
Paper-based Transwell assays: an inexpensive alternative to study cellular invasion†
Rachael M.
Kenney‡
a,
Adam
Loeser‡
a,
Nathan A.
Whitman
a and
Matthew R.
Lockett
 *ab
*ab
aDepartment of Chemistry, University of North Carolina at Chapel Hill, 125 South Road, Chapel Hill, NC 27599-3290, USA. E-mail: mlockett@unc.edu
bLineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Chapel Hill, NC 27599-7295, USA
First published on 10th October 2018
Abstract
Cellular movement is essential in the formation and maintenance of healthy tissues as well as in disease progression such as tumor metastasis. In this work, we describe a paper-based Transwell assay capable of quantifying cellular invasion through an extracellular matrix. The paper-based Transwell assays generate similar datasets, with equivalent reproducibility, to commercially available Transwell assays. With different culture configurations, we quantify invasion: upon addition of an exogenous factor or in the presence of medium obtained from other cell types, in an indirect or direct co-culture format whose medium composition is dynamically changing, and in a single-zone or parallel (96-zone) format.
1. Introduction
Cellular movement plays an essential role in the development and maintenance of tissues. In angiogenesis, new vessels capable of perfusing tissues with oxygen and nutrients are formed.1 In wound healing, a number of cell types move into the affected site to tackle inflammation and reconstruct damaged tissue structures.2,3 In the context of cancer progression, the metastatic cascade requires tumorigenic cells to first invade neighboring healthy tissue.4The Transwell assay is commonly used to identify and evaluate the chemical cues that modulate cell migration and invasion.5–7 This setup consists of two chambers separated by a porous membrane. In a typical experiment, cells are seeded on one side of a membrane, both chambers filled with culture medium, and a chemokine of interest placed in the chamber opposite the cells. At the experiment's end, cells that moved through the membrane are stained and enumerated. In migration assays the porous membrane is unmodified. In invasion assays the membrane is first coated with a naturally derived or synthetically prepared extracellular matrix (ECM).7,8 The ECM results in a more representative in vivo response than migration assays, as cells must remodel their extracellular environment before movement.9
Commercially available Transwell inserts contain pore sizes suitable for cell culture (0.4–1.0 μm) or migration assays (3.0–8.0 μm). Each insert supports an individual experiment. When combined with a commercial well plate, up to 96 assays can be performed in parallel. Despite their simplicity, these devices are costly. Individual inserts can cost as much as $12.00 and 96-well formats as much as $300 per plate.10 Insert prices increase considerably if the membranes are pre-coated with ECM components.
Paper-based cultures are an emerging and highly modular platform capable of quantifying cellular phenotypes, invasion, and drug metabolism in tissue-like environments.11–14 In this platform, wax-patterned paper scaffolds provide a culture environment with a defined volume. The cellulose fibers provide structural integrity to thin slabs of cell-laden hydrogel, which are prone to cracking upon tactile manipulation.
We previously described invasion assays in which a single cell-containing scaffold was sandwiched between scaffolds that contained only ECM.15–17 At the end of these assays, the paper scaffolds were separated, stratifying cells by their level of invasiveness.
The paper-based Transwell assay we present here separates cells into two populations: invasive and non-invasive. The decreased resolution of separation results in an easier setup that requires a paper scaffold and a standard 96-well plate, making the format accessible and the readout an experimental analog to commercially available Transwell assays. The ability to prepare the paper scaffolds in the laboratory offers the flexibility to readily change experimental parameters such as the volume of the cell containing region, the composition of the matrix, cell type(s), culture configuration, as well as paper thickness and porosity.
With a series of experimental setups, we show that both the paper-based and traditional Transwell assays have similar levels of performance when comparing the trends and reproducibility of cellular invasion in the presence of increasing concentrations of fetal bovine serum (FBS). We also highlight a number of culture configurations that are possible with the paper-based assays as well as the different methods of analysis to quantify both the invasive and non-invasive cell populations.
2. Materials and methods
2.1 Cell lines and culture maintenance
All reagents were used as received unless otherwise noted. All cell culture medium and additives were purchased from Gibco (Life Technologies) except for FBS (VWR).MDA-MB-231 (M231) and MCF7 cell lines were purchased from ATCC. Both cell lines were engineered, to constitutively express the mCherry protein, with LPP-MCHR-Lv105-025 lentiviral particles (Genecopoeia) according to the manufacturer's protocol. Reduction mammoplasty fibroblasts (RMF) were provided by Dr Melissa Troester. Hepatocyte growth factor (HGF)-overexpressing reduction mammary fibroblasts (RMF-HGF) were provided by Dr Bonnie Sloane. Each of these cell lines was described and characterized previously.18,19
M231-mCherry cells were maintained in RPMI 1640 medium supplemented with fetal bovine serum (FBS, 10% v/v), penicillin–streptomycin (1% v/v), and HEPES (25 mM). The MCF7-mCherry, RMF, and RMF-HGF cells were maintained in DMEM supplemented with FBS (10% v/v), penicillin–streptomycin (1% v/v), and HEPES (25 mM). All cells were maintained as monolayers at 37 °C and 5% CO2, culture medium exchanged every 48 h, and cells passed when 70–80% confluent.
