
Advances in crosslinking strategies of biomedical hydrogels
Weikang
Hu†
ab,
Zijian
Wang†
cd,
Yu
Xiao
cd,
Shengmin
Zhang
a and
Jianglin
Wang
 *ab
*ab
aAdvanced Biomaterials and Tissue Engineering Center, Huazhong University of Science and Technology, Wuhan 430074, P. R. China. E-mail: jwang520@hust.edu.cn
bWuhan Institute of Biotechnology, Wuhan 430075, P. R. China
cDepartment of Urology, Zhongnan Hospital of Wuhan University, Wuhan 430071, China
dDepartment of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan 430071, China
First published on 26th December 2018
Abstract
Biomedical hydrogels as sole repair matrices or combined with pre-seeded cells and bioactive growth factors are extensively applied in tissue engineering and regenerative medicine. Hydrogels normally provide three dimensional structures for cell adhesion and proliferation or the controlled release of the loading of drugs or proteins. Various physiochemical properties of hydrogels endow them with distinct applications. In this review, we present the commonly used crosslinking method for hydrogel synthesis involving physical and chemical crosslinks and summarize their current progress and future perspectives.
Introduction
Extracellular matrix (ECM) plays a pivotal role in manipulating multiple cellular fates and largely influences the repair and regeneration of injured organs and tissues.1,2 Up to now, the structures, components and properties of natural ECMs have been clearly illustrated, and most of natural ECMs exhibit porous network matrices that normally consist of some nanofiber materials like collagen, fibronectin and laminin.3 Like natural ECMs, hydrogels as artificial ECMs with 3D network structures are fabricated to provide microenvironments4 for cell adhesion, proliferation and migration, and promote the exchange of nutrients and signalling molecules.5 Hydrogels are hydrophilic polymers with high affinities towards water that are prevented from dissolution, owing to their chemical or physical crosslinking networks.6 Mechanical and biochemical properties of hydrogels are highly associated with their crosslinking methods, and even the hydrogels with the same constituents but different crosslinking structures can present various functions.7Hydrogels are mainly fabricated by the crosslinking formation of the stable polymeric networks. Among various crosslinking methods, physical and chemical crosslinks are two basic strategies.8–10 Physical crosslinks include ionic/electrostatic interaction, hydrogen bonding, crystallization/stereo-complex and hydrophobic interactions of thermal induction based on LCST (Lower Critical Solution Temperature)11–13/UCST (Upper Critical Solution Temperature)14,15 and ultrasonication16–18 mediated sol-to-gel phase transition. Chemical crosslinks comprise photo-polymerization,19,20 enzyme-induced crosslink,21–24 and “click” chemistry25–27 including Michael type-addition,28–30 Diels–Alder “click” reaction,31–33 oxime formation,34–38 and Schiff base formation.39–41 In this review, we summarize current physical and chemical crosslinking strategies of biomedical hydrogels (Fig. 1). These crosslinking methods may inspire us to design and fabricate novel hydrogels with superior structures and desirable properties.
 | ||
| Fig. 1 Crosslinking strategies for hydrogel construction. In this diagram, the most widely used crosslinking methods are presented, including physical crosslinking, such as ionic crosslink, LCST/UCST induced hydrogel formation, crystallization induced hydrogel formation and chemical crosslinking, such as UV-induced crosslink and Enzyme catalyzed crosslink. | ||
Physically crosslinked hydrogels
Physically crosslinked hydrogels are usually created by intermolecular reversible interactions,42 such as ionic/electrostatic interaction,43,44 hydrogen bonds, polymerized entanglements, hydrophobic/hydrophilic interactions, crystallization/stereo-complex formation, metal coordination and π–π stacking. The prominent advantage of a physical crosslink is biomedical safety owing to the absence of chemical crosslinking agents, thus, avoiding potential cytotoxicity from unreacted chemical crosslinkers.45 More importantly, physically crosslinked hydrogels are stimuli-responsible with self-healing and injectable properties under room temperature. These hydrogels can be designed as bioactive hydrogels for the encapsulation of living cells and drug delivery of therapeutic molecules.46Hydrogel crosslinking by ionic/electrostatic interactions
The ionic/electrostatic interaction that has been extensively applied to the construction of hydrogels is the basis of a routine physical crosslink with 2 molecules of opposite electric charges. For example, alginate, a naturally derived polysaccharide with mannuronic and glucuronic acid residues, can be crosslinked by divalent cations, such as calcium (Ca2+), barium (Ba2+) and magnesium (Mg2+).47 Divalent cations can solely bind to guluronate blocks from the alginate chains with a high degree of coordination of the divalent ions. The guluronate blocks of one polymer then form junctions with the guluronate blocks of the adjacent polymer chains, resulting in a gel structure. So far, alginate hydrogels have been explored in wound healing, drug delivery, and tissue engineering.48,49Electrostatic interactions occur between the opposite charged macromolecules and those macromolecules interact with each other to yield polyelectrolyte complexes.50 Chitosan is a natural polycationic biopolymer that consists of β-[1-4]-linked 2-acetamido-2-deoxy-D-glucopyranose and 2-amino-2-deoxy-dglucopyranose. Thus, chitosan easily forms polyelectrolyte complexes (PECs) via electrostatic interactions between its cationic amino groups and anionic groups from various anionic polyelectrolytes in nature, such as pectin, chondroitin sulfate and alginate.45,51 Also, chitosan can similarly interact with some synthetic polymers, like polylactic acid, polyacrylic acid and polyphosphoric acid.52 The hydrogels generated from those polyelectrolyte complexes can be modulated with a number of factors, including the charge density of the polymers, the mixed ratio and the amount of each polymer, as well as the soluble microenvironment of polymer. If the net charge of the formed complex is zero, it will influence the solubility and the complex will precipitate.53 Ye et al.54 prepared a fibrils-reinforced polysaccharide composite hydrogel that was formable in situ (Fig. 2). A maleilated chitosan (CS-MA) and a thiol derivatized sodium alginate (SA-SH) were separately synthesized, and the hydrogel was subsequently self-crosslinked via Michael addition and ionic interaction between CS-MA and SA-SH. The resultant hydrogels exhibited dual healing ability with good cytocompatibility as well as excellent mechanical properties.
 | ||
| Fig. 2 Schematic illustration of the fibrils-reinforced polysaccharide-based composite hydrogels. The hydrogels were generated by self-crosslinking of CS-MA and SA-SH. The ionic interaction between CS-MA and SA-SH was able to improve mechanical properties of hydrogels. Reproduced with permission from Compos. Sci. Technol., 2018, 156, 238–246. Copyright 2018 Elsevier.54 | ||
The specific advantage of an ionic/electrostatic interaction is its self-healing ability as a result of which the physical network of hydrogels can be broken at high stress and reform once the stress is removed. However, the mechanical strength of hydrogel is extremely limited owing to the crosslinking strategy of the ionic/electrostatic interaction.
