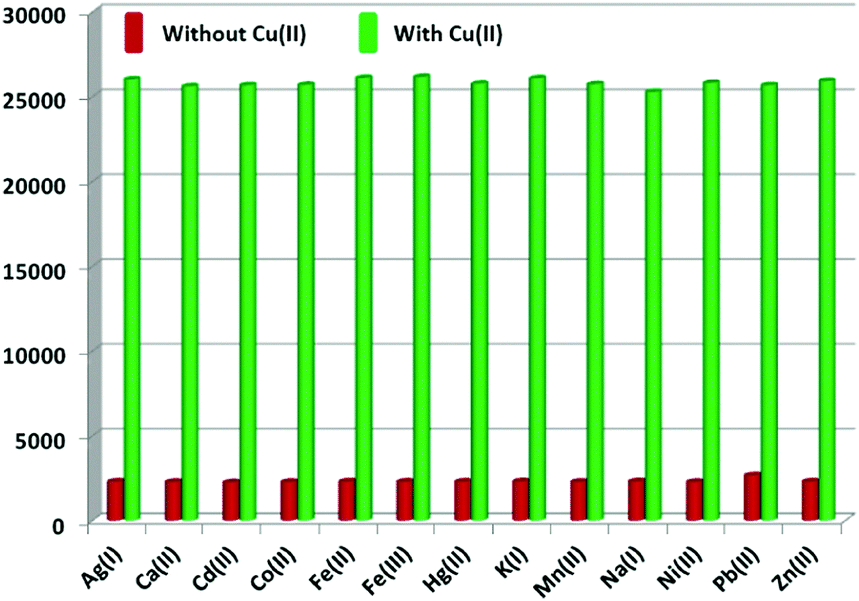 | ||
| Fig. 4 Bar diagram indicating the selective enhancement of the Eu3+ emission intensity of the Eu–guanine–DNSA CPMFs by Cu2+ ions in the presence of other metal ions. | ||
The binding of guanine with DNSA facilitates the decrease in the intramolecular energy transfer from DNSA to Eu3+, leading to the decrease in Eu3+ luminescence. It is worth mentioning that the typical characteristic of the PET mechanism is that the luminescence associated with the PET process commonly enhances with the decrease in the pH value of the solution.49 As shown in Fig. 5 and Fig. S13 (ESI†), the luminescence intensity of the Eu–guanine–DNSA CPMFs increased with the decrease in pH, indicating an efficient PET action between guanine and DNSA molecules. Under the acidic conditions, the protonation of guanine and DNSA deactivates the PET process, leading to the recovery of the ET process from DNSA to Eu3+ and subsequently an enhancement in the emission signal from Eu–guanine–DNSA CPMFs occurs. Furthermore, the pH of the solution affects the stability of the Eu–guanine–DNSA CPMFs. When the solution is highly acidic, the Eu–guanine–DNSA CPMFs gradually dissociate due to the protonation of the ligand molecules and the dissociation increases with gradual decrease in the pH of the solution. However, when the pH value of the solution reached to 5.06, the Eu–guanine–DNSA CPMFs become stable and could be stored for several days. Moreover, from Fig. 5, it is clearly noted that the PL emission intensity of the Eu–guanine–DNSA CPMFs decreased with the increase in pH, indicating the activation of PET behavior between guanine and DNSA. In the basic medium, the guanine becomes highly deprotonated and a rich electron density resides on the nucleus of the guanine molecule. The di-nitro aromatic (for example DNSA), as an electron acceptor, can favorably interact with electron donors like guanine. In the present case, the electron-rich amine or amide groups of guanine have a charge transfer (CT) interaction with the electron-deficient aromatic nucleus of DNSA coordinated to the Eu3+ ion in the Eu–guanine–DNSA CPMFs. In addition, hydrogen bonding between O atoms (–NO2 groups of DNSA) and H atoms (–NH2 