2.2 Paper-based scaffolds
Wax-patterned Whatman 105 paper-based scaffolds were prepared as described previously.28 Prior to use, each scaffold was sterilized under ultraviolet light. Two designs were used in this work: (1) a single-zone scaffold, which fits directly into the well of a 96-well plate and (2) a 96-zone scaffold, which matches the dimensions and spacing of a standard well plate. Each zone was 3.0 mm in diameter and surrounded by a border of wax. Detailed schematics and photographs of each scaffold are available in the ESI (Fig. S1–5†).2.3 Paper-based Transwell assays
For each single-zone Transwell assay, a cell-laden scaffold was placed in a well of a clear bottom well plate containing 300 μL of culture medium (Fig. 1) and incubated for 48 h. | ||
| Fig. 1 Representative photographs of a single zone, paper-based Transwell assay format. (a) Seeding a single zone paper scaffold with cells suspended in collagen, using a 2.5 μL pipette. (b) Top-view of single zone scaffolds floating at the air-liquid interface of individual wells of a commercially available 96-well plate. Scale bar 10 mm. | ||
In the monoculture format, scaffolds were seeded with 0.5 μL of collagen I containing 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 serum-starved M231-mCherry cells and added to wells containing medium with 0.0, 0.5, 1.0, or 5.0% v/v of FBS. In the indirect co-culture format, scaffolds were seeded with 0.5 μL of collagen I containing 75000 serum-starved MCF7-mCherry cells and added to wells containing either: (1) conditioned medium taken from confluent fibroblast cultures, or (2) a monolayer of 32000 fibroblasts (RMF or RMF-HGF) in DMEM with 1% FBS. In the direct co-culture format, scaffolds were seeded with 0.5 μL of collagen I containing either a 1:1 or 1:3 ratio of MCF7-mCherry cells to fibroblasts (75000 cells total). Seeded scaffolds were added to wells containing DMEM with 1.0% FBS.
000 serum-starved M231-mCherry cells and added to wells containing medium with 0.0, 0.5, 1.0, or 5.0% v/v of FBS. In the indirect co-culture format, scaffolds were seeded with 0.5 μL of collagen I containing 75000 serum-starved MCF7-mCherry cells and added to wells containing either: (1) conditioned medium taken from confluent fibroblast cultures, or (2) a monolayer of 32000 fibroblasts (RMF or RMF-HGF) in DMEM with 1% FBS. In the direct co-culture format, scaffolds were seeded with 0.5 μL of collagen I containing either a 1:1 or 1:3 ratio of MCF7-mCherry cells to fibroblasts (75000 cells total). Seeded scaffolds were added to wells containing DMEM with 1.0% FBS.
In the mono- and co-culture formats, the mCherry expressing cells that remained in the paper scaffolds as well as at the bottom of the well plates were imaged with a Typhoon 9400 scanner (GE Life Sciences) at a 100 μm resolution, as described previously.15 Images were analyzed with ImageJ software.20 In the monoculture formats, cell number was also measured with the Cell Titer-Glo assay (Promega) according to the manufacturer's protocol. Luminescence values were measured on a SpectraMax M5 plate reader (Molecular Devices).
Representative fluorescence images of the cell-containing scaffolds and well plate are shown in Fig. S6.† Cells in the paper scaffolds and collected in the well plates were also imaged with an Olympus IX-70 fluorescence microscope equipped with a 10× objective. Representative brightfield and fluorescence micrographs of these cells are shown in Fig. S7 and S8.†
2.4 Traditional Transwell invasion assays
Traditional Transwell assays were performed in a Corning HTS Transwell 96-well plate containing polycarbonate membranes with 8 μm-diameter pores. Prior to usage, the membranes were coated with 50 μL of collagen I (1.2 mg mL−1) and allowed to dry overnight. A suspension of 25000 serum-starved M231-mCherry cells in 50 μL of RPMI medium was added to each insert. The corresponding wells contained 150 μL of RPMI medium with 0.0, 0.5, 1.0, or 5.0% v/v of FBS. The assembled assays were incubated for 48 h.
The number of invasive cells on the bottom of the inserts was enumerated with a light microscope. Before counting the cells, each insert was fixed in ice cold 100% methanol for 10 min, dried, and then stained with crystal violet for 10 min.