Hydrogel crosslinking by hydrophobic interactions
Hydrophobic interactions for hydrogel crosslinking exist in water-soluble polymers with hydrophobic end groups, side chains or monomers. Two methods create hydrophobic interaction. One is thermal induction based on LCST (Lower Critical Solution Temperature) or UCST (Upper Critical Solution Temperature), the other is the ultrasonic treatment. Generally, hydrogels can be produced using both methods by promoting sol-to-gel transition under certain conditions.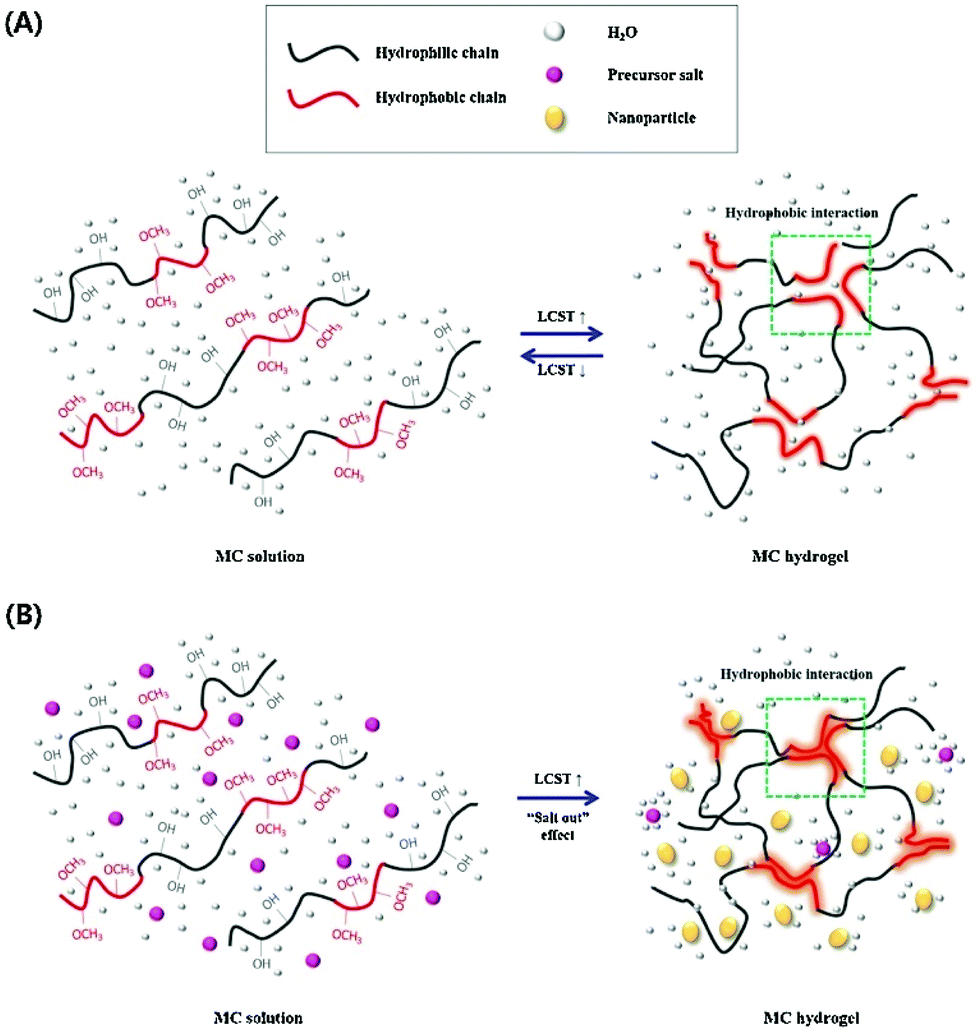 | ||
| Fig. 3 The sol-to-gel transition process of MC, (A) pure MC solution and (B) MC solution with salt. As the temperature increases, hydrogen bonds are broken, which results in hydrogel formation. Addition of salts to the MC solution destroys hydrogen bonds and reduces the sol–gel transition temperature and time. Reproduced with permission from Carbohydr. Polym., 2017, 15, 775–783. Copyright 2017 Elsevier.58 | ||
In contrast, the UCST (upper critical solution temperature) induced hydrogel is prepared during the cooling of a polymer solution, and the cooling temperature is termed as UCST.59 Hydrogels form by the micelle aggregation below UCST and disintegrate when the temperature restores UCST because the hydrophobic micelle cores become water-soluble.60 Fu et al.61 prepared a thermo-reversible physically crosslinked hydrogel from UCST-type thermosensitive ABA linear triblock copolymers, which were composed of hydrophilic poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMMA) middle block and UCST-type thermosensitive poly(acrylamide-co-acrylonitrile) (P(AAm-co-AN)) outer block. Those triblock copolymers exhibited cooling-induced, reversible sol–gel transitions at concentrations of 3% and 5%. The transition temperature of sol–gel elevated along with the increased content of P(AAm-co-AN) segment as well as polymer concentration.
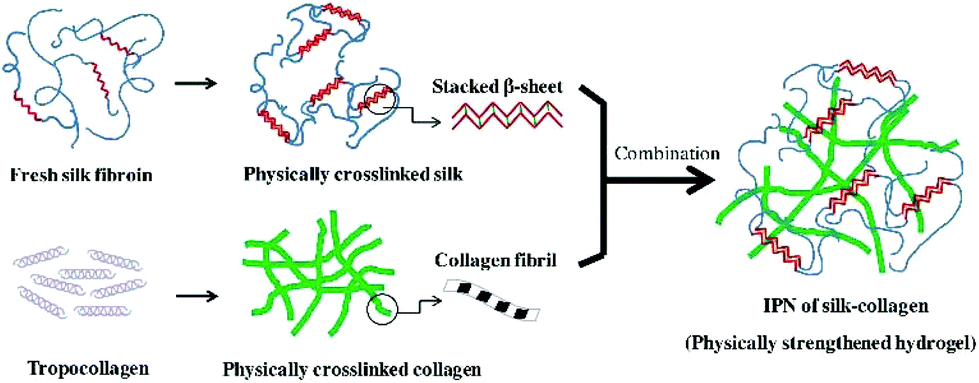 | ||
| Fig. 4 The schematic illustration of ultrasonic induced gelation of fibroin/collagen IPNs. Silk fibroin solution was placed in an ultrasound device for inducing β-sheet formation, and the ultrasonicated solution was added to the collagen solution. The mixed solution was aliquoted into a mold and incubated at 37 °C for complete gelation. Reproduced with permission from Acta Biomater., 2018, 69, 218–233. Copyright 2018 Elsevier.63 | ||
Hydrogel crosslinking by crystallization
The crystallites of polymer chains act as physically crosslinking sites in the network, resulting in the hydrogel formation. For instance, the hydrogel will form when the aqueous solution of PVA repeatedly undergoes freeze-thawing. The properties of the above hydrogel depend on molecular weight, aqueous solution concentration, freezing temperature and time, and the number of freeze-thawing cycles.64 A poly(vinyl alcohol) (PVA)/sodium alginate (SA) hydrogel was fabricated by the freeze-thawing technique, and the saturated NaCl solution was used to enhance the gel strength and conductivity.65 Zhang et al.66 prepared a physical double network (PDN) hydrogel which was composed of a physically crosslinked PVA and a hydrophobically associated polyacrylamide (HAPAM) by one-pot in situ polymerization and subsequent freeze-thawing cycling (Fig. 5). Mechanical strength of PDN gel was improved owing to strong crystallization of PVA and the presence of hydrogen bonds between PVA and PAM chains.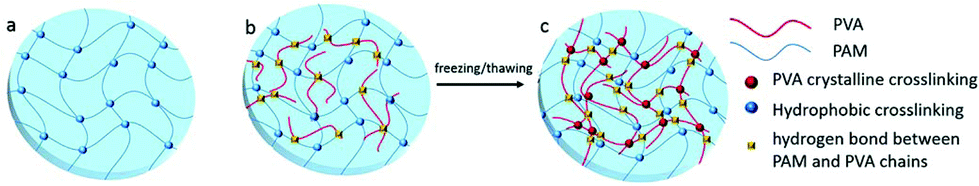 | ||
| Fig. 5 Schematic illustration of hydrogel network structure: (a) HAPAM gel, (b) semi-IPN HAPAM/PVA gel, (c) PDN gel after freezing/thawing. The Semi-IPN HAPAM/PVA hydrogels were prepared by one-pot in situ polymerization in the PVA solution, and then the PDN hydrogel was fabricated via the freezing/thawing treatment. Reproduced with permission from RSC Adv., 2016, 6, 112468–112476. Copyright 2016 RSC.66 | ||
The stereo-complex interaction can form racemic crystallization between 2 enantiomeric polymers, which can be used for the hydrogel preparation. Compared with homopolymer crystallite, the racemic crystallite exhibits more compact side by side crystallization.67 Poly(L-lactic acid) (PLLA) and poly(D-lactic acid) (PDLA) are enantiomeric polymers, which can form a stereo-complex based on racemic crystallite. In a recent study,68 an enantiomeric mixture of PDLA/poly(ethylene glycol) (PEG) di-block copolymer and PLLA/PEG triblock copolymer was used as a novel stereo-complex system. Hydrogel formed via unique gel-sol-gel multiple transitions upon heating, and the stereo-complexation of PLLA/PDLA segment played an important role in the phase transition of gel-sol-gel.
