
A hybridization-triggered DNAzyme cascade assay for enzyme-free amplified fluorescence detection of nucleic acids†
Huizhen
Wang
,
Dinggeng
He
*,
Ri
Wu
,
Hong
Cheng
,
Wenjie
Ma
,
Jin
Huang
,
Hongchang
Bu
,
Xiaoxiao
He
* and
Kemin
Wang
 *
*
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, College of Chemistry and Chemical Engineering, Hunan University, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Changsha 410082, China. E-mail: hedinggeng@hnu.edu.cn; xiaoxiaohe@hnu.edu.cn; kmwang@hnu.edu.cn
First published on 27th October 2018
Abstract
An enzyme-free and ultrasensitive fluorescence assay for the detection of nucleic acids was successfully established by a hybridization-triggered DNAzyme cascade (HTDC). This simple and cost-effective sensor has good sensitivity, selectivity and the capability for detection of target DNA from complex fluids.
The presence and concentrations of nucleic acids inside the human body have been proven to be associated with the occurrence and development of certain diseases.1 For example, variations in nucleic acid sequences are found to be the sources of several genetic diseases,2,3 which have been identified by genome-wide linkage analysis and positional cloning.4 Due to the low levels of target nucleic acids in the human body, signal amplification is necessary for achieving their sensitive detection. Most of the traditional amplification methods are based on enzyme amplification technology, including ligation chain reaction (LCR),5 polymerase chain reaction (PCR),6 rolling circle amplification (RCA)7 and ligation-mediated strand displacement.8 Although protein enzymes, as biocatalysts, play an important role in these reported signal amplification assays, the use of enzymes also introduces some issues, such as high cost, not easy to store, irreversible denaturation under harsh conditions (e.g., temperature, strong acid and strong alkali, and heavy metal ions), and a complicated operation process, which greatly restrict their wide applications in biological studies and medical diagnostics. To overcome these above-mentioned issues, enzyme-free signal amplification strategies have been employed for highly sensitive analysis of nucleic acids. These enzyme-free strategies involve hybridization chain reaction (HCR),9,10 target-catalyzed hairpin assembly (CHA),11,12 entropy-driven reaction,13etc. Despite the fact that these reports have featured the highly sensitive detection of DNA by enzyme-free amplification strategies, there is still a lack of nonenzymatic signal amplification techniques with high efficiency and simple sequence design.
Catalytic nucleic acids (DNAzymes or ribozymes)14,15 have gained growing interest as amplifying labels for DNA sensing events. The flexibility to encode in the base sequences of DNA functional and structural information, together with the reduced nonspecific binding properties of nucleic acids, provides unique features for bioanalytical applications.16–18 DNAzymes are single-stranded DNA that can catalyze the cleavage of specific substrate strands with a RNA base.19 Owing to their good stability, high catalytic efficiency and easy synthesis,20 DNAzymes have recently shown wide application in constructing biomimetic DNA nanodevices,21,22 gene silencing and biosensing,23–26 especially in living cells.27–29 Most of the developed DNAzyme sensors for nucleic acids are based on Mg2+-dependent DNAzymes, because they can be designed as split DNAzymes with an inactive configuration that can be activated by the target DNA sequence to catalyze the cleavage of ribonucleobase (rA)-containing substrates. Unfortunately, the Mg2+-dependent DNAzymes show a low catalytic activity with a maximum catalytic rate of 0.08 min−1.30 Previous reports have demonstrated that Zn2+-dependent DNAzymes (8–17) have high catalytic cleavage activity (a catalytic cleavage rate of 1.35 min−1), which is 16 times as much as that of the Mg2+-dependent DNAzymes.31–34 However, only a little attention has been paid to the designing of biosensors based on an 8–17 DNAzyme.