groups of guanine) is also favorable. On the basis of the above characteristics, we presume that, upon photoexcitation, guanine molecules are able of transferring an electron to DNSA molecules in Eu–guanine–DNSA CPMFs. To further support the PET action between guanine and DNSA, we varied the concentration of guanine to form Eu–guanine–DNSA CPMFs. From Fig. S14a–c (ESI†), it is quite clear that the lifetime of the 5D0 state of Eu3+ ion decreases with increase in guanine concentration. Upon photoexcitation, the presence of more guanine facilitates more electron transfer to the DNSA molecule, leading to a more pronounced activation of PET behavior in between them and subsequently the lifetime of the 5D0 state of Eu3+ in the Eu–guanine–DNSA CPMFs decreases. These results clearly suggest that the PET behavior became more efficient with the increase in guanine concentration in the Eu–guanine–DNSA CPMFs. Moreover, from an analysis of the emission decay curves as a function of Cu2+ concentration (2–10 μM) (Fig. S15, ESI†), it is obvious that the luminescence lifetime of the 5D0 state of Eu3+ is slightly changed in the presence of Cu2+. Based on the lifetime data presented in Fig. S14 and S15 (ESI†), we infer that the inherent transition processes of Eu3+ induced by the ligand field are at least slightly affected by Cu2+. So we consider that the coordination of Cu2+ mainly occurred with the guanine nitrogen, although the anion nature of the Eu–guanine–DNSA from DNSA may also have some contribution to the bonding of Cu2+. A schematic of the proposed mechanism is shown in Fig. 6.
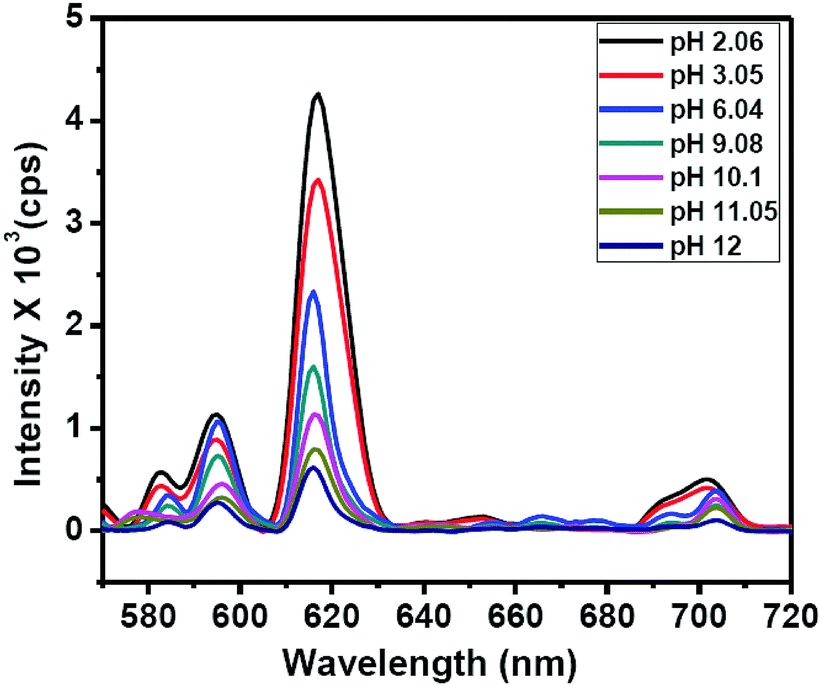 | ||
| Fig. 5 PL emission spectra of Eu–guanine–DNSA CPMFs with different pH of the solution. All the measurements were carried out in the CH3OH:H2O (3:1, v/v) mixture. | ||
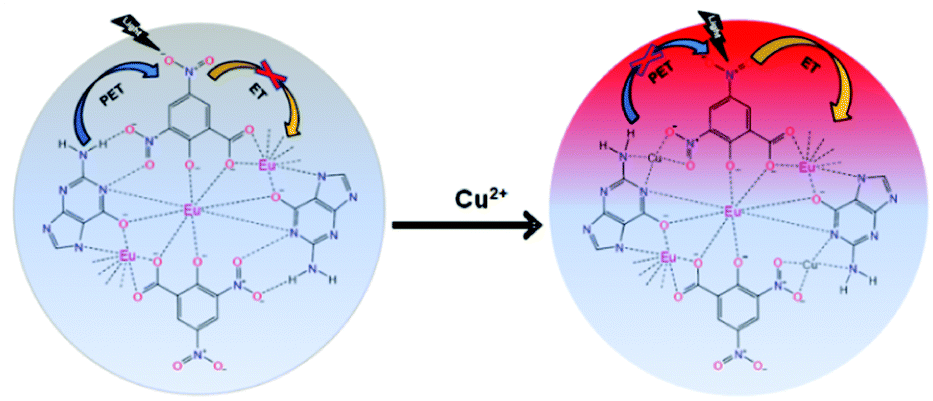 | ||
| Fig. 6 Scheme illustrating the PET behavior between guanine and DNSA (before the Cu2+ addition) and the ET mechanism between DNSA and Eu3+ ions (after Cu2+ addition). | ||