Hydrogel crosslinking by hydrogen bonding
Hydrogen bond is one of the most important noncovalent interactions. For example, hydrogen bond could stabilize a secondary structure during a peptide or agarose based hydrogel formation. For hydrogel formation, amide, urea, carboxylic acid, pyrrole, carbazole and hydroxyl groups could form hydrogen bonds among themselves or interact with electron donor groups, such as pyridine and imidazole groups. However, a single hydrogen bond is generally not strong enough to support hydrogel formation. By creating multiple multivalent hydrogen bonds, for example, a strong network can be formed using ureidopyrimidinone (UPy).69For example, a novel self-healing hydrogel was fabricated based on UPy–UPy interactions.70 The DMAEMA (2-(dimethylamino)ethyl methacrylate) was copolymerized with 2-(3-(6-methyl-4-oxo-1,4dihydropyrimidin-2-yl)ureido)ethyl methacrylate (SCMHBMA) containing UPy to produce DMAEMA–SCMHBMA copolymer. The polymer formed hydrogels above pH 8, and the hydrogels exhibited dynamic assembly and disassembly in response to damage along the stretching direction.
A multiblock copolymer with PEG and UPy was synthesized, and the polymer adopted nanoscopic physical cross-links between UPy–UPy dimers embedded in hydrophobic domains within the PEG matrix.71 The UPy units with self-complementary properties could assemble into dimers via 4-fold hydrogen bonds (H-bonding), which reinforced networks. The formed hydrogels exhibited high strength and resilience upon deformation as well as shape memory behaviour (Fig. 6).
 | ||
| Fig. 6 The PEG-UPy copolymers with multiblock architectures and self-complementary quadruple H-bonding interaction between 2 UPy segments. The descriptive morphology of dry semicrystalline polymer and a reversible transition to hydrogel. Reproduced with permission from J. Am. Chem. Soc., 2014, 136, 6969–6977. Copyright 2014 ACS.71 | ||
A mechanically strong supramolecular hydrogel with self-healable property was created and the poly(N-acryloyl glycinamide) (PNAGA) containing glycinamide-conjugated monomer with dual-amide in one side group was able to reconstruct and amplify the hydrogen bonding interactions between amino acid residues in a polymer hydrogel.72 Among the dual amide motifs, the self-recognizing H-bonded supramolecular interaction occured, and the hydrogel was rewarded with high mechanical performance, thermoplasticity and self-healing ability.
Hydrogel crosslinking by metal coordination
Metal coordination (metal–ligand interaction) between metal ions and functional groups in polymer chains is frequently used as a physical crosslinking method to fabricate hydrogels. The metal–ligand interaction can be regarded as a special Lewis acid–base interaction, which is stronger than most noncovalent interactions, but weaker than typical covalent bonding interactions.73 Therefore, metal–ligand interactions can dynamically occur or disappear owing to their moderate bond energy that is responsible for the self-healing property.74–77For example, hydrogels derived from side-chain bipyridyl-functionalized poly(2-oxazoline)s could be crosslinked with Fe(II), Ru(II), Ni(II) or Co(III).78,79 The extent of hydrogel swelling is dependent on the degree of functionalization of a polymer. The metal–ligand interactions of Fe(II) and Co(III) were relatively stable at room temperature, but the interactions would disintegrate when the temperature reached 30 °C. That was caused by the exchange from intermolecular crosslinks to intramolecular crosslinks. In contrast, hydrogels crosslinked by Ru(II) were quite stable even in boiling water. When the liquid was evaporated from hydrogels, all dry gels could recover gelation in water.
In addition, Harrington et al.80 discovered that mussel byssal threads originated from coordination between ferric ions and catechol ligands. The extent of catecholato-iron chelation influenced the stiffness, toughness, and self-healing capacity of the mussel byssal threads. Following the ferric ions and catechol interaction, a mussel-based hydrogel was fabricated using linear and branched PEG, which was end-functionalized with 1 to 4 3,4-dihydroxyphenylalanine (dopa) groups.81,82 Hydrogels were formed upon mixing with oxidizing agents. The 4-armed PEG was end-functionalized with dopa and presented a similar structure as catechol that could bind to Fe(III) ions to form catechol-Fe3+ complexes. The complex structure (mono-, bis-, or tris-) is controlled by pH via the deprotonation of the catechol hydroxyls. In detail, the metal–ligand interaction formed mono-complexes at pH below 5, bis-complexes about pH 8 and tris-complexes above pH 8. These results indicated that it was possible to control the gelation and crosslink density by pH variation.
Zheng's group synthesized a series of poly(acrylamide-co-acrylic acid) (P(AAm-co-AAc)) copolymers.83 A specifically tough hydrogel was subsequently prepared by swelling a cast film of poly(acrylamide-co-acrylic acid) (P(AAm-co-AAc)) in FeCl3 aqueous solution. The supramolecular networks were cross-linked by carboxyl-Fe3+ coordination bonds and the hydrogels possessed high stiffness and toughness, fatigue resistance, and stimulation-triggered healing along with shape memory and processing abilities. Furthermore, the strength of the coordination bond was controlled by varying pH, and the mechanical properties as well as shape memory abilities of hydrogels could be tuned (Fig. 7).
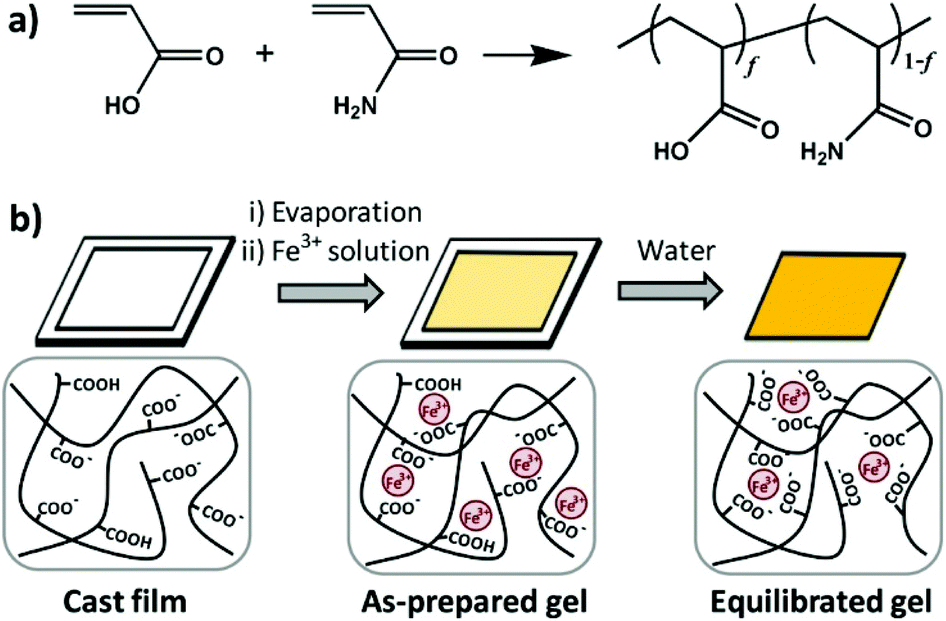 | ||
| Fig. 7 Diagrams of synthesis of copolymers (a) and physical crosslinked hydrogels (b). Hydrogels were formed by carboxyl-Fe3+ coordination. The dynamic nature of coordination bonds was rate dependent and the mechanical performance of hydrogels could be tuned by controlling the composition of copolymers, concentration of metallic ions, and pH values of solutions. Reproduced with permission from Macromolecules, 2016, 49, 9637–9646. Copyright 2016 ACS.83 | ||
Hydrogel crosslinking by host–guest interactions
Among various noncovalent interactions for hydrogel fabrication, the host–guest interaction is quite an important crosslinking strategy. By host–guest inclusion, polymers can be integrated in a facile and reversible way, promoting construction of hydrogels.84 Usually, a host is a molecule with a large cavity volume, such as cyclodextrins (CDs), cucurbiturils (CBs), calixarenes (CAs) and crown ethers, while guests with complementary shapes can interact with the hosts. Various noncovalent interactions can facilitate the host–guest inclusion, such as hydrogen bonding, electrostatics, and van der Waals, hydrophobic interactions; molecular shape is also important. Furthermore, the host–guest interaction is reversible and could be selective between one host and one guest.85In aqueous solutions, host–guest interactions usually occur by encapsulating hydrophobic guest molecules into hydrophobic cavities. This host–guest inclusion exhibits strong binding, and, more importantly, fixed geometry and directionality of the host–guest inclusion can respond to pH86 or other physical stimuli.87,88 Therefore, it can be useful for drug delivery. Among various host molecules, cyclodextrins (CD) are widely used because of the hydrophilic surface outside and the hydrophobic cavity inside.