Herein, we describe a hybridization-triggered DNAzyme cascade (HTDC) assay using the 8–17 DNAzyme with high catalytic activity, which can realize the enzyme-free amplified fluorescence detection of target DNA. The detection mechanism of the HTDC assay integrates the 8–17 DNAzyme activity and a hybridization-initiated conformational change of DNA (Scheme 1). Oligonucleotide sequences used here are shown in Fig. S1 and Table S1, ESI.† The assay consists of an 8–17 DNAzyme and a substrate: the DNAzyme (D) is a DNA molecule that can cleave a ribonucleobase (rA)-containing substrate (S) with sequence specificity. The 8–17 DNAzyme contains a catalytic core, two binding arms and a recognition sequence. The two binding arms that can bind to a substrate via Watson–Crick base-pairing are asymmetrical by design: one arm is 9 bases long and the other is 3 bases long. The short binding arm (3 bases) is not able to stably bind to the substrate in the absence of target DNA, thus showing an inactive substrate–DNAzyme (S–D) complex. Target DNA can simultaneously hybridize with the recognition sequences of the DNAzyme and substrate, resulting in a stable binding of the short arm and substrate and an activated DNAzyme structure (Scheme 1). The target-activated DNAzyme can cleave the FAM-labeled substrate strand and cause the recovery of the fluorescence. When the RNA substrate was cleaved, the hybridization of the target DNA and the long arm containing 9 bases remained stable under the experimental conditions, which can maintain the high catalytic activity of the DNAzyme and act as scissors resulting in the cleavage of the FAM-labeled substrate strand one after another. Therefore, an amplified fluorescent signal was achieved during the cyclic process, and a trace amount of the target DNA can initiate the cleavage of a large number of substrate strands.
 | ||
| Scheme 1 Schematic representation of the hybridization-triggered DNAzyme cascade (HTDC) assay for enzyme-free amplified fluorescence detection of nucleic acids. | ||
We evaluated the feasibility of the HTDC assay in buffer (50 mM Tris-HCl, 100 mM NaCl, pH 7.4). As shown in Fig. 1A, a remarkable fluorescence enhancement (curve d) was observed in the presence of target DNA, which was nearly 6 times greater than that in the absence of target DNA (curve c), indicating that the cleaved product containing FAM has been released from the rupture substrate strands containing a BHQ-1 quencher. We also made a mutant DNAzyme (one mismatch in the catalytic core) (curve e) and selected a random DNA (curve f), and both of them present a very low fluorescence intensity. Gel electrophoresis was also performed to demonstrate the occurrence of HTDC (Fig. S2, ESI†). A brighter band (lane 4) was formed in the absence of the target DNA, and an assembly (the top band in lane 5) with significantly slow migration between the substrate strand (lane 1) and DNAzyme (lane 2) can be observed in the presence of target DNA (lane 3) (Fig. S2A, ESI†). 15% denaturing polyacrylamide gel (Fig. S2B, ESI†) was also used to validate the cleavage of the substrate strands. A larger extent of cleavage can be observed in the presence of target DNA (lane 2), while no cleavage was observed in the absence of target DNA (lane 1). Based on the above results, we can conclude that the target DNA acted as an allosteric effector that triggered a conformation switching from the inactive DNAzyme to an active DNAzyme structure in a Zn2+-dependent fashion and exhibited the specific cleavage ability.