2.5 Proliferation assays
A suspension of 2500 M231-mCherry cells in RPMI medium containing 0.0, 0.5, 1.0, or 5.0% FBS was added to a 96-well plate. The cells were incubated at 37 °C and 5% CO2, and culture medium (100 μL) exchanged every day. Cell numbers were assessed with the Cell Titer-Glo assay (Promega) according to the manufacturer's protocol. Luminescence values were measured on a SpectraMax M5 plate reader (Molecular Devices).3. Results and discussion
3.1 Paper-based assays have similar trends and reproducibility as traditional Transwell assays
To compare the paper-based and traditional Transwell assays, we quantified the invasion of M231-mCherry cells through a collagen matrix, in the presence of increasing concentrations of FBS. We chose the M231 cells because they are highly invasive, with a mesenchymal phenotype.21,22 We selected collagen over other matrices because it is readily purified, allowing us to probe cellular responses to chemokines in a relatively growth factor-free environment. We selected FBS because it is a common chemokine that is known to promote cellular movement.23In the traditional assay format (Fig. 2a), cells were placed atop a collagen-coated membrane, and the number of cells that crossed the porous membrane counted on the underside of the membrane (Fig. 2b). In the paper-based assay format, we used single-zone scaffolds (Fig. 2c) that fit directly into the well of a standard 96-well plate. Each scaffold was 6.5 mm in diameter and contained a 3.0 mm seeding region surrounded by a wax border. The low-density wax border ensured the scaffold remained at the air–water interface throughout the experiment.
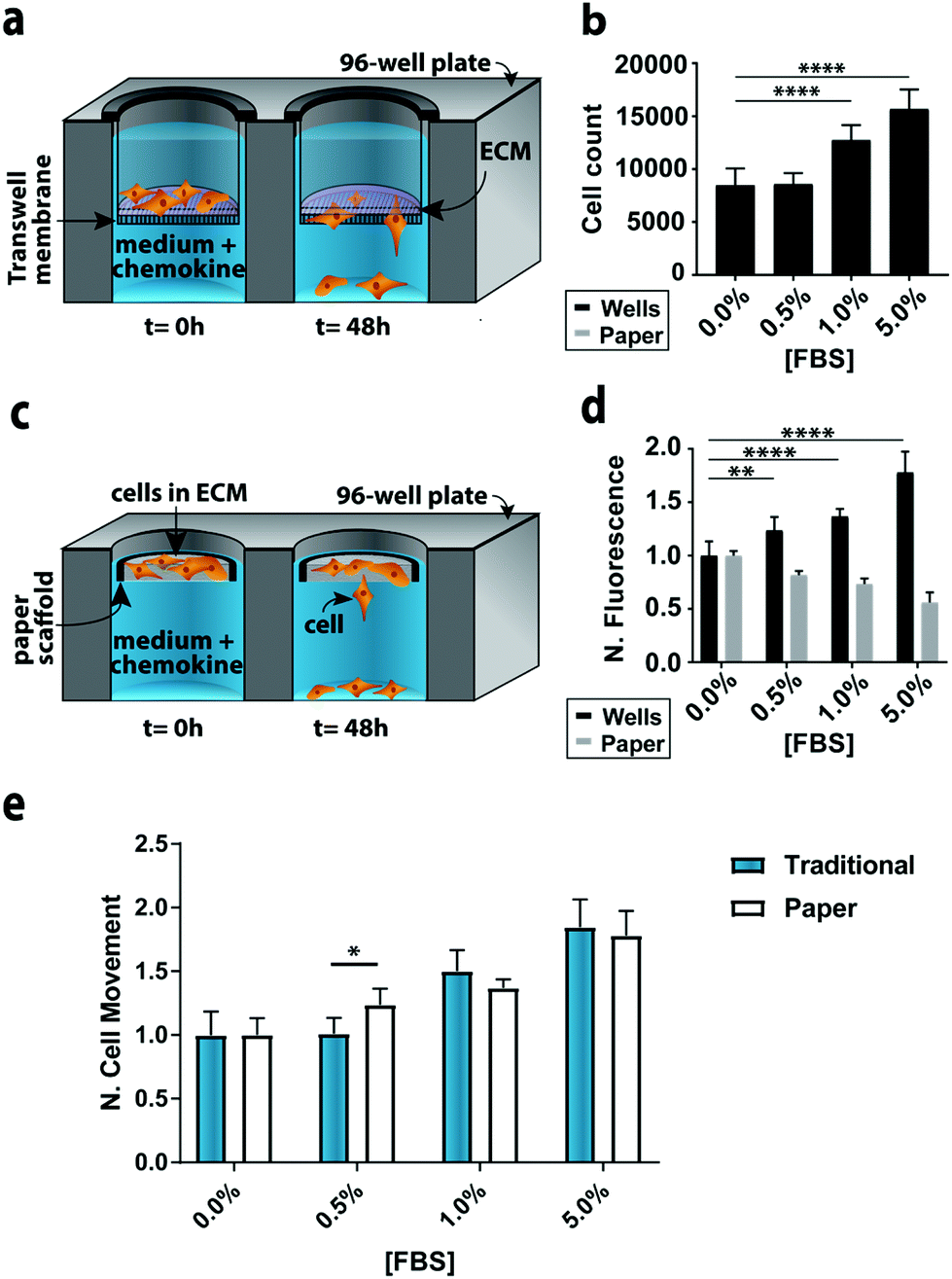 | ||
| Fig. 2 Representative monoculture assay setups. (a) Schematic of a traditional Transwell invasion assay used in this work. (b) M231 cell invasion through collagen-coated inserts after a 48 h exposure to medium containing increasing concentrations of FBS (n = 15). (c) Schematic of the paper-based assay used in this work. (d) M231 cell invasion from collagen-containing paper scaffolds after a 48 h exposure to medium containing increasing concentrations of FBS (n = 5). All signals were normalized to 0.0% FBS. Each bar represents the average and standard deviation. (e) Comparison of M231 invasion in paper-based and traditional Transwell invasion assays (n = 5). * p < 0.05, ** p < 0.01, and **** p < 0.0001. | ||
A suspension of M231-mCherry cells in collagen were seeded directly into the paper scaffolds. Each scaffold was then placed into the well of a clear bottom 96-well plate and incubated for 48 h in culture medium. During the course of the experiment, the highly invasive cells escaped from the scaffold and attached to the bottom of the 96-well plate. At the end of the experiment, the number of invasive cells at the bottom of the plate and in the paper scaffolds was determined with the Cell Titer-Glo assay or from images collected with a fluorescence flatbed scanner (Fig. 2d). We have shown that there is a linear relationship between fluorescence intensity and cell number in the paper scaffolds for cell densities ranging from 3000 to 100000 cells per zone.17