For example, PEG can form host–guest complexes with α-cyclodextrin (α-CD), and PEG can crosslink to form larger aggregates or thread among a series of CD molecules. When PEG is mixed with α-CD, the inclusion complexes of PEG chains in α-CD cavities are formed after several days. The time and concentration of PEG required to form α-CD inclusion complexes were dependent on the molecular weight of PEG.89–91
PEG-CD inclusion complexes are always designed as injectable hydrogels and used for a controlled drug release. A hydrogel was prepared by mixing PEG-b-poly[(R)-3-hydroxybutyrate]-b-PEG with α-CD.92 The inclusion of PEG segments with α-CD, as well as the hydrophobic interactions between poly[(R)-3-hydroxybutyrate] blocks, resulted in the formation of a strong macromolecular network, which could be used for a long-term sustained controlled release of macromolecular drugs. Wang et al.93 prepared a branched polyrotaxane hydrogel based on α-CD inclusion with low-molecular-weight four-arm PEG. The hydrogel was injectable and exhibited shear-thinning and thixotropic properties. Drug release was controlled by shear stress.
Recently, to improve the mechanical properties the PEG-CD based hydrogels, Liu et al.94 synthesized nucleobase guanine/cytosine (G/C)-terminated PEGs (G-PEG-G and C-PEG-C), where the base-pairing interaction between G and C enhanced the storage moduli of the PEG-CD inclusion complex. The hydrogels exhibited better mechanical properties, because the G–C base pair formation acted as an additional network junction (Fig. 8). The hydrogels were thermo-responsible and could be used as thermo-controlled drug delivery systems.
 | ||
| Fig. 8 Synthesis of supramolecular hydrogels by host–guest inclusion between guanine/cytosine (G/C)-terminated PEG and α-cyclodextrin. The G–C base pairing acts as additional network junctions and enhances the hydrogel machinal properties. These hydrogels also exhibit excellent cytocompatibility and temperature-responsivity. Reproduced with permission from Mater. Sci. Eng., C, 2018, 82, 25–28. Copyright 2016 Elsevier.94 | ||
Chemically crosslinked hydrogels
Covalent bonds are normally formed among polymer chains in chemically crosslinked hydrogels, and most of their linkages are strong and permanent when they are compared with those of the physically crosslinked hydrogels. Up to now, several crosslinking methods have been reported,95 and they typically involve free radical polymerization induced crosslink, enzymatic induced crosslink, Diels–Alder “click” reaction, Schiff base formation, oxime formation and Michael type-addition. Compared with physically crosslinked hydrogels, chemically crosslinked hydrogels usually exhibit enhanced stability under physiological conditions and excellent mechanical properties as well as tuneable degradation behaviour.Hydrogel crosslinking by photopolymerization
Photo-activated crosslinking has been widely used for hydrogel formation in the field of therapeutics or cytokines encapsulation.96–99 The advantage of this method is the rapid formation of hydrogel networks at ambient temperature under mild conditions, and the mechanical properties of hydrogels can be tuned by controlling the crosslinking reactions.100 The crosslinked site is also ready to be accurately selected, because the photo-initiated polymerization takes place under light exposure and only the irradiated areas are involved in hydrogel crosslinking.101Photo-initiated polymerization is related to the presence of unsaturated groups, in most situations, the (meth)acrylates. The double bonded carbons in these groups are highly reactive and promote a free radical chain-growth polymerization when they are exposed to photo irradiation. Conventionally, water-soluble polymers with hydroxyl, carboxyl and amino groups can react with acryloyl chloride, glycidylmethacrylate (GMA) and N-(3-aminopropyl) methacrylamide to introduce vinyl groups.102 Formation of biomedical hydrogels usually requires the presence of cytocompatible photoinitiators, such as Irgacure 2959,103 Irgacure 1173,104 Irgacure 819,105 Irgacure 651,106 riboflavin phsphnate,107 camphorquinone,108 eosin Y,109 and so on. Those photo-initiators can absorb specific light at different wavelengths, including UV (250 nm–370 nm), visible blue & purple (405 nm–550 nm) and red light (750 nm–810 nm) and either decompose (Type I) or abstract hydrogen from a donor molecule (Type II) to form radical initiating species.100 However, type II initiators always require a co-initiator. For example, camphorquinone (CQ) requires ethyl 4-N,N-dimethylaminobenzoate (4EDMAB), triethanolamine (TEA) and the photosensitizer iso-propyl thioxanthone when the hydrogel is crosslinked by visible light.110 Another example is eosin Y, where the hydrogel crosslinking relies on triethanolamine and N-vinyl pyrrolidone (NVP) to promote the visible light activated photopolymerization.109
For all photo-initiators, cytocompatibility of light-activated hydrogel systems should be initially considered when they are designed to encapsulate cells and drugs. Bryant group110 systemically investigated the cytocompatibility of various photo-initiators with different photo-initiator concentrations and intensities. The 3T3 cells were exposed to photo-initiators under varying concentrations from 0.01% (w/w) to 0.1% (w/w). At low photo-initiator concentrations (≤0.01% (w/w)), most of the initiator molecules showed excellent biocompatibility, while CQ, Irgacure 651 and 4EDMAB exhibited a relatively low survival. In the presence of low intensity initiating light (365 nm UV light about 6 mW cm−2, and about 470–490 nm visible light about 60 mW cm−2), the Irgacure 2959 at concentrations −0.05% (w/w) and CQ at concentrations −0.01% (w/w) were the most promising cytocompatible UV and visible light initiating systems.
For ultraviolet light (UV-light) induced polymerization, the initiator, such as Irgacure 2959,103 is prevailing to prepare the functional hydrogels with well-designed patterned structure in situ. More importantly, cytotoxicity of UV-induced free radicals is relatively low. In general, the photo-initiator Irgacure 2959 is well tolerated by many cell types and it is the least cytotoxic.103 The long-wave UV light centred around 365 nm is mostly applied, because this long-wave UV exposure at intensities less than 10 mW cm−2 is well tolerated by most cell types for exposure times less than 3–5 min, providing cell viability following photo encapsulation typically greater than 90%. For example, a methacrylated γ-PGA (mPGA) was prepared111via crosslinking between γ-PGA and glycidyl methacrylate (GMA). The hydrogel synthesized by UV-light radiation exhibited ionic- and pH-sensitive properties and low cytotoxicity towards bovine chondrocytes.112 A chitosan hydrogel with a cell-loading pattern was fabricated by incorporating UV crosslinking method.113 This hydrogel exhibited low cytotoxicity based on the acute inflammatory response (Fig. 9).
 | ||
| Fig. 9 UV irradiation induced radical polymerization for hydrogel synthesis. Water-soluble N-methacryloyl chitosan (N-MAC) was synthesized and able to fabricate cell-loading microgels with the desired patterns. The hydrogels could be utilized in tissue engineering as a platform for sustained protein delivery. Reproduced with permission from Acta Biomater., 2015, 22, 59–69. Copyright 2015 Elsevier.113 | ||
However, there are still some concerns about DNA damage caused by UV radiation.114 Some studies claimed that UV radiation posed a potential risk of accelerating organ/tissue aging115 and cancer onset.116 Thus, visible light photoinitiation might be an alternative candidate. Blue light (visible) photo-initiators have been used, such as camphorquinone108 and eosin Y,117 lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP),118 riboflavin119 and ruthenium.120 These photo-initiators were able to effectively photo encapsulate various cells with high viability. Besides, visible light more readily penetrates tissue, which creates a pre-polymer solution forming hydrogel after subcutaneous injection. For example, Shih et al.117 reported a visible-light-mediated thiol–ene hydrogel using eosin-Y as the only photo-initiator. The hydrogel exhibited a rapid and tunable step-growth gelation by PEG-norbornene crosslinking with dithiothreitol under visible light exposure. Recently, chemical modification of photo-initiator appears to be more exciting as it could improve the photo-initiation effect. A carboxylated camphorquinone was synthesized to increase water solubility and photo reactivity of camphorquinone. The resultant hydrogel based on carboxylated-camphorquinone showed a significantly higher photoactivity and crosslink density as well as better mechanical properties.121
Light attenuation by the initiator restricts the maximum attainable cure depth to a few millimetres.122 To overcome this issue, polymerization of co-monomers containing complementary reactive groups, such as thiol-acrylates/enes, offers a better choice. The advantage of this strategy is the presence of a more homogeneous structure, greater tensile strength and increased strain at break. For thiol-acrylate/ene polymerization, the photo-initiator radical abstracts a hydrogen atom from a thiol and forms a thiyl radical. Polymerization was caused by the presence of the thiyl radicals and the corresponding thiol-acrylate/ene photopolymerization can rapidly occur with a low light intensity.123 More interesting, this approach can be employed in the absence of a photo-initiator, thereby, allowing curing to greater depths even in the lack of light intensity due to the photo-initiator attenuation.