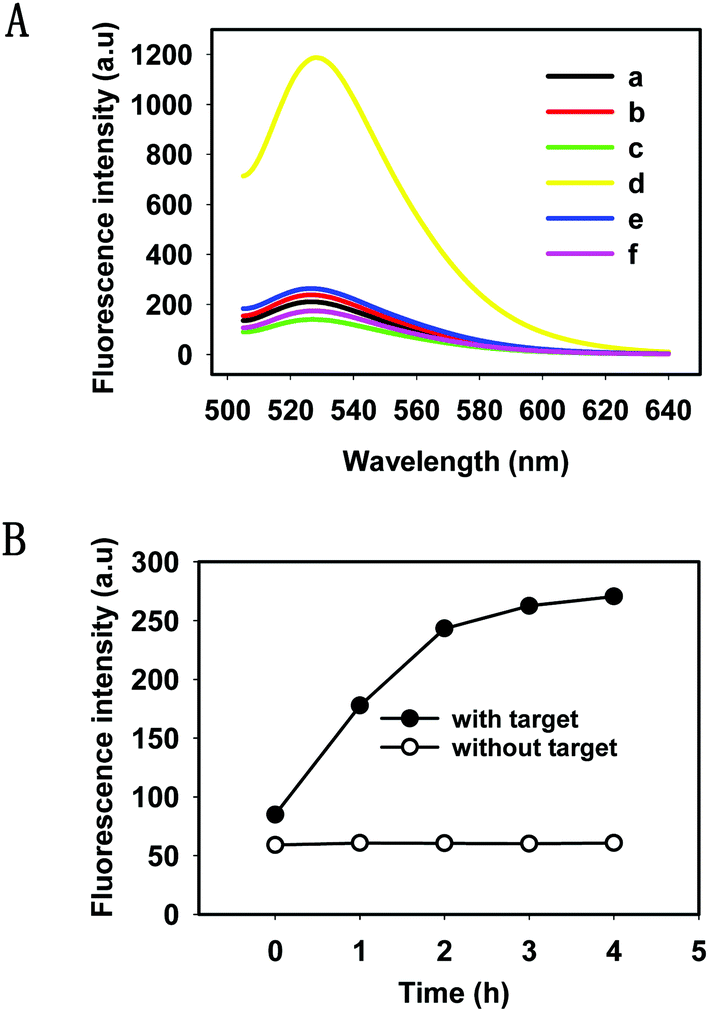 | ||
| Fig. 1 (A) Fluorescence spectra of the feasibility of the HTDC assay in Tris-HCl buffer (100 mM NaCl, pH 7.4) at 37 °C (a) only the substrate strand, (b) the S–D complex, (c) the S–D complex with cofactor Zn2+, (d) the S–D complex in the presence of target DNA (100 nM) with cofactor Zn2+, (e) the mutant S–D complex in the presence of target DNA (100 nM) with cofactor Zn2+, (f) the S–D complex in the presence of random DNA (100 nM) with cofactor Zn2+. (B) Fluorescence intensities of the HTDC assay without and with target DNA (10 nM) over different time points. The concentrations of the substrate and DNAzyme were fixed at 200 nM and 100 nM, respectively. | ||
To gain a better understanding of the detection method, a kinetic study of the HTDC assay for amplified detection of nucleic acids was characterized by measuring the fluorescence intensities over different time points (Fig. 1B). The fluorescence intensities increased with the increase of reaction time, and the fluorescence signal reached a plateau in 4 h. Since DNA hybridization and DNAzyme catalytic efficiency were influenced by the buffer solution, we measured the fluorescence intensities of the HTDC assay in the presence of target DNA in different buffer solutions including PBS, Tris-HCl, and HEPES (Fig. S3, ESI†). A better signal-to-background ratio (SBR) was obtained in Tris-HCl buffer than that in PBS and HEPES (SBR = F/F0, where F is the fluorescence emission intensity of FAM at 520 nm in the presence of target DNA, and F0 is the background fluorescence signal). Hence, Tris-HCl was used as an ideal buffer for further detection assay.
To investigate the multiple turnover capability of the 8–17 DNAzyme and the signal amplification effect, we keep the concentration of the 8–17 DNAzyme at 100 nM and then recorded the SBR after the addition of the substrate strands. As depicted in Fig. 2A, by changing the ratio of the substrate strand to DNAzyme, an increasing SBR can be reached until it was 2.
 | ||
| Fig. 2 (A) Optimization of the concentration ratio of the substrate to DNAzyme in Tris-HCl buffer (100 mM NaCl, 500 μM Zn2+, pH 7.4) at 37 °C. The concentrations of the DNAzyme and target were fixed at 100 nM and 50 nM, respectively. (B) Optimization of Zn2+ concentrations in Tris-HCl buffer (100 mM NaCl, pH 7.4) at 37 °C. The concentrations of the substrate, DNAzyme and target were fixed at 200 nM, 100 nM and 50 nM, respectively. Error bars represent the standard deviations from three independent experiments. | ||
DNAzyme catalytic cleavage efficiency was greatly affected by bivalent metal ions. To achieve the best sensing performance, it is necessary to study the influence of bivalent metal ions. According to the previous report,35 the cleavage reaction of the DNAzyme required a high concentration of Zn2+ or Pb2+. In view of the toxicity of Pb2+, we chose Zn2+ as a cofactor of the 8–17 DNAzyme. Fig. 2B shows that the SBR increased with an increasing concentration of Zn2+ and reached a plateau at 300 μM. When the concentration exceeded 300 μM, the catalytic cleavage efficiency of the DNAzyme was obviously decreased. As a result, we selected 300 μM Zn2+ for the subsequent experiments.