In our work, we carried out the Cu2+-induced enhancement reaction at pH 7.03. Cu2+ is an effective quencher for the Eu–DNSA complex because of its more stable binding to DNSA than that of Eu3+ which leads to the replacement of Eu3+ in the complex by Cu2+. In our case, the presence of Cu2+ enhanced the luminescence of CPMFs and thus indicated that Cu2+ was not only directly bound to the DNSA molecule alone, but also to guanine molecules. In particular, the binding of Cu2+ to guanine prevented the PET process resulting in luminescence enhancement from the Eu–guanine–DNSA CPMFs. To get more insight into the mechanism, we propose an electronic diagram shown in Fig. S16 (ESI†). Upon 355 nm excitation, the DNSA molecule gets excited but the close proximity between DNSA and guanine due to the existence of H-bonding results in subsequent electron transfer from guanine to the DNSA ground state followed by the migration of the DNSA excited electron to the guanine ground state. Due to this PET process, the excited singlet states of DNSA are unable to transfer the excitation energy to their triplet state, from where energy would subsequently transfer to the electronic states of Eu3+ ion to produce intense red emission. Thus, in the absence of Cu2+ ions, the Eu3+ luminescence signal from the CPMFs is found to be weak due to the existence of PET from guanine to DNSA. However, in the presence of Cu2+ ions, H-bonding between guanine and DNSA breaks down. The increased separation between the molecules inhibits PET. As a result, upon 355 nm excitation, DNSA gets excited to the lowest energy excited singlet state from where it crosses over to the lowest energy triplet state via intersystem crossing (ISC). After an encounter with the Eu3+ ion which has an absorption level of slightly lower energy than the energy of the triplet state of the donor (DNSA in this case), the donor loses its energy by a radiationless deactivation. Subsequently, Eu3+ accepts energy lost by DNSA and gets raised to its luminescent electronic level. Eu3+ loses its energy by emitting photons. Moreover, the closer proximity between Eu3+ and DNSA in the presence of Cu2+ facilitates charge transfer to a small extent, leading to the lowering of the triplet state of the donor (DNSA) and at the same time increase in the ligand-metal charge transfer (LMCT) level. As a result, the gap between these two energy levels decreases resulting in increased red emission from Eu3+.