In the process of thiol-acrylate photopolymerization, the competing Michael-type addition reaction between the thiol and acrylates takes place at the same time, resulting in a mixed-mode polymerization. Such mixed-mode polymerizations can lead to tuneable hydrogel properties, such as network structure and degradation by adjusting the ratio of thiol and acrylate. Furthermore, the residual thiol groups remain to be utilized to functionalize post-polymerization of the hydrogel. One elegant approach for the fast polymer post-functionalization and step-growth polymerization has been suggested, and it is based on the visible light photocatalytic thiol–ene “click” reaction.124 Two model polymers, polybutadiene and poly(allyl methacrylates), were modified with a large range of functional thiols. Subsequently, photoredox thiol–ene was successfully used to prepare linear polymers by step-growth addition reactions, which was highly efficient under low-energy (blue LED, 4.8 W) and eco-friendly visible light in the presence of air.
Hydrogel crosslinking by enzyme catalyzed reactions
Enzymatic crosslinking is an attractive method, as it offers the possibility for kinetic manipulation of gel formation in situ by controlling the enzyme concentration.125 The advantage of an enzymatic crosslink is the strongly covalent bonding as well as rapid gelation (always no more than 10 min) under physiological conditions. So far, there are many kinds of enzymatically catalyzed crosslinked methods for hydrogel formation in situ. For instance, the transglutaminase (TG) along with calcium ions as cofactors can promote formation of amide linkages between carboxamide and amine groups.126 Another crosslinking method is the horseradish peroxidase (HRP)-catalyzed crosslinking reaction. HRP catalyzes the coupling of aniline,21 phenol127and their derivative tyramine22,128–130 in the presence of hydrogen peroxide (H2O2). Kuo group constructed a novel vascularized tissue using an injectable cell-loaded enzymatically crosslinked collagen hydrogel. Collagen was conjugated with tyramine (collagen-Ph) and the hydrogel was formed in the presence of HRP and H2O2. The human blood-derived endothelial colony-forming cells (ECFCs) and bone marrow-derived mesenchymal stem cells (MSCs) were incorporated in the hydrogels. The results of cell evaluation indicated that human ECFCs-lined vascular networks were able to be generated within 7 days. The animal evaluation further proved that cell-combined collagen-Ph hydrogel could improve long-term differentiation of MSCs into osteoblasts as well as the increase the number of adipocytes in the mouse model after 1 month of implantation131 (Fig. 10). | ||
| Fig. 10 Schematic illustration of fabricating collagen-Ph hydrogel and in vivo experiment. Collagen was conjugated with tyramine and the injectable hydrogel containing ECFs and MSCs formed in the presence of HRP/H2O2. Cell-loaded hydrogels could form vascular networks after 7 days and promote adipose and bone-like tissue formation. Reproduced with permission from Acta Biomater., 2015, 27, 151–168. Copyright 2015 Elsevier.131 | ||
Hydrogel crosslinking by “click chemistry”
Utilization of “click chemistry” for design and fabrication of functional hydrogels grew rapidly since 2001.25 Chemical reactions that are termed click reactions possess advantages such as high yields under mild conditions, fewer by-products, high specificity and selectivity. A wide variety of functional groups can act as attractive candidates for the fabrication of complex polymeric materials.132 Here, we illustrate several classical crosslinking methods, including (1) Diels–Alder, (2) Schiff base, (3) oxime and (4) Michael-type addition.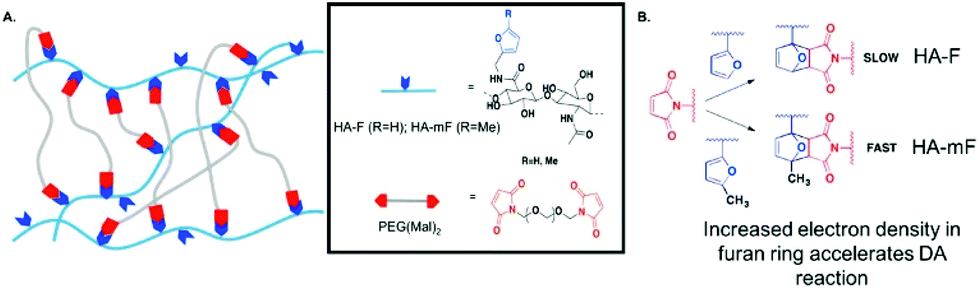 | ||
| Fig. 11 Schematic illustration of Diels–Alder (DA) reaction. When the furan group is replaced by methylfuran, DA reaction accelerates and hydrogels can be synthesized efficiently at pH 7.4 since the methylfuran group provided a more electron-rich diene. Reproduced with permission from Biomacromolecules, 2018, 19, 926–935. Copyright 2018 ACS.134 | ||
 | ||
| Fig. 12 Schematic illustration of Schiff base formation and self-healing process. The self-crosslinked hydrogel was synthesized via Schiff base linkage between the amino group of acrylamide-modified chitin (AMC) and the dialdehyde group (ADA) of oxidized alginate. The self-healing capability relied on the molar ratio of AMC and ADA as well as pH. Reproduced with permission from Soft Matter, 2015, 11, 3971–3976. Copyright 2015 RSC.137 | ||
In another case, the reaction of Schiff base can also occur between aldehyde and hydrazide groups. Ma et al.138 developed a novel injectable hydrogel for protein delivery by self-crosslinking of aldehyde hyaluronic acid (HA-CHO) and hydrazide-modified poly (γ-glutamic acid) (γ-PGA-ADH). The gelation time of hydrogel was as fast as 9s with high swelling ratios. Simultaneously, the hydrogel exhibited favorable mechanical properties and biocompatibility.
 | ||
| Fig. 13 Schematic illustration of self-healing behaviour. Two previously cut pieces of hydrogels (a) were placed in contact for 2 h (b) and the repaired gels were stretched with tweezers (c and c′); the healed gels were suspended under gravity (d and d′). When the gel was self-healed for 24 h, the hydrogel was cut again (e). Reproduced with permission from Soft Matter, 2015, 11, 6152–6161. Copyright 2015 RSC.34 | ||
A chitosan-based hydrogel with high mechanical strength was prepared via Michael addition.141 Hydrogels were fabricated from poly(ethylene glycol) diacrylate (PEGDA) and 3 thiolated natural polymers (chitosan, gelatin and heparin) via Michael addition under physiological conditions. Poly(ethylene glycol-b-caprolactone-b-ethylene glycol) (PECL) micelles with double bonds (bi-acrylated PECL micelles) were also incorporated to form mechanically reinforced chitosan-based hydrogels. Drugs were trapped inside the PECL micelles and hydrogels containing these drug-loading micelles exhibited excellent delivery performance (Fig. 14).