In Fig. 2d, the black bars represent the number of invasive cells collected at the bottom of the well plate; these cells correspond to the number of cells that crossed the porous membrane in the traditional Transwell setup; the gray bars correspond to the number of cells that remain in the scaffold. We did not measure the number of cells that did not cross the porous membrane in the traditional Transwell assay.
In accordance with previous studies,23 the number of invasive cells increased with increasing concentrations of FBS. When normalized to the 0.0% FBS, the paper-based and traditional Transwell assay produced similar results (Fig. 2e). Fold changes between 0 and 5% FBS are statistically indistinguishable: a 1.84 ± 0.21 fold increase for the traditional format and a 1.78 ± 0.17 increase in the paper-based format (p = 0.90). A comparison of the number of cells retained in the paper scaffolds and at the bottom of the well is also in agreement (Fig. 2D).
We also quantified the number of cells at the bottom of the wells and in the paper scaffolds with fluorescence images, but found that the sensitivity of the images (Δ fluorescence intensity/Δ cell number) differed for monolayer and 3D cultures (Fig. S9†). A summary of these findings is available in the ESI.† These results highlight the importance of generating calibration curves when comparing cultures with different dimensionalities.
To confirm that the invasion trends observed in both assays were not confounded by cellular proliferation, we measured the doubling times of monolayers of M231 cells at each of the FBS concentrations tested (Fig. S10†). We found that the number of cells in medium containing: ≤0.5% FBS did not increase significantly over a 5-day period; 1% FBS doubled every 161 h, and; 5% FBS doubled every 30 h. Given these rates of proliferation, we are confident the trends observed after a 48 h incubation are indicative of cellular movement and not due to proliferation.
We also compared the reproducibility of both assay formats to account for the variable size and density of pores present in both Whatman 105 paper scaffolds and the commercially available Transwell inserts.24 In the paper scaffolds, these variations arise from a non-uniform placement of cellulose fibers across a single sheet. Despite the heterogeneity within and across sheets of paper, our previous studies of cellular invasion through stack- or channel-based formats have yielded highly reproducible invasion results.15,17,25 Both formats had similar variances for n = 15 replicates. For 0% FBS, the relative standard deviation (RSD) of invasive cell numbers was 13.4% for the paper-based format and 18.3% for the traditional format. For 5% FBS, the paper-based format had an RSD of 10.4% and the traditional Transwell assay 11.7%.