The Eu3+ emission signals of Eu–Gu–DNSA CPMFs as function of Cu2+ concentration are shown in Fig. 7. It is clearly noted that the PL emission of Eu3+ ions in Eu–guanine–DNSA CPMFs enhanced with an increase in Cu2+ concentration from 0 to 12 μM. There is a linear relationship between the emission signal (615 nm) and Cu2+ concentration in the 2 to 12 μM range. The sensitivity of the Eu–guanine–DNSA CPMFs to Cu2+ is evaluated on the basis of a signal-to-noise ratio of 3:1. The detection limit is found to be 1.42 μM. For comparison, the linear range and detection limit (LOD) obtained from our method are listed in Table S1 (ESI†) along with the corresponding values obtained from other methods, Förster resonance energy transfer (FRET), ET and aggregation-induced fluorescence quenching, for instance. It is clear from the data that Cu2+ detection using Eu–guanine–DNSA CPMFs via competition between PET and ET is comparable to other methods. The change in Eu3+ emission signal may also possibly arise from (i) the difference in Eu3+ content of the samples and/or (ii) the change in local environment of the Eu3+ ions upon Cu2+ addition. Hypothesis (i) is ruled out since the Eu3+ loading of the samples is the same (0.5 wt%) as confirmed by ICP-MS analysis. To test hypothesis (ii), we calculated the asymmetric ratio, I(5D0 → 7F2)/I(5D0 → 7F1), both for Eu–guanine–DNSA (as pristine sample) and Eu–guanine–DNSA (as real sample) CPMFs for different concentrations of Cu2+ ions (Tables S2 and S3, ESI†). Unlike a linear increase in intensity of Eu3+ emission as a function of Cu2+ ions, we observed a random change in the asymmetric ratio values rather than a gradual decrease or increase. These results clearly indicate that the enhancement of Eu3+ emission signals (upon Cu2+ addition) occurs primarily due to the efficient intramolecular energy transfer from DNSA to Eu3+ ions rather than due to a change in the local environment of the Eu3+ ion.
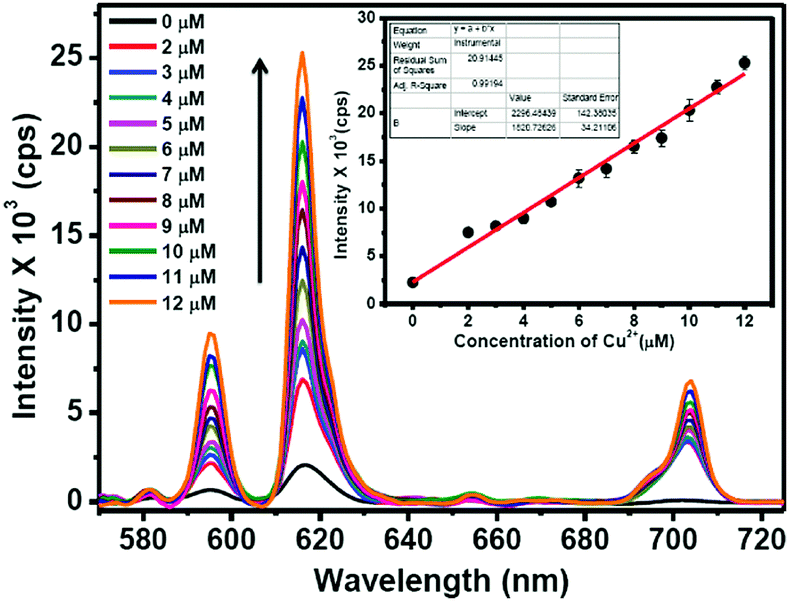 | ||
| Fig. 7 PL emission spectra of Eu–guanine–DNSA CPMFs in the presence of different concentrations of Cu2+ ions. The inset shows the linear relationship of the luminescence intensity of Eu–guanine–DNSA CPMFs at 615 nm against different Cu2+ concentrations. The error bars correspond to the standard deviation (SD) for triplicate measurements. | ||
Moreover, since the 5D0 → 7F0 transition can provide information regarding the number of sites occupied by the Eu3+ ion, we analyzed the effect of change in Cu2+ concentration and pH on this transition. As is evident from Fig. 7, no splitting of this transition is observed with change in Cu2+ concentration. This indicates that for any concentration of Cu2+, there is only one type of site occupied by Eu3+. However, with the increase in Cu2+ concentration, a very small red-shift is observed. It is well known that any change in this peak position reflects a change in covalence.50 We believe that the interaction between Cu2+ and guanine changes the nature of bonding between them in CPMFs. This in turn leads to a change in covalence between Eu3+ and the ligands. As a result, the 5D0 → 7F0 transition slightly shifts toward the red region. Similarly, the change in pH may alter the binding of ligands with Eu3+. However, we would like to point out that the change in peak position or peak broadening does not follow any particular trend as a function of pH. Although a change in peak position definitely indicates an alteration in binding, the randomness inhibits any further evaluation.