 | ||
| Fig. 14 Schematic illustration of Michael addition of chitosan-based hydrogels cross-linked with PECL micelles. Chitosan, gelatin and heparin were thiolated and crosslinked with PEGDA and bi-acrylated PECL micelles at different molar ratios. Reproduced with permission from J. Mater. Chem. B, 2017, 5, 961–971. Copyright 2017 RSC.141 | ||
Another in situ-forming Michael addition hydrogel was synthesized by mixing dithiothreitol (DTT) and glycidyl methacrylate derivatized dextran (Dex-GMA) in phosphate buffered saline (PBS). By changing the pH of PBS, the mechanical properties, gelation and the degree of swelling of the hydrogels could be easily tuned. The cell-loaded hydrogel exhibited good viability and cytocompatibility.142
Hydrogel crosslinking by dynamic covalent chemistry
The reversibility in hydrogels can be achieved via both physical crosslinks and reversible covalent bonds. The advantage of a reversible crosslink is its ability to make the hydrogels not only maintain the robust integrity of covalently cross-linked materials, but also employ the intrinsic reversibility of physical crosslinking.143 Boronic ester, as an example of a molecule containing dynamic covalent bonds, has a great potential for preparation of self-healing hydrogels. Recently, reversible boronate esters prepared from boronic acids and 1,2- or 1,3-diols have been explored to create self-healing hydrogels.144 In aqueous media, the strength and reversibility of a boronate ester are largely dependent on the pH value of solution and the pKa of the boronic acid component. Formation of the boronate ester bond is favoured at pH values above the pKa of the boronic acid, whereas the free boronic acid and diol are favoured at pH values below the pKa. This dynamic equilibrium between diol/boronic acid and boronate ester governs bond rearrangement to exhibit a self-healing behaviour. The newly formed bonds could span the damaged interface between 2 boronate ester-cross-linked materials. Smithmyer's group145 developed new boronic acid-based hydrogels that exhibited good viability in MDA-MB-231 breast cancer cells and CCL151 pulmonary fibroblasts. Huang et al. synthesized injectable hydrogels based on boronated PEG coupling with various linkers to form dynamic boronate ester bonds, such as plant derived polyphenols ellagic acid (EA), epigallocatechin gallate (EGCG), tannic acid (TA), nordihydroguaiaretic acid (NDGA), rutin trihydrate (RT), rosmarinic acid (RA) and carminic acid (CA).146 Among all polyphenol linkers, the ellagic acid (EA), epigallocatechin gallate (EGCG) and tannic acid (TA) were proved to form stable hydrogels under physiological conditions. Chen et al.147 prepared 2 kinds of cell-membrane mimicking copolymers based on 2-methacryloyloxyethyl phosphorylcholine (MPC), which were modified with benzoxaborole and catechol pendant groups. Hydrogel gelation occurred rapidly at pH 7.4 and the hydrogels exhibited self-healing ability as well as pH/sugar duel responses. Also, hydrogels containing dynamic bonds between boronic acid and alginate were prepared,148 the carboxyl groups of alginate were conjugated with the amine group of boronic acid (alginate-BA) and the reversible boronate-cis-diol complexation took place between boronated groups and the intrinsic cis-diol along alginate backbones. Seidler et al. applied a molecular recognition strategy based on boronic acid/salicyl hydroxamate, where an apoptosis enzyme, cytochrome c, was encapsulated into a hydrogel via dynamic covalent interactions (Fig. 15).149 This strategy facilitated the structural integration of native enzymes into the hydrogel scaffold and provided a stimulus-controlled enzyme release. | ||
| Fig. 15 Schematic illustration of a smart hydrogel with dynamic covalent bonds based on boronic acid/salicyl hydroxamate, which incorporated an apoptosis enzyme, cytochrome c. The hydrogel is bioactive and responsive to controlled administration. Reproduced with permission from Tetrahedron, 2017, 73, 499–4987. Copyright 2017 Elsevier.149 | ||
Perspectives
Comprehensive hydrogel crosslinking strategies
Hydrogel properties are highly associated with their crosslinking methods, so functional improvement of hydrogels is extremely dependent on the innovation of fabrication strategies. Normally, chemical methods are apt to achieve a stable hydrogel with suitable mechanical properties, whereas a physical method takes advantage of biocompatibility owing to the absence of chemical crosslinking agents. Consequently, a combination of physical and chemical approaches would be benefitable for attaining balanced properties of hydrogels. Actually, several pioneering studies have reported applying the combined crosslinking strategy to produce self-healing hydrogels.150 For instance, a double crosslinked hydrogel was prepared via physical crosslinking of hydrogen bonds and chemical crosslinking of imine bonds.151 This type of hydrogel exhibited an excellent self-healing capabilty. Besides, the physical crosslinking can act as the secondary crosslinking for enhancing the mechanical strength of a hydrogel. Consequently, a comprehensive crosslinking strategy of hydrogel generation paves a new way to construct self-healing smart hydrogels.Hierarchical structures of hydrogels
Ideally, bio-inspired hydrogels should have anisotropic and highly ordered architecture because natural soft tissues or organs consist of multiscale hierarchical structures.152 Hydrogel crosslinking methods play a pivotal role on determining the inner structure of hydrogels. So far, a bi-layered composite hydrogel with a well-designed hierarchical structure was successfully developed to promote osteochondral tissue regeneration.153,154 Thus, the hierarchically biomimetic structure of hydrogels should be considered as a crucial factor prior to designing and constructing new functional hydrogels. For example, the double crosslinking and interpenetrating polymer network (IPN) hydrogels usually have hierarchical structures as they are prepared using step by step crosslinking procedures.Intelligent stimuli-responsibility of hydrogels
Hydrogels, particularly injectable ones, have been widely applied in drug delivery systems. However, designing an intelligent hydrogel for drug delivery that can be modulated by physical and chemical stimulation is still a major challenge in the field of hydrogel-based drug delivery. This kind of drug delivery system is capable of releasing an appropriate amount of drug whenever required by the inflammatory tissue. To realize the on-demand drug release therapeutics, hydrogels should be synthesized via physical or chemical stimuli-responsive interactions.155 The method of drug-loaded hydrogel preparation influences the stimuli-responsive drug delivery behaviour and more innovative crosslinking methods should be proposed.Conclusions
In this review, we summarized the typical crosslinking methods of hydrogel synthesis involving physical and chemical strategies and illustrated their specific mechanisms and conditions of hydrogel formation. We also suggested potential prospects of hydrogel development based on a balanced combination between physical and chemical approaches, construction of hierarchical hydrogels and stimuli-responsive hydrogels for drug delivery systems. In the future, hydrogels with desirable mechanics and favourable biocompatibility could be useful for tissue engineering and regenerative medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017YFC1103900, 2018YFC1105700), the National Natural Science Foundation of China (31670968, 81601610, 81461148032, 31430029, and 31781240266), and HUST Key Innovation Team Project for Interdisciplinary Advancement (2016JCTD101).References
- F. Gattazzo, A. Urciuolo and P. Bonaldo, Biochim. Biophys. Acta, 2014, 1840, 2506–2519 CrossRef CAS PubMed.
- F. M. Watt and W. T. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467–473 CrossRef CAS PubMed.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- B. P. Hung, J. N. Harvestine, A. M. Saiz, T. Gonzalez-Fernandez, D. E. Sahar, M. L. Weiss and J. K. Leach, Biomaterials, 2019, 189, 1–10 CrossRef CAS PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- S. Q. Liu, R. Tay, M. Khan, P. L. R. Ee, J. L. Hedrick and Y. Y. Yang, Soft Matter, 2010, 6, 67–81 RSC.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- A. S. Hoffman, J. Controlled Release, 1987, 6, 297–305 CrossRef CAS.
- X. S. Wu, A. S. Hoffman and P. Yager, J. Polym. Sci., Polym. Chem., 1992, 30, 2121–2129 CrossRef CAS.
- L. C. Dong and A. S. Hoffman, J. Controlled Release, 1986, 4, 223–227 CrossRef CAS.
- M. Boustta, P. E. Colombo, S. Lenglet, S. Poujol and M. Vert, J. Controlled Release, 2014, 174, 1–6 CrossRef CAS PubMed.
- H. Y. Ye, C. Owh and X. J. Loh, RSC Adv., 2015, 5, 48720–48728 RSC.
- X. Hu, Q. Lu, L. Sun, P. Cebe, X. Wang, X. Zhang and D. L. Kaplan, Biomacromolecules, 2010, 11, 3178–3188 CrossRef CAS PubMed.
- X. Wang, J. A. Kluge, G. G. Leisk and D. L. Kaplan, Biomaterials, 2008, 29, 1054–1064 CrossRef CAS PubMed.