3.2 Paper-based Transwell assays support direct and indirect co-culture formats
In a second set of experiments we quantified MCF7 invasion in indirect and direct co-culture formats (Fig. 3). Co-cultures can account for different aspects of cell–cell signaling present in vivo. We chose MCF7 cells because they have an epithelial phenotype,22,26 but undergo an epithelial-to-mesenchymal transition (EMT) and adopt an invasive phenotype in the presence of chemokines such as hepatocyte growth factor (HGF) and transforming growth factor beta (TGFβ).27–29 We chose fibroblasts as the second cell type in these experiments because they produce both of these chemokines.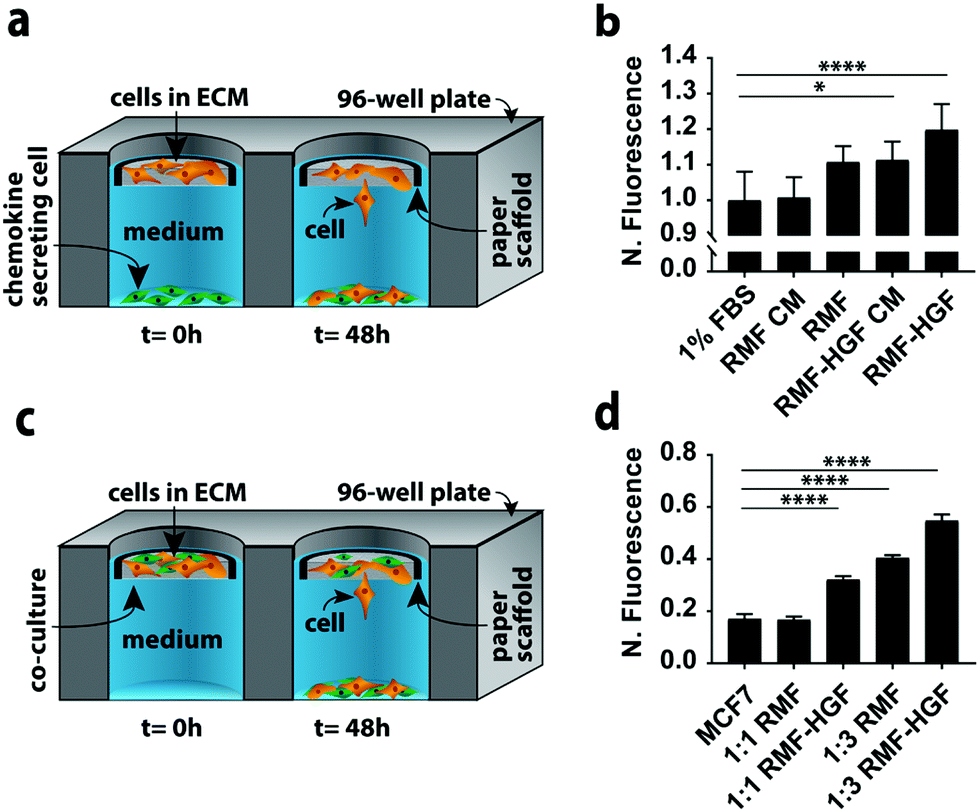 | ||
| Fig. 3 Indirect and direct co-cultures in the paper-based format. (a) Schematic of the indirect co-culture setup used in this work. (b) MCF7 (orange) invasion from collagen-containing paper scaffolds after a 48 h exposure to conditioned medium (CM) or a monolayer of 32000 fibroblasts (green). All signals were normalized to 1% FBS. (c) Schematic of the direct co-culture setup used in this work. (d) MCF7 invasion from collagen-containing paper scaffolds after a 48 incubation in the presence of a 1:1 or 1:3 ratio with either RMF or RMF-HGF cells. All signals were normalized to their starting fluorescence signal. Each bar represents the average and standard deviation of n = 15 replicates. ** p < 0.05, and **** p < 0.0001. | ||
The simplest version of an indirect co-culture exposes cells to conditioned medium, which contains growth factors and other metabolites excreted from cells. In these experiments, MCF7-containing scaffolds were placed in wells containing medium collected from a confluent monolayer of either RMF or RMF-HGF cells. The invasiveness of the MCF7 cells in the presence of the RMF medium was indistinguishable from control experiments with medium containing 1% FBS (Fig. 3b). The RMF-HGF conditioned medium increased MCF7 invasiveness significantly, and supports earlier works that showed HGF is a primary contributor to cellular movement of breast cancer cells.27–29
While conditioned medium assays are easily setup, they do not account for temporal changes in factor secretion that arise from intercellular signaling. Continuous communication is important for molecules such as HGF, which degrade quickly in culture medium.29Fig. 3a is a schematic of the indirect co-culture setup, which was prepared by placing MCF7-laden scaffolds into wells containing monolayers of either 32000 RMF or RMF-HGF cells. The physical separation of the two cell types mimics the paracrine-signaling environment that occurs in a ductal carcinoma in situ, where a basement membrane separates cancerous and stromal cells. Fig. 3b further supports the importance of HGF in promoting invasiveness as only the indirect cultures containing the HGF-RMF cells were statistically different from the control setups, that exposed monocultures of MCF7 cells to medium with 1% FBS.
Fig. 3c is a schematic of the direct co-culture setup in which scaffolds contained different ratios of MCF7 cells and fibroblasts. The localization of both cell types mimics the juxtacrine signaling that occurs in an invasive ductal carcinoma, where the basement membrane is compromised and stromal cells invaded the cancerous mass. Increasing the number of fibroblasts from 1:1 to 1:3 resulted in a statistically significant increase in the number of invasive MCF7 cells (Fig. 3d). Similar to the results observed in the indirect co-cultures, the RMF-HGF cells more readily promoted invasion than the RMF cells. When compared to the MCF7 monoculture, the 1:3 ratio with RMF cells increased invasion by 2.4-fold and the RMF-HGF by 3.2-fold.