Under the optimal conditions, the kinetics of the HTDC assay with different concentrations of target DNA were tested by monitoring the fluorescence intensities at different time points (Fig. S4, ESI†). The results showed that the reaction rate of fluorescence enhancement was dependent on the target concentrations. Then, the sensitivity of the target DNA detection was evaluated. The fluorescence intensities with different concentrations of target DNA and the detectable minimum concentration were shown in Fig. 3A. There was a gradual increase in the fluorescence intensities by varying the concentrations of target DNA from 1 pM to 70 nM, which was nearly a 6-fold enhancement. Fig. 3B showed the relationship between the intensity change and the concentrations of the target DNA. A good linear relationship (R2 = 0.991) was obtained in the range from 1 pM to 250 pM for target DNA (see the inset in Fig. 3B). When the minimum concentration of target DNA was 1 pM, there was still a distinguishable change from the background signal. Therefore, 1 pM was defined as the detectable minimum target concentration. The detection capability of our strategy was comparable to or even better than some enzyme-based methods for DNA amplified detection.36–38
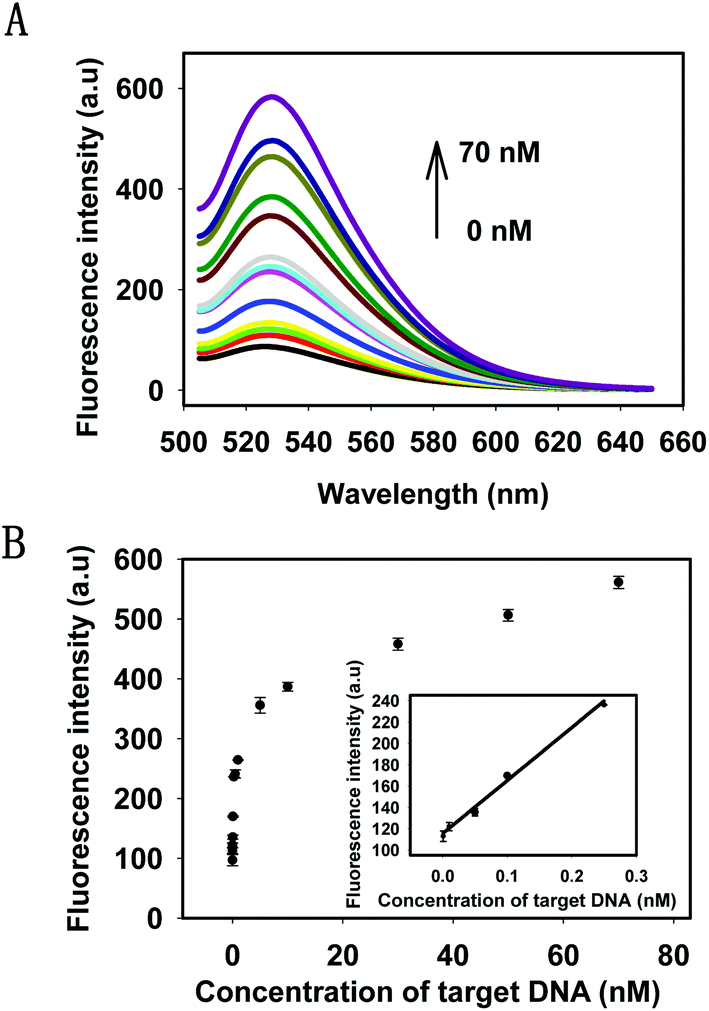 | ||
| Fig. 3 (A) Fluorescence spectra of the assay with different concentrations of target DNA (from bottom to top: 0 pM, 1 pM, 10 pM, 50 pM, 100 pM, 250 pM, 500 pM, 1 nM, 5 nM, 10 nM, 50 nM, 70 nM). (B) The relationship between the fluorescence intensities and the target DNA concentrations. The inset shows the linear range from 1 pM to 250 pM. Error bars represent the standard deviations from three independent experiments. | ||
A series of control experiments, including one base mismatch target DNA, two bases mismatch target DNA, three bases mismatch target DNA and random DNA, were performed to evaluate the selectivity of the proposed assay for target DNA detection (Fig. 4). The fluorescence intensity in the presence of target DNA was obviously stronger than that of control experiments, which indicated that the developed strategy has a high ability to detect perfectly matched target DNA from mismatched target DNA. Furthermore, to evaluate the selectivity of the method for the detection of various one-base mismatch targets in detail, we chose six main one base mismatch positions: two were on the different positions of the DNAzyme arm binding with target DNA (mis D-arm 1 and mis D-arm 2), two were on the different joints of the substrate and DNAzyme (mis SD-joint 1 and mis SD-joint 2) and two were on the different positions of the target binding with the DNAzyme arm (mis T1 and mis T2). Negligible changes in fluorescence intensities were observed with different one-base mismatch target DNAs (Fig. S5, ESI†), which indicated that the proposed method also had the potential to distinguish single nucleotide polymorphism (SNP).