- W. Zhang, X. Wang, S. Wang, J. Zhao, L. Xu, C. Zhu, D. Zeng, J. Chen, Z. Zhang, D. L. Kaplan and X. Jiang, Biomaterials, 2011, 32, 9415–9424 CrossRef CAS PubMed.
- S. L. Bourke, M. Al-Khalili, T. Briggs, B. B. Michniak, J. Kohn and L. A. Poole-Warren, AAPS PharmSci., 2003, 5, E33 CrossRef PubMed.
- I. Mironi-Harpaz, D. Y. Wang, S. Venkatraman and D. Seliktar, Acta Biomater., 2012, 8, 1838–1848 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- K. S. Kim, S. J. Park, J. A. Yang, J. H. Jeon, S. H. Bhang, B. S. Kim and S. K. Hahn, Acta Biomater., 2011, 7, 666–674 CrossRef CAS PubMed.
- R. Jin, L. S. Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Biomaterials, 2010, 31, 3103–3113 CrossRef CAS PubMed.
- L. S. Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 40, 2004–2021 CrossRef CAS.
- M. van Dijk, D. T. Rijkers, R. M. Liskamp, C. F. van Nostrum and W. E. Hennink, Bioconjugate Chem., 2009, 20, 2001–2016 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. X. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- Z. Wei, J. H. Yang, X. J. Du, F. Xu, M. Zrinyi, Y. Osada, F. Li and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464–1470 CrossRef CAS PubMed.
- S. Mukherjee, M. R. Hill and B. S. Sumerlin, Soft Matter, 2015, 11, 6152–6161 RSC.
- J. Kalia and R. T. Raines, Angew. Chem., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- F. Lin, J. Yu, W. Tang, J. Zheng, A. Defante, K. Guo, C. Wesdemiotis and M. L. Becker, Biomacromolecules, 2013, 14, 3749–3758 CrossRef CAS PubMed.
- J. Collins, Z. Y. Xiao, M. Mullner and L. A. Connal, Polym. Chem., 2016, 7, 3812–3826 RSC.
- W. Tao, T. Mahir and G. Sundaram, Polym. Int., 2004, 53, 911–918 CrossRef.
- Y. Xin and J. Y. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- Y. Jia and J. Li, Chem. Rev., 2015, 115, 1597–1621 CrossRef CAS PubMed.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CrossRef CAS PubMed.
- C. K. Kuo and P. X. Ma, Biomaterials, 2001, 22, 511–521 CrossRef CAS PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 35–52 CrossRef CAS PubMed.
- H. J. Chung, Y. Lee and T. G. Park, J. Controlled Release, 2008, 127, 22–30 CrossRef CAS PubMed.
- P. Gacesa, Carbohydr. Polym., 1988, 8, 161–182 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- H. Lin, Q. Li, Q. Du, O. Wang, Z. Wang, L. Akert, M. A. Carlson, C. Zhang, A. Subramanian, C. Zhang, M. Lunning, M. Li and Y. Lei, Biomaterials, 2019, 189, 23–36 CrossRef CAS PubMed.
- X. Wang, F. Liu, X. Zheng and J. Sun, Angew. Chem., 2011, 50, 11378–11381 CrossRef CAS PubMed.
- J. Ostrowska-Czubenko and M. Gierszewska-Druzynska, Carbohydr. Polym., 2009, 77, 590–598 CrossRef CAS.
- J. H. Hamman, Mar. Drugs, 2010, 8, 1305–1322 CrossRef CAS PubMed.
- F. Ahmadi, Z. Oveisi, S. M. Samani and Z. Amoozgar, Res. Pharm. Sci., 2015, 10, 1–16 CAS.
- B. H. Ye, S. Y. Zhang, R. W. Li, L. H. Li, L. Lu and C. R. Zhou, Compos. Sci. Technol., 2018, 156, 238–246 CrossRef CAS.
- S. J. Lue, C. H. Chen and C. M. Shih, J. Macromol. Sci., Part B: Phys., 2011, 50, 563–579 CrossRef CAS.
- M. A. Haq, Y. Su and D. Wang, Mater. Sci. Eng., C, 2017, 70, 842–855 CrossRef PubMed.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed.
- H. Park, M. H. Kim, Y. I. Yoon and W. H. Park, Carbohydr. Polym., 2017, 157, 775–783 CrossRef CAS PubMed.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS PubMed.
- H. Zhang, S. W. Guo, S. Y. Fu and Y. Zhao, Polymers, 2017, 9, 238 CrossRef.
- W. X. Fu and B. Zhao, Polym. Chem., 2016, 7, 6980–6991 RSC.
- X. Hu, D. Kaplan and P. Cebe, Macromolecules, 2006, 39, 6161–6170 CrossRef CAS.
- J. O. Buitrago, K. D. Patel, A. El-Fiqi, J. H. Lee, B. Kundu, H. H. Lee and H. W. Kim, Acta Biomater., 2018, 69, 218–233 CrossRef CAS PubMed.
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS.
- X. Jiang, N. Xiang, H. Zhang, Y. Sun, Z. Lin and L. Hou, Carbohydr. Polym., 2018, 186, 377–383 CrossRef CAS PubMed.
- Y. L. Zhang, M. W. Song, Y. F. Diao, B. W. Li, L. Y. Shi and R. Ran, RSC Adv., 2016, 6, 112468–112476 RSC.
- D. W. Lim, S. H. Choi and T. G. Park, Macromol. Rapid Commun., 2000, 21, 464–471 CrossRef CAS.
- H. L. Mao, C. Wang, X. H. Chang, H. Q. Cao, G. R. Shan, Y. Z. Bao and P. J. Pan, Mater. Chem. Front., 2018, 2, 313–322 RSC.
- H. Hofmeier, R. Hoogenboom, M. E. Wouters and U. S. Schubert, J. Am. Chem. Soc., 2005, 127, 2913–2921 CrossRef CAS PubMed.
- J. Cui and A. del Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- H. Li, P. Yang, P. Pageni and C. Tang, Macromol. Rapid Commun., 2017, 38, 1700109 CrossRef PubMed.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., 2002, 41, 2892–2926 CrossRef CAS.
- C. A. Fustin, P. Guillet, U. S. Schubert and J. F. Gohy, Adv. Mater., 2007, 19, 1665–1673 CrossRef CAS.
- J. Brassinne, F. D. Jochum, C. A. Fustin and J. F. Gohy, Int. J. Mol. Sci., 2015, 16, 990–1007 CrossRef CAS PubMed.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1993, 26, 6315–6319 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Polym. J., 1993, 25, 599 CrossRef CAS.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216–220 CrossRef CAS PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106–2115 CrossRef CAS PubMed.
- H. Frisch and P. Besenius, Macromol. Rapid Commun., 2015, 36, 346–363 CrossRef CAS PubMed.
- Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 CrossRef CAS PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- K. M. Huh, T. Ooya, W. K. Lee, S. Sasaki, I. C. Kwon, S. Y. Jeong and N. Yui, Macromolecules, 2001, 34, 8657–8662 CrossRef CAS.
- J. Li, A. Harada and M. Kamachi, Polym. J., 1994, 26, 1019 CrossRef CAS.
- M. Guo, M. Jiang, S. Pispas, W. Yu and C. Zhou, Macromolecules, 2008, 41, 9744–9749 CrossRef CAS.
- J. Li, X. Li, X. Ni, X. Wang, H. Li and K. W. Leong, Biomaterials, 2006, 27, 4132–4140 CrossRef CAS PubMed.
- J. Wang, G. S. Williamson and H. Yang, Colloids Surf., B, 2018, 165, 144–149 CrossRef CAS PubMed.
- L. Liu, X. Feng, Y. Pei, J. Wang, J. Ding and L. Chen, Mater. Sci. Eng., C, 2018, 82, 25–28 CrossRef CAS PubMed.
- S. Li, Y. Xia, Y. Qiu, X. Chen and S. Shi, J. Appl. Polym. Sci., 2018, 135, 45761 CrossRef.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Biomaterials, 1993, 14, 1008–1016 CrossRef CAS PubMed.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS PubMed.