To understand the suitability of the Eu–guanine–DNSA CPMFs to detect Cu2+ in real water samples, we mixed 1–10 μM concentrations of Cu2+ with tap water samples. The Cu2+ spiked water samples were added to the Eu–guanine–DNSA CPMFs and the Eu3+ luminescence was measured. A gradual enhancement in the PL emission intensity of Eu3+ is observed for the Cu2+ spiked tap water samples (shown in Fig. 8) whereas hardly any change in the PL emission intensity is noted for the pure tap water sample. The calibration curve for real sample analysis is given in the inset in Fig. 8. A linear relationship is observed between the PL emission intensity of Eu–guanine–DNSA CPMFs and the concentration of Cu2+ ions in the range of 1–10 μM in the Cu2+ spiked tap water sample. The sensitivity of the Eu–guanine–DNSA CPMFs is evaluated on the basis of a signal-to-noise ratio of 3:1. The detection limit is found to be 1.15 μM. To check the feasibility of this method toward Cu2+ detection, we also calculated % recovery and RSD for samples with different Cu2+ concentrations (1–10 μM) and the values are shown in Table S4 in the ESI.†
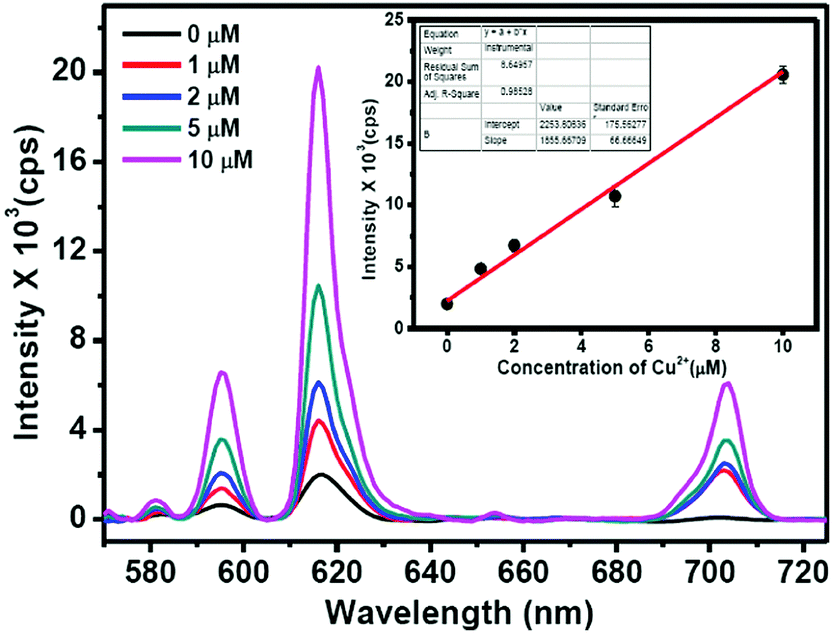 | ||
| Fig. 8 PL emission spectra of Eu–guanine–DNSA CPMFs at variable concentrations of Cu2+ in spiked tap water samples. The inset shows the linear calibration curve. The error bars correspond to the standard deviation (SD) for triplicate measurements. | ||
Alternatively, we could also obtain an almost linear calibration curve by taking the overall Eu3+ emission or integrated emission intensity (570–720 nm) for both the pristine Eu–guanine–DNSA (Fig. S17, ESI†) and spiked Eu–guanine–DNSA (Fig. S18, ESI†) samples. The obtained experimental errors in Fig. S17 and S18 (ESI†) are not significantly deviated from that obtained from Fig. 7 and 8, respectively. The limit of detection values for the pristine and spiked Eu–guanine–DNSA samples as calculated from Fig. S16 and S17 (ESI†) considering the overall Eu3+ emission were found to be 1.99 and 1.88 μM, respectively. Finally, we studied the photostability of our fibers (with and without Cu2+ addition) toward UV irradiation (Fig. S19, ESI†). The only slight decrease in the absorbance values observed in the spectra of both samples upon exposure to UV light for different intervals of time suggests that the prepared fibers exhibit sufficient stability under UV irradiation. These results clearly suggest the feasibility of the developed Eu–guanine–DNSA CPMFs for the detection of Cu2+ in the micromolar regime.