 | ||
| Fig. 4 The SBR for the detection assay in the presence of perfectly matched target DNA (100 nM), one (100 nM), two (100 nM), and three (100 nM) mismatched target DNA and random DNA (100 nM) in Tris-HCl buffer (100 mM NaCl, 300 μM Zn2+, pH 7.4) at 37 °C. The concentrations of the substrate and DNAzyme were fixed at 200 nM and 100 nM, respectively. Error bars represent the standard deviations from three independent experiments. | ||
There are numerous interference factors in complex fluids, which affect the hybridization efficiency of DNA and the catalytic efficiency of the DNAzyme, making the target DNA detection difficult. In order to evaluate the practical application capability of the proposed assay, we chose the RPMI 1640 cell medium containing 15% fetal bovine serum as a model complex fluid. Different amounts of target DNA were spiked into complex fluids, and the results showed satisfactory recovery and acceptable standard deviations (see in Table 1), which indicated that the assay provided the reliability and practicality of target detection in complex fluids. Furthermore, the HTDC assay can also be a versatile platform for sensitive detection of various target DNAs (Fig. S6, ESI†).
| Added (pM) | Detection (pM) | Recovery (%) | SD (%), n = 3 |
|---|---|---|---|
| 50 | 46.59 | 93.19 | 4.53 |
| 100 | 100.5 | 100.5 | 2.26 |
| 250 | 241.24 | 96.49 | 1.20 |
In summary, we have developed a HTDC assay for enzyme-free amplified fluorescence detection of certain nucleic acids. The assay we designed has a good sensitivity, selectivity and application in complex fluids. Moreover, it is a simple and cost-effective amplification method only through hybridization reaction, without the need for additional enzymes, intricate instrumentations, and expensive modifications. The ubiquitous disadvantage of a DNAzyme-based method is, however, the long reaction time of the analytical process. To decrease the reaction time, generally, we can use the following three main ways: (1) appropriate length design of substrate binding arms, (2) chemical modification of substrate binding arms (e.g., 2O-methyl RNA or LNA modified DNAzymes) and (3) optimization of the reaction conditions (e.g., pH, temperature and ionic concentration). We will use these ways to decrease the reaction time of the DNAzyme-based method in the subsequent experiments. As a novel HTDC assay, we infer that the proposed assay may lead to useful development of fundamental research and clinical diagnostics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Key Project of the Natural Science Foundation of China (21775036, 21675046, 21735002, 21521063 and 21874035), the Hunan Provincial Natural Science Foundation (2018JJ2033) and the Key Point Research and Invention Program of Hunan Province (2017DK2011).Notes and references
- J. W. Liu, Z. H. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- K. Irizarry, V. Kustanovich, C. Li, N. Brown, S. Nelson, W. Wong and C. J. Lee, Nat. Genet., 2000, 26, 233–236 CrossRef CAS PubMed.
- R. Sachidanandam, D. Weissman, S. C. Schmidt, J. M. Kakol, L. D. Stein and G. Marth, Nature, 2001, 409, 928–933 CrossRef CAS PubMed.
- F. S. Collins, Clin. Res., 1991, 39, 615–623 CAS.
- W. Shen, H. M. Deng and Z. Q. Gao, J. Am. Chem. Soc., 2012, 134, 14678–14681 CrossRef CAS PubMed.
- I. A. Nanayakkara, W. D. Cao and I. M. White, Anal. Chem., 2017, 89, 3773–3779 CrossRef CAS PubMed.
- A. P. Cao and C. Y. Zhang, Anal. Chem., 2012, 84, 6199–6205 CrossRef CAS PubMed.
- H. Q. Wang, W. Y. Liu, Z. Wu, L. J. Tang, X. M. Xu and R. Q. Yu, Anal. Chem., 2011, 83, 1883–1889 CrossRef CAS PubMed.
- Y. Chen, J. Xu, J. Su, Y. Xiang, R. Yuan and Y. Q. Chai, Anal. Chem., 2012, 84, 7750–7755 CrossRef CAS PubMed.
- D. Chen, H. B. Feng and J. H. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- E. G. Ju, X. J. Yang, Y. H. Lin, F. Pu, J. S. Ren and X. G. Qu, Chem. Commun., 2012, 48, 11662–11664 RSC.
- C. P. Ma, W. S. Wang, Z. X. Li, L. J. Cao and Q. Y. Wang, Anal. Chem., 2012, 429, 99–102 CAS.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- F. A. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295–299 CrossRef CAS PubMed.
- F. A. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, K. Quan, H. Wang, L. Ying, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2016, 88, 5981–5887 CrossRef CAS PubMed.
- Y. J. Yang, J. Huang, X. H. Yang, X. X. He, K. Quan, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2017, 89, 5850–5856 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, X. W. Cao, J. Wei, N. Wu, J. Li, L. H. Wang, C. H. Fan and Y. X. Zhao, ACS Nano, 2017, 11, 11908–11914 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS PubMed.
- X. H. Zhao, L. Gong, X. B. Zhang, B. Yang, T. Fu, R. Hu, W. H. Tan and R. Q. Yu, Anal. Chem., 2013, 85, 3614–3620 CrossRef CAS PubMed.
- Y. Chen, M. S. Wang and C. D. Mao, Angew. Chem., Int. Ed., 2010, 43, 3554–3557 CrossRef PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. J. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS PubMed.
- H. H. Fan, Z. L. Zhao, G. B. Yan, X. B. Zhang, C. Yang, H. M. Meng, Z. Chen, H. Liu and W. H. Tan, Angew. Chem., Int. Ed., 2015, 54, 4801–4805 CrossRef CAS PubMed.
- J. W. Liu and Y. Lu, Anal. Chem., 2004, 6, 1627 CrossRef PubMed.
- Y. Qian, C. Y. Wang and F. L. Gao, Anal. Chim. Acta, 2014, 845, 1–6 CAS.
- Y. Qian, C. Y. Wang and F. L. Gao, Biosens. Bioelectron., 2015, 63, 425–431 CrossRef CAS PubMed.
- L. Lu, J. Feng, Y. Y. Fan and B. Tang, Anal. Chem., 2015, 87, 4829–4835 CrossRef PubMed.
- P. H. Zhang, Z. M. He, C. Wang, J. N. Chen, J. J. Zhao, X. N. Zhu, C. Z. Li, Q. H. Min and J. J. Zhu, ACS Nano, 2015, 9, 789–798 CrossRef CAS PubMed.
- F. L. Gao, J. Wu, Y. Yao, Y. Zhang, X. J. Liao, D. Q. Geng and D. Q. Tang, RSC Adv., 2018, 8, 28161–28171 RSC.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS PubMed.
- A. K. Brown, J. Li, C. M. B. Pavot and Y. Lu, Biochemistry, 2003, 42, 7152–7161 CrossRef CAS PubMed.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS.
- D. C. D. Faulhammer and P. D. M. Famulok, Angew. Chem., Int. Ed., 2010, 35, 2837–2841 CrossRef.
- J. Li, W. C. Zheng, A. H. Kwon and Y. Lu, Nucleic Acids Res., 2000, 28, 481–488 CrossRef CAS PubMed.
- J. W. Liu and Y. Lu, Anal. Chem., 2004, 76, 1627–1632 CrossRef CAS PubMed.
- H. Chen, J. Q. Wang, G. H. Liang, P. Zhang and J. L. Kong, Chem. Commun., 2012, 48, 269–271 RSC.
- V. Pavlov, Y. Xiao, R. Gill, A. Dishon, M. Kotler and I. Willner, Anal. Chem., 2004, 76, 2152–2156 CrossRef CAS PubMed.
- Z. X. Zhang, E. Sharon, R. Freeman, X. Q. Liu and I. Willner, Anal. Chem., 2012, 84, 4789–4797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary results. See DOI: 10.1039/c8an01796d |
| This journal is © The Royal Society of Chemistry 2019 |