The differences in invasion observed in the co-culture setups are likely due to increased concentrations of signaling factors that results from bringing the two cell types into contact. This proximity especially increased cellular response for signaling factors with short half-lives29 because the rate of diffusion to the targeted receptor outpaces the rate of HGF degradation. Others have shown that direct co-cultures result in gene signatures that are more predictive of tumor subtype than indirect co-cultures with similar cell types.28
3.3 A 96-zone, paper-based Transwell assay
To demonstrate the scalable nature of the paper-based format, we constructed a device capable of supporting 96 assays in parallel. Fig. 4 contains a schematic of the device, which is comprised of: (i) two acrylic 96-well inserts containing through-holes that enable delivery of medium to the bottom of the device; (ii) a sheet of Whatman 105 paper patterned with 96 individual zones; (iii) three silicone gaskets to prevent leaking between adjacent wells, and; (iv) an acrylic base plate. Once assembled, the components were compressed with a set of stainless steel screws. Detailed schematics of each component are provided in the ESI.†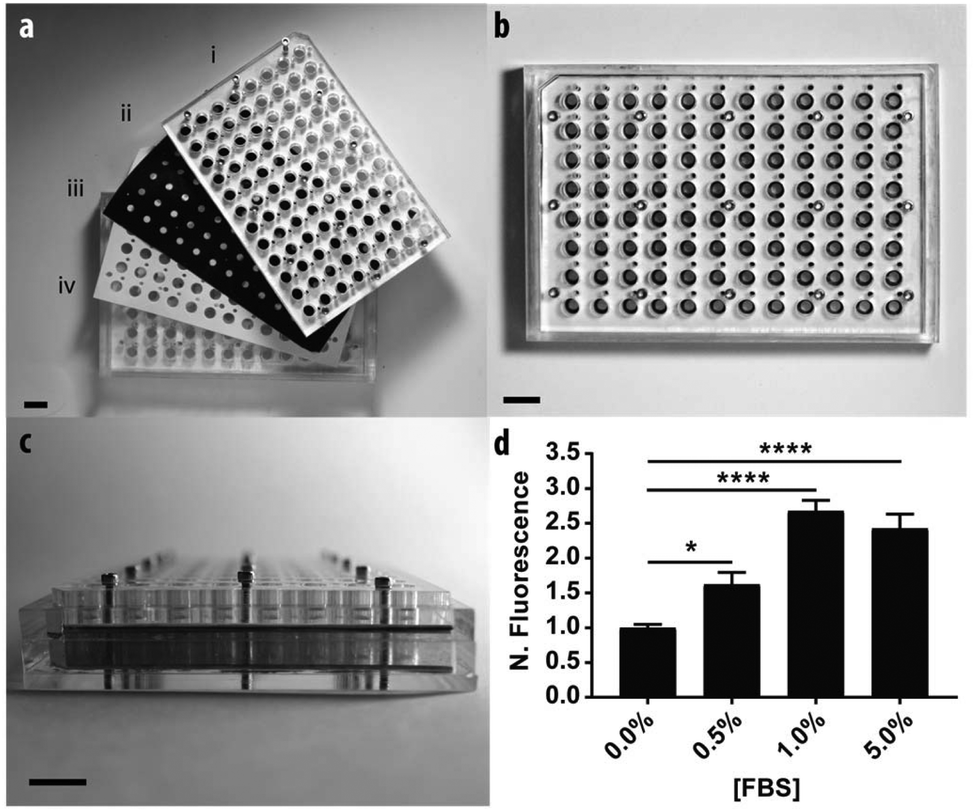 | ||
| Fig. 4 The 96-zone, paper-based Transwell assay format. The individual components of the device pictured in (a) include: (i) an acrylic top insert with a silicone gasket, (ii) a paper-based scaffold, (iii) a silicone gasket, and (iv) an acrylic bottom insert with a silicone gasket and bottom sheet of acrylic. (b) Top-view of the assembled device. (c) Side-view of the assembled device. (d) M231 cell invasion from collagen-containing paper scaffolds after a 48 h exposure to medium containing increasing concentrations of FBS. Each bar represents the average and standard error of the mean of n = 21 zones from a single setup. *p < 0.05, and ****p < 0.0001. | ||
We measured the invasion of n = 21 replicate zones containing M231-mCherry cells for each of the following conditions: 0.0, 0.5, 1.0, or 5.0% FBS. Before device assembly, each of the 96 zones was seeded with cell-laden collagen, this time with a multichannel pipette. After assembly, 300 μL of culture medium was added to the bottom wells through access ports in the acrylic.
Similar to the single zone assays, we observed a statistically significant increase in the numbers of invasive cells with increased concentrations of FBS (Fig. 3b). The fold-increase of invasive cells from the control was substantially higher for each FBS concentration in the 96-zone format when compared to the single zones. One possible cause of this variance is an increased inconsistency in cell density that arose from seeding the scaffolds with a multi-channel pipette. We recently showed that 96-zone paper scaffolds could be loaded consistently with a multichannel pipette with RSD values of 10.6%–21.4% across a single deposition; 8.6%–19.9% across multiple depositions with the same pipette tip, and; 15.1% across an entire plate.30 While reproducible seeding is user dependent and improves with practice, inconsistencies in cell density can be eliminated with the incorporation of liquid handlers capable of accurately and reproducibly seeding each zone.
A second potential cause for this variance is differences in medium composition throughout the incubation period, arising from chemical cross talk between neighboring wells. To assess this possibility, we assembled a pre-soaked 96-zone paper scaffold into the device. The bottom wells were filled with a checkerboard pattern of fluorescein- or fluorescein-free culture medium. The fluorescence intensity of each well was measured immediately after assembly and again after a 48 h incubation at 37 °C (Fig. S11†). The fluorescence intensity of the fluorescein-containing wells decreased by 7% after 48 h but was not statistically significant. There was a slight, but significant, increase in the fluorescence intensity of fluorescein-free wells. Similar cross-talk problems were observed in other multi-zone paper-based devices,31 and will be addressed through gasket optimization and if necessary an alternative design.
4. Conclusions
The paper-based Transwell format offers both advantages and disadvantages over the traditional format. One advantage is that the paper-based approach is a cost-effective and accessible platform. Using the single-zone scaffolds and a commercially available well plate, the material costs for the paper-based format are approximately $0.09 per assay,32 a substantial saving compared to the $1.22 per assay in the traditional format.33 The paper scaffolds also provide experimental modularity, enabling the selection of matrix type, cell type(s), culture configuration, as well as paper thickness and porosity in-house. The 96-well format also has the modularity of the single-zone formats but requires the fabrication of a reusable holder. The holder presented here can be prepared with a laser cutter, although other fabrication methods (e.g., 3D printing) could achieve a similar outcome.Both the single- and 96-zone formats also highlight the ability of the paper-based scaffolds to easily separate two populations of cells that can be re-cultured or analyzed for secondary experiments. Furthermore, single zone scaffolds enable tactile manipulation of 3D cultures for experiments completed in traditional 96-well plates. Our results also support previous experimental findings in migration and invasion assays carried out in traditional Transwell assays. First, epithelial-like breast cancer cells are more invasive when cultured with HGF overexpressing RMF cells.27 Second, direct co-cultures promote higher levels of invasion than indirect co-cultures.29
A perceived disadvantage of the single zone format is that cells are exposed to a single culture condition throughout the experiment as opposed to the gradients formed in traditional Transwell assay format. We note that cellular movement in the traditional assay formats is likely not due to this initial gradient, as they are transient and dissipate over the course of a few hours.34 In both setups, cellular movement is most likely the result of an overall increase in cellular movement and not a directional response (i.e., chemokinetic and not chemotactic in nature).
We envision the paper-based Transwell assay as an alternative format for labs that study the role of intercellular communication in promoting cellular invasion, and note that the cell-laden scaffolds introduce experimental flexibility that the traditional format could not afford.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported with funds provided by Eli Lilly and Company's Young Investigator Award, the National Center for Advancing Translational Science (NCATS) under award number UL1TR001111, and the National Institute of General Medical Sciences (NIGMS) under award number R35GM129697. This work was performed in part at the UNC BeAM Makerspace and the UNC Microscopy Services Laboratory. We would also like to extend our thanks Mr Zhi-Wei Lin for providing helpful feedback and discussions.References
- A. Czirok, B. J. Rongish and C. D. Little, Genes Cancer, 2011, 2, 1072–1080 CrossRef PubMed.
- K. J. Sonnenmann and W. M. Bement, Annu. Rev. Cell Dev. Biol., 2011, 27, 237–263 CrossRef PubMed.
- N. D. Evans, R. O. C. Oreffo, E. Healy, P. J. Thurner and Y. H. Man, J. Mech. Behav. Biomed. Mater., 2013, 28, 397–409 CrossRef PubMed.
- A. W. Lambert, D. R. Pattabiraman and R. A. Weinberg, Cell, 2017, 168, 670–691 CrossRef CAS PubMed.
- K. I. Hulkower and R. L. Herber, Pharmaceutics, 2011, 3, 107–124 CrossRef CAS PubMed.
- N. Kramer, A. Walzl, C. Unger, M. Rosner, G. Krupitza, M. Hengstschlager and H. Dolznig, Mutat. Res., Rev. Mutat. Res., 2012, 752, 10–24 CrossRef PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- C. C. Lin, RSC Adv., 2015, 5, 39844–39853 RSC.
- K. M. Hakkinen, J. S. Harunaga, A. D. Doyle and K. M. Yamada, Tissue Eng., Part A, 2011, 17, 713–724 CrossRef CAS PubMed.
- Prices based on Corning™ Costar™ HTS Transwell™ setups, found on the Fisher Scientific website in October 2018.
- R. M. Kenney, C. C. Lloyd, N. A. Whitman and M. R. Lockett, Chem. Commun., 2017, 53, 7194–7210 RSC.
- K. Ng, B. Gao, K. W. Yong, Y. H. Li, M. Shi, X. Zhao, Z. D. Li, X. H. Zhang, B. Pingguan-Murphy, H. Yang and F. Xu, Mater. Today, 2017, 20, 32–44 CrossRef CAS.
- D. Lantigua, Y. N. Kelly, B. Unal and G. Camci-Unal, Adv. Healthcare Mater., 2017, 6, 1700619 CrossRef PubMed.
- D. Rodenhizer, T. Dean, E. D'Arcangelo and A. P. McGuigan, Adv. Healthcare Mater., 2018, 7, 1701174 CrossRef PubMed.
- C. C. Lloyd, M. W. Boyce and M. R. Lockett, Curr. Protoc. Chem. Biol., 2017, 9, 1–20 Search PubMed.
- B. Mosadegh, M. R. Lockett, K. T. Minn, K. A. Simon, K. Gilbert, S. Hillier, D. Newsome, H. Li, A. B. Hall, D. M. Boucher, B. K. Eustace and G. M. Whitesides, Biomaterials, 2015, 52, 262–271 CrossRef CAS PubMed.
- A. S. Truong, C. A. Lochbaum, M. W. Boyce and M. R. Lockett, Anal. Chem., 2015, 87, 11263–11270 CrossRef CAS PubMed.
- C. Kuperwasser, T. Chavarria, M. Wu, G. Magrane, J. W. Gray, L. Carey, A. Richardson and R. A. Weinberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4966–4971 CrossRef CAS PubMed.
- P. Casbas-Hernandez, J. M. Felming and M. A. Troester, J. Biomed. Biotechnol., 2011, 2011, 520987 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- P. A. Kenny, G. Y. Lee, C. A. Myers, R. M. Neve, J. R. Semeiks, P. T. Spellman, K. Lorenz, E. H. Lee, M. H. Barcellos-Hoff, O. W. Petersen, J. W. Gray and M. J. Bissell, Mol. Oncol., 2007, 1, 84–96 CrossRef CAS PubMed.
- A. Voulgari and A. Pintzas, Biochim. Biophys. Acta, Rev. Cancer, 2009, 1796, 75–90 CrossRef CAS PubMed.
- M. A. Khajah, S. Al Saleh, P. M. Matthew and Y. A. Luqmani, PLoS One, 2012, 7, e41847 CrossRef CAS PubMed.
- M. Y. Kim, D. J. Li, L. K. Pham, B. G. Wong and E. E. Hui, J. Membr. Sci., 2014, 452, 460–469 CrossRef CAS PubMed.
- R. M. Kenney, M. W. Boyce, A. S. Truong, C. R. Bagnell and M. R. Lockett, Analyst, 2016, 141, 661–668 RSC.
- E. Ziegler, M. T. Hansen, M. Haase, G. Emons and C. Grundker, Breast Cancer Res. Treat., 2014, 148, 269–277 CrossRef CAS PubMed.
- C. Jedeszko, B. C. Victor, I. Podgorski and B. F. Sloane, Cancer Res., 2009, 69, 9148–9155 CrossRef CAS PubMed.
- J. T. Camp, F. Elloumi, E. Roman-Perez, J. Rein, D. A. Stewart, J. C. Harrell, C. M. Perou and M. A. Troester, Mol. Cancer Res., 2011, 9, 3–13 CrossRef CAS PubMed.
- P. Casbas-Hernandez, M. D'Arcy, E. Roman-Perez, H. A. Brauer, K. McNaughton, S. M. Miller, R. K. Chhetri, A. L. Oldenburg, J. D. Fleming, K. D. Amos, L. Makowski and M. A. Troester, Breast Cancer Res., 2013, 15, R82 CrossRef PubMed.
- N. A. Whitman, Z. W. Lin, T. J. DiProspero, J. C. McIntosh and M. R. Lockett, Anal. Chem., 2018 DOI:10.1021/acs.analchem.8b02486.
- F. Deiss, A. Mazzeo, E. Hong, D. E. Ingber, R. Derda and G. M. Whitesides, Anal. Chem., 2013, 85, 8085–8094 CrossRef CAS PubMed.
- Cost calculated from prices of each material listed on the Fisher Scientific website on October 2, 2018. A breakdown of this calculation is enumerated in the ESI.†.
- Price per assay based on Fisher Scientific price fo Corning™ Costar™ HTS Transwell™ -96 Permeable Support with Polyester Membrane on October 2, 2018: $235.11/two 96-well plates.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Photographs and detailed schematics of the paper-based scaffolds and 96-zone culture assembly; proliferation datasets for increasing concentrations of FBS; results from the instrument sensitivity, and; results from the chemical cross talk experiments in the 96-zone culture assembly. See DOI: 10.1039/c8an01157e |
| ‡ Indicates shared-authorship. |
| This journal is © The Royal Society of Chemistry 2019 |