Conclusions
Novel guanine-based europium(III) coordination polymer sub-micron fibers (CPMFs) were successfully synthesized via a hydrothermal method using guanine, europium ions (Eu3+) and 3,5-dinitrosalicylic acid (DNSA) for the first time. We show how the interplay between energy transfer (ET) and electron transfer (PET) can be exploited to design novel and selective sensors for Cu2+ ions. In the CPMFs, the PET process from guanine to DNSA supressed the intermolecular ET process from DNSA to Eu3+ ions, leading to the quenching of the luminescence emission signal from the CPMFs. Upon the addition of Cu2+ ions, however, a significant enhancement in the luminescence signal from the CPMFs was noticed which is attributed to the inhibition of the PET process by the coordination of Cu2+ with guanine. This luminescence enhancement is very selective as the addition of other metal ions hardly affects the intensity of the PL emission. We consider that our findings open a new avenue for the detection of Cu2+ ions upto the 1.42 μM level, which is well below the safety limit.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
C. Hazra and S. Ullah acknowledge financial support from FAPESP (The São Paulo Research Foundation, Brazil) under fellowship grant number 2015/18733-0 and 2015/22875-4, respectively. The authors are also thankful to the Brazilian National Institute of Photonics (INFO) and the Brazilian funding agencies CAPES (Coordination for the Improvement of Higher Education Personnel) and CNPq (The Brazilian National Council for Scientific and Technological Development) for financial assistance.Notes and references
- M. Oh and C. A. Mirkin, Nature, 2005, 438, 651–654 CrossRef CAS PubMed.
- D. Liu, C. Poon, K. Lu, C. He and W. Lin, Nat. Commun., 2014, 5, 4182 CAS.
- D. J. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef PubMed.
- H. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- J. Y. Lee, D. H. Olson, L. Pan, T. J. Emge and J. Li, Adv. Funct. Mater., 2007, 17, 1255–1262 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Soc. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC.
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata and C. Adachi, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS PubMed.
- F. Beuerle, Nature, 2016, 540, 529–531 CrossRef CAS.
- V. Baumann, M. A. H. Muhammed, A. J. Blanch, P. Dey and J. R. Fernández, Isr. J. Chem., 2016, 56, 195–213 CrossRef CAS.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- B. Charma, A. Mahata, S. Mandani, T. K. Sarma and B. Pathak, RSC Adv., 2012, 6, 62968–62973 Search PubMed.
- N. V. Hud, Nucleic Acid-Metal Ion Interactions, RSC Publishing, Cambridge, 2009, 10.1039/9781847558763.
- A. Sigel, H. Siegel and R. K. O. Sigel, Cadmium: From Toxicity to Essentiality, Springer, Dordrecht, 2012 Search PubMed.
- I. Imaz, M. Rubio-Martinez, J. An, I. Sole-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC.
- K. Ghosh and T. Sen, Beilstein J. Org. Chem., 2010, 6, 8 Search PubMed.
- K. Ghosh, T. Sen and R. Fröhlich, Tetrahedron Lett., 2007, 48, 7022–7026 CrossRef CAS.
- K. Ghosh and T. Sen, Tetrahedron Lett., 2008, 49, 7204–7208 CrossRef CAS.
- H. Tan, B. Liu and Y. Chen, ACS Nano, 2012, 6, 10505–10511 CrossRef CAS PubMed.
- R. R. Su, P. Tao, Y. Han, C. H. Zeng and S. L. Zhong, J. Nanomater., 2016, 2016, 5 Search PubMed.
- C. H. Zeng, J. L. Wang, Y. Y. Yang, T. S. Chu, S. L. Zhong, S. W. Ng and W. T. Wong, J. Mater. Chem., 2014, 2, 2235–2242 CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer Verlag, Berlin, 1st edn, 1994 Search PubMed.
- J. C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- Q. Su, W. Feng, D. Yang and F. Li, Acc. Chem. Res., 2017, 50, 32–40 CrossRef CAS PubMed.
- S. Sarkar, M. Chatti, V. N. K. B. Adusumalli and V. Mahalingam, ACS Appl. Mater. Interfaces, 2015, 7, 25702–25708 CAS.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan, X. Liu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- S. Wilhelm, M. D. Barrio, J. Heiland, S. F. Himmelsto, J. Galbán, O. S. Wolfbeis and T. Hirsch, ACS Appl. Mater. Interfaces, 2014, 6, 15427–15433 CAS.
- P. Harvey, C. Oakland, M. D. Driscoll, S. Hay and L. S. Natrajan, Dalton Trans., 2014, 43, 5265–5268 RSC.
- Z. Qi, Q. You and Y. Chen, Anal. Chim. Acta, 2015, 902, 168–173 CrossRef PubMed.
- G. Tang, J. Gao, C. Wang and H. Tan, Sens. Actuators, B, 2017, 244, 571–576 CrossRef CAS.
- H. H. Zeng, W. B. Qiu, L. Zhang, R. P. Liang and J. D. Qiu, Anal. Chem., 2016, 88, 6342–6348 CrossRef CAS PubMed.
- H. Tan, C. Ma, Q. Li, L. Wang, F. Xu, S. Chen and Y. Song, Analyst, 2014, 139, 5516–5522 RSC.
- D. G. Barceloux, J. Toxicol., Clin. Toxicol., 1999, 37, 217–230 CrossRef CAS PubMed.
- X. B. Zhang., J. Peng., C. L. He., G. L. Shen and R. Q. A. Yu, Anal. Chim. Acta, 2006, 567, 189–195 CrossRef CAS.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10–11 CrossRef CAS PubMed.
- S. Mare, S. Penugond, S. M. Robinson, S. Dohgu, W. A. Banks and N. Ercal, Peptides, 2007, 28, 1424–1432 CrossRef CAS PubMed.
- J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2008, 130, 6898–6899 CrossRef PubMed.
- Y. Zhou, F. Wang, Y. Kim, S. J. Kim and J. Yoon, Org. Lett., 2009, 11, 4442–4445 CrossRef CAS PubMed.
- W. Lin, L. Yuan, W. Tan, J. Feng and L. Long, Chem. – Eur. J., 2009, 15, 1030–1035 CrossRef CAS PubMed.
- N. Pourreza and R. Hoveizavi, Anal. Chim. Acta, 2005, 549, 124–128 CrossRef CAS.
- J. S. Becker, A. Matusch, C. Depboylu, J. Dobrowolska and M. V. Zoriy, Anal. Chem., 2007, 79, 6074–6080 CrossRef CAS PubMed.
- A. A. Ensafi, T. Khayamian, A. Benvidi and E. Mirmomtaz, Anal. Chim. Acta, 2006, 561, 225–232 CrossRef CAS.
- M. Koneswaran and R. Narayanaswamy, Sens. Actuators, B, 2009, 139, 104–109 CrossRef CAS.
- Y. H. Chan, J. Chen, Q. Liu, S. E. Wark, D. H. Son and J. D. Batteas, Anal. Chem., 2010, 82, 3671–3678 CrossRef CAS PubMed.
- W. Chen, X. Tu and X. Guo, Chem. Commun., 2009, 1736–1738 RSC.
- X. Guo, X. Qian and L. A. Jia, J. Am. Chem. Soc., 2004, 126, 2272–2273 CrossRef CAS PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley and Sons Ltd, West Sussex, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures of guanine and DNSA, histogram, EDS spectrum, FTIR, excitation and absorption spectra, PL spectra, digital images, bar diagram, and PL lifetime. See DOI: 10.1039/c7tc02638b |
| This journal is © The Royal Society of Chemistry 2018 |