- Y. An and J. A. Hubbell, J. Controlled Release, 2000, 64, 205–215 CrossRef CAS PubMed.
- O. Jeon, C. Powell, L. D. Solorio, M. D. Krebs and E. Alsberg, J. Controlled Release, 2011, 154, 258–266 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed.
- H. Y. Yao, J. Q. Wang and S. L. Mi, Polymers, 2018, 10, 27 Search PubMed.
- W. N. E. van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Biomaterials, 2010, 31, 6511–6518 CrossRef CAS PubMed.
- A. Urrios, C. Parra-Cabrera, N. Bhattacharjee, A. M. Gonzalez-Suarez, L. G. Rigat-Brugarolas, U. Nallapatti, J. Samitier, C. A. DeForest, F. Posas, J. L. Garcia-Cordero and A. Folch, Lab Chip, 2016, 16, 2287–2294 RSC.
- M. B. Mellott, K. Searcy and M. V. Pishko, Biomaterials, 2001, 22, 929–941 CrossRef CAS PubMed.
- H. J. Lee, G. M. Fernandes-Cunha and D. Myung, React. Funct. Polym., 2018, 131, 29–35 CrossRef CAS.
- J. Hu, Y. Hou, H. Park, B. Choi, S. Hou, A. Chung and M. Lee, Acta Biomater., 2012, 8, 1730–1738 CrossRef CAS PubMed.
- Y. D. Park, N. Tirelli and J. A. Hubbell, Biomaterials, 2003, 24, 893–900 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- W. Zeng, W.-k. Hu, H. Li, Y.-h. Jing, H. Kang, Q. Jiang and C. Zhang, Chin. J. Polym. Sci., 2014, 32, 1507–1514 CrossRef CAS.
- W. Hu, X. Feng, X. Liu, S. Dai, W. Zeng, Q. Jiang, B. Chen, C. Quan, K. Sun and C. Zhang, J. Biomater. Sci., Polym. Ed., 2016, 27, 1–13 CrossRef PubMed.
- B. Li, L. Wang, F. Xu, X. Gang, U. Demirci, D. Wei, Y. Li, Y. Feng, D. Jia and Y. Zhou, Acta Biomater., 2015, 22, 59–69 CrossRef CAS PubMed.
- J. H. Hoeijmakers, N. Engl. J. Med., 2009, 361, 1475–1485 CrossRef CAS PubMed.
- U. Panich, G. Sittithumcharee, N. Rathviboon and S. Jirawatnotai, Stem Cells Int., 2016, 2016, 7370642 Search PubMed.
- P. Dakup and S. Gaddameedhi, Photochem. Photobiol., 2017, 93, 296–303 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Macromol. Rapid Commun., 2013, 34, 269–273 CrossRef CAS PubMed.
- H. Lin, D. Zhang, P. G. Alexander, G. Yang, J. Tan, A. W. Cheng and R. S. Tuan, Biomaterials, 2013, 34, 331–339 CrossRef CAS PubMed.
- G. M. Fernandes-Cunha, H. J. Lee, A. Kumar, A. Kreymerman, S. Heilshorn and D. Myung, Biomacromolecules, 2017, 18, 3185–3196 CrossRef CAS PubMed.
- J. W. Bjork, S. L. Johnson and R. T. Tranquillo, Biomaterials, 2011, 32, 2479–2488 CrossRef CAS PubMed.
- E. A. Kamoun, A. Winkel, M. Eisenburger and H. Menzel, Arabian J. Chem., 2016, 9, 745–754 CrossRef CAS.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS PubMed.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- J. J. Sperinde and L. G. Griffith, Macromolecules, 1997, 30, 5255–5264 CrossRef CAS.
- M. K. McHale, L. A. Setton and A. Chilkoti, Tissue Eng., 2005, 11, 1768–1779 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314, 10.1039/b506989k.
- R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, J. Controlled Release, 2011, 152, 186–195 CrossRef CAS PubMed.
- K. C. Kuo, R. Z. Lin, H. W. Tien, P. Y. Wu, Y. C. Li, J. M. Melero-Martin and Y. C. Chen, Acta Biomater., 2015, 27, 151–166 CrossRef CAS PubMed.
- S. Yigit, R. Sanyal and A. Sanyal, Chem.– Asian J., 2011, 6, 2648–2659 CrossRef CAS PubMed.
- F. Yu, X. Cao, J. Du, G. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 24023–24031 CrossRef CAS PubMed.
- L. J. Smith, S. M. Taimoory, R. Y. Tam, A. E. G. Baker, N. Binth Mohammad, J. F. Trant and M. S. Shoichet, Biomacromolecules, 2018, 19, 926–935 CrossRef CAS PubMed.
- N. L. Morozowich, J. L. Nichol and H. R. Allcock, J. Polym. Sci., Polym. Chem., 2016, 54, 2984–2991 CrossRef CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- F. Ding, S. Wu, S. Wang, Y. Xiong, Y. Li, B. Li, H. Deng, Y. Du, L. Xiao and X. Shi, Soft Matter, 2015, 11, 3971–3976 RSC.
- X. Ma, T. Xu, W. Chen, H. Qin, B. Chi and Z. Ye, Carbohydr. Polym., 2018, 179, 100–109 CrossRef CAS PubMed.
- X. Sui, L. van Ingen, M. A. Hempenius and G. J. Vancso, Macromol. Rapid Commun., 2010, 31, 2059–2063 CrossRef CAS PubMed.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS PubMed.
- Y. Wen, F. Li, C. G. Li, Y. J. Yin and J. J. Li, J. Mater. Chem. B, 2017, 5, 961–971 RSC.
- Z. Q. Liu, Z. Wei, X. L. Zhu, G. Y. Huang, F. Xu, J. H. Yang, Y. Osada, M. Zrinyi, J. H. Li and Y. M. Chen, Colloids Surf., B, 2015, 128, 140–148 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- C. C. Deng, W. L. A. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef CAS.
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS.
- Z. Huang, P. Delparastan, P. Burch, J. Cheng, Y. Cao and P. B. Messersmith, Biomater. Sci., 2018, 6, 2487–2495 RSC.
- Y. J. Chen, D. Diaz-Dussan, D. Wu, W. D. Wang, Y. Y. Peng, A. B. Asha, D. G. Hall, K. Ishihara and R. Narain, ACS Macro Lett., 2018, 7, 904–908 CrossRef CAS.
- S. H. Hong, S. Kim, J. P. Park, M. Shin, K. Kim, J. H. Ryu and H. Lee, Biomacromolecules, 2018, 19, 2053–2061 CrossRef CAS PubMed.
- C. Seidler, D. Y. W. Ng and T. Weil, Tetrahedron, 2017, 73, 4979–4987 CrossRef CAS.
- T. J. Long, Y. X. Li, X. Fang and J. Q. Sun, Adv. Funct. Mater., 2018, 28, 9 Search PubMed.
- R. Wang, Q. Li, B. Chi, X. Wang, Z. Xu, Z. Xu, S. Chen and H. Xu, Chem. Commun., 2017, 53, 4803–4806 RSC.
- M. T. I. Mredha, Y. Z. Guo, T. Nonoyama, T. Nakajima, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1704937 CrossRef PubMed.
- K. Kim, J. Lam, S. Lu, P. P. Spicer, A. Lueckgen, Y. Tabata, M. E. Wong, J. A. Jansen, A. G. Mikos and F. K. Kasper, J. Controlled Release, 2013, 168, 166–178 CrossRef CAS PubMed.
- S. Lu, J. Lam, J. E. Trachtenberg, E. J. Lee, H. Seyednejad, J. van den Beucken, Y. Tabata, M. E. Wong, J. A. Jansen, A. G. Mikos and F. K. Kasper, Biomaterials, 2014, 35, 8829–8839 CrossRef CAS PubMed.
- N. Joshi, J. Yan, S. Levy, S. Bhagchandani, K. V. Slaughter, N. E. Sherman, J. Amirault, Y. Wang, L. Riegel, X. He, T. S. Rui, M. Valic, P. K. Vemula, O. R. Miranda, O. Levy, E. M. Gravallese, A. O. Aliprantis, J. Ermann and J. M. Karp, Nat. Commun., 2018, 9, 1275 CrossRef PubMed.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |