
Alkenylation of unactivated alkyl bromides through visible light photocatalysis†
Quan-Quan
Zhou‡
a,
Simon Josef Siegfried
Düsel‡
b,
Liang-Qiu
Lu
 a,
Burkhard
König
*b and
Wen-Jing
Xiao
*a
a,
Burkhard
König
*b and
Wen-Jing
Xiao
*a
aCCNU-uOttawa Joint Research Centre, Hubei International Scientific and Technological Cooperation Base of Pesticide and Green Synthesis, Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: wxiao@mail.ccnu.edu.cn
bFaculty of Chemistry and Pharmacy, University of Regensburg, Universitätsstraße 31, 93053, Regensburg, Germany. E-mail: burkhard.koenig@ur.de
First published on 26th November 2018
Abstract
Two visible-light driven alkenylation reactions of unactivated alkyl bromides, which were enabled by the use of Ir(dF(CF3)ppy)2(dtbbpy)PF6 as the photocatalyst and (TMS)3SiH as the atom transfer reagent to activate the alkyl bromides, were described for the first time. These protocols can be used to produce a variety of alkenes from easily available feedstock with good reaction efficiency and high chemoselectivity under mild reaction conditions. To further demonstrate the applicability of the present strategy, the alkenylation of bioactive molecules and glycosyl bromides, as well as the alkynylation of unactivated alkyl bromides, was proven to be feasible.
Alkenes are a class of fundamentally important compounds, as they are broadly applied in synthetic chemistry and materials science.1 Tremendous research efforts have been devoted to the synthesis of alkenes, including the well-known Wittig-type reactions,2 semi-reduction of alkynes,3 Heck-type reactions4 and many others.5 Inherently, the cross coupling of an alkene moiety with the pre-functionalized alkane partner through transition metal catalysis has been considered as an ideal tool for this purpose with anticipated chemo- and regioselectivity.4,5 Although being a great achievement, this valuable protocol still has some drawbacks. For example, organometallic reagents, which are widely used in these reactions, are usually sensitive to water and air.6 In addition, the oxidative addition of alkyl halides to a metal catalyst is difficult and the generated transient species might undergo undesired β-hydride elimination.4,5 Moreover, elevated temperatures are frequently required.
Visible light photocatalysis has attracted increasing interest from the synthetic community, because it enables the generation of reactive intermediates under mild conditions.7 This strategy has been gradually applied in alkenylation reactions during the last decade.8 In this context, alkyl fluoroborate salts, which have a photocatalytically addressable oxidation potential, have been widely used as potent radical precursors for many coupling reactions,9 including alkenylation with alkenyl sulfones (Scheme 1a).9a However, the preparation of these reagents produces stoichiometric amounts of by-products and usually requires three consecutive steps, including the reaction using oxygen- and water-sensitive organometallic reagents under low temperatures, somewhat limiting its application. Thus, many other alkyl precursors have been developed to improve the photocatalytic alkenylation reaction.8 Among them, easily available halides10 have been proven to be an efficient feedstock for the alkenylation. Usually, activated alkyl halides, such as alkyl iodides or electron-deficient alkyl bromides, are required due to their higher reduction potential, which favors the generation of alkyl radicals via photocatalytic SET processes. Hence, the direct use of unactivated alkyl bromides in alkenylation reactions through visible light photocatalysis remains a challenge, despite their advantages of low price, bench stability and easy availability.11 Inspired by the well-established process using organosilicon radicals to activate alkyl bromides,12 we therefore envision that alkyl bromides can also be utilized as efficient alkyl radical precursors to participate in new alkenylation reactions through the combination of visible light photocatalysis and silicon radical debromination. In this work, we plan to develop two kinds of coupling reactions of non-activated alkyl bromides with phenyl vinyl sulfones (method A) or bromides (method B) as shown in Scheme 1b. If successful, these protocols will provide two competitive and alternative methods for the preparation of alkenes from easily available chemicals under mild conditions.
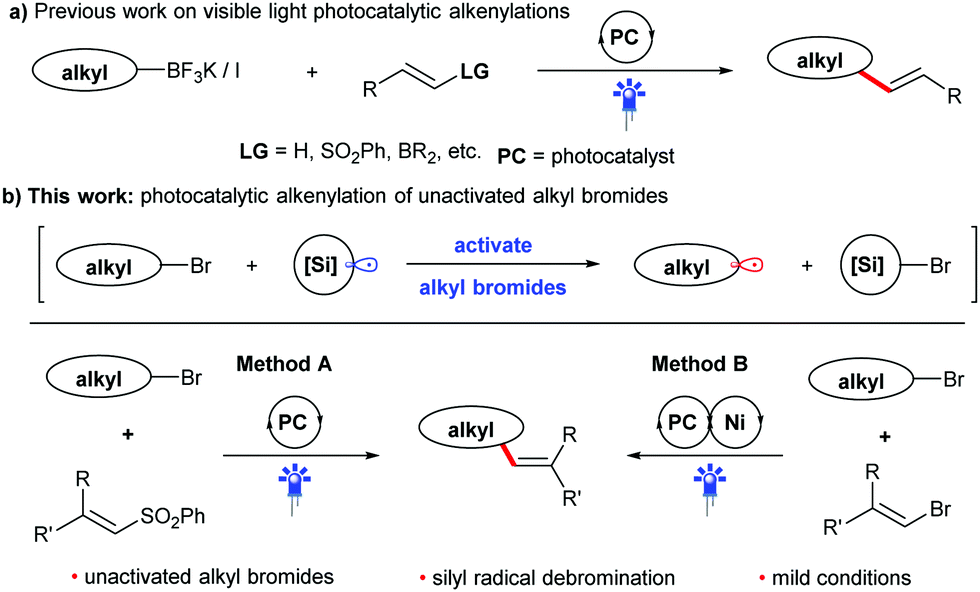 | ||
| Scheme 1 Reaction design: intermolecular alkyl–alkenyl coupling reactions via visible light photocatalysis. | ||
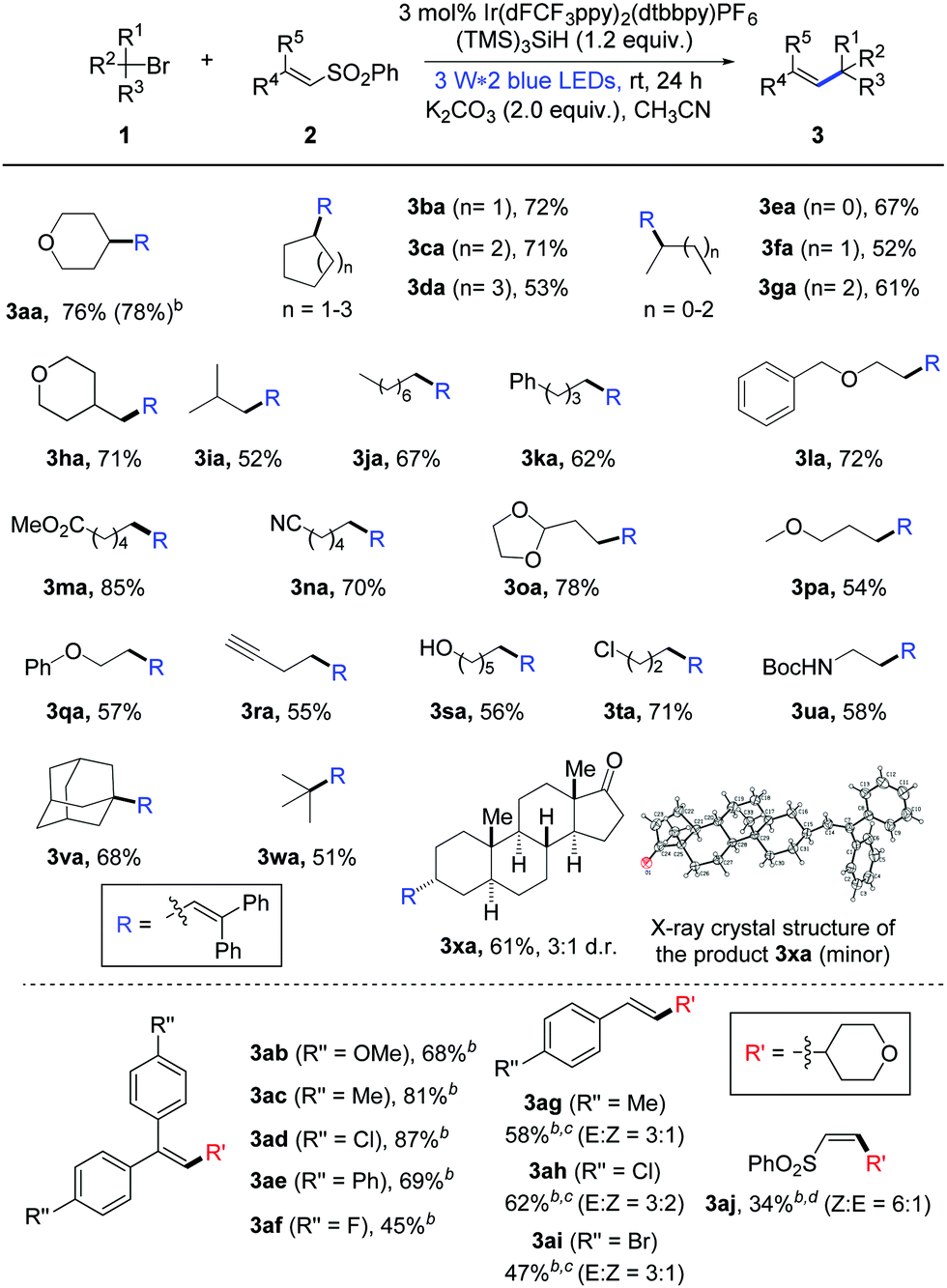
Initially, we began our investigations with the coupling of bromotetrahydropyran (1a) and diphenyl vinyl sulfone (2a) as the model reaction (please see the ESI,† Table S1).13 In the presence of photocatalyst Ir(dF(CF3)ppy)2(dtbbpy)PF6, silicon radical precursor tris(trimethylsilyl)silane (TTMSS) and inorganic base Na2CO3, a high amount of the desired alkene product 3aa was detected after blue light irradiation at room temperature (rt) for 24 hours (Table S1, entry 1:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 83% NMR yield, ESI†). Then, routine optimization of the reaction parameters including bases, solvents, hydrosilanes and concentration (Tables S1–S4, ESI†) improved the result, in which the combination of K2CO3 and CH3CN (0.05 M) stood out as the best choice (for optimal conditions, see the footnote in Table 1).13 Moreover, the reaction efficiency was not obviously affected by the air atmosphere (Table S1, entry 17, ESI†). Control experiments showed that visible light and a suitable photoredox catalyst were necessary for the transformation.13
83% NMR yield, ESI†). Then, routine optimization of the reaction parameters including bases, solvents, hydrosilanes and concentration (Tables S1–S4, ESI†) improved the result, in which the combination of K2CO3 and CH3CN (0.05 M) stood out as the best choice (for optimal conditions, see the footnote in Table 1).13 Moreover, the reaction efficiency was not obviously affected by the air atmosphere (Table S1, entry 17, ESI†). Control experiments showed that visible light and a suitable photoredox catalyst were necessary for the transformation.13
| a Reaction conditions: 1 (0.6 mmol), 2 (0.2 mmol), TTMSS (0.24 mmol) Ir(dF(CF3)ppy)2(dtbbpy)PF6 (3 mol%), K2CO3 (2.0 equiv.) in CH3CN (4.0 mL) at rt under irradiation with 3 W × 2 blue LEDs for 24 h, isolated yield. b 1.5 equiv. of TTMSS. c (E)-Phenyl vinyl sulfone was used. d cis-1,2-Bis(phenylsulfonyl)ethane was used. |
|---|
|
Having established the optimal reaction conditions, we started to examine the generality of this photocatalytic alkenylation reaction. As highlighted in Table 1, the mild protocol enables the alkenylation of different unactivated bromides. The coupling with secondary cyclic alkyl bromides proceeded smoothly, providing the corresponding alkene products in good yields (3aa–3da: 53–78% yields). Acyclic secondary alkyl bromides can readily undergo this transformation too; the alkenylation products were obtained in 52–67% yields (3ea–3ga). In addition, primary and tertiary alkyl bromides were also converted into the desired products with satisfied results (3ha–3xa: 51–85% yields). To our delight, this protocol shows good functional group tolerance. Many alkyl bromides bearing ether, ester, alkyne, acetal and several other moieties can participate well in the photocatalytic alkenylation reaction (1l–1r). More importantly, the reaction with substrate 1s containing a free hydroxyl group also proceeds smoothly with moderate yield. Besides, the reaction with dihalide 1t showed excellent chemoselectivity between the bromide and the chloride. Moreover, this protocol allows the debrominative alkenylation of bioactive molecule 1x under mild reaction conditions, delivering product 3xa in good yield.14
Next, we probed the scope of phenyl vinyl sulfones under photocatalysis conditions. As summarized in Table 1, a variety of 2,2′-diaryl-substituted vinyl sulfones can be applied as efficient substrates and the electronic characteristic of aryls somewhat affect the reaction results. In the case of the chlorinated product 3ad, a high yield of 87% was obtained and the phenyl chloride moieties were not affected by the silyl radical. In addition to 2,2′-diaryl-substituted vinyl sulfones, styrene-substituted sulfones can participate in this photochemical transformation as well. For example, when para-Me-, Cl-, or Br-substituted styrene sulfones were subjected to the standard reaction conditions, the desired alkenylation products 3ag–3ai were obtained in good yields with modest E/Z ratios. Moreover, this protocol can be successfully extended to the alkenylation of alkyl-substituted vinyl sulfones (3aj, Z:E = 6:1), but in 34% yield.
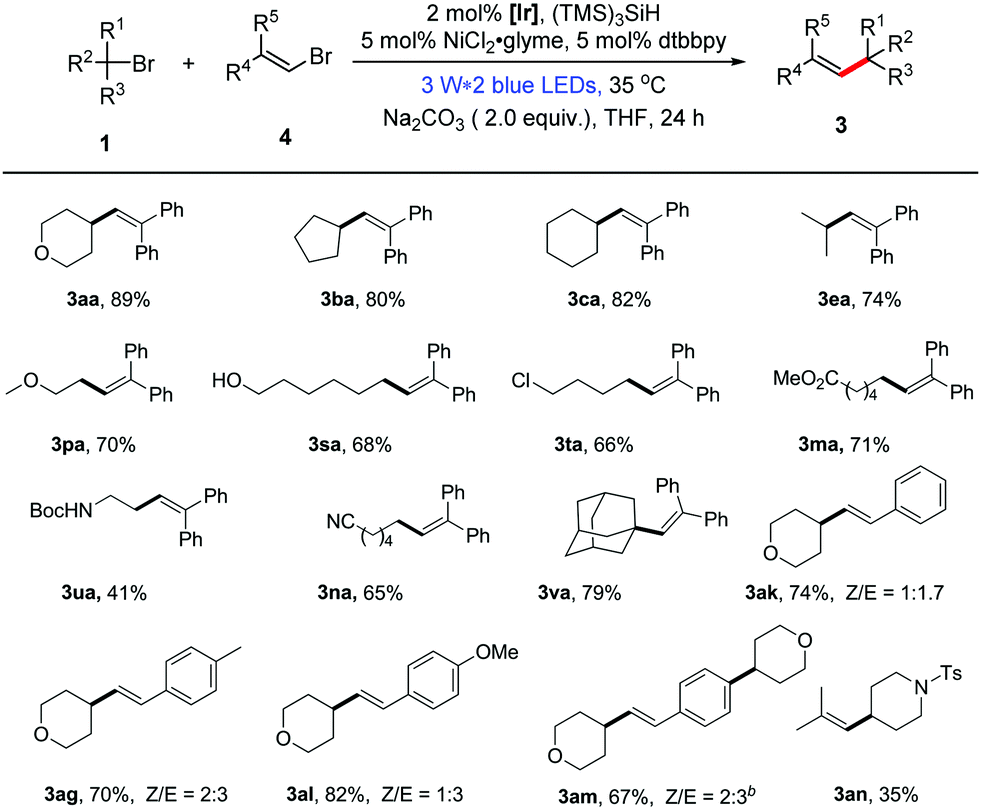
Recently, the use of nickel catalysis in combination with photoredox catalysis has been proven to be feasible for the coupling reaction of aryl halides.15 However, examples for the dual catalytic coupling reactions of vinyl halides are rare.16 In order to be more comprehensive, we present an additional method for the reaction of unactivated alkyl bromides with vinyl bromides through dual photoredox/nickel catalysis. With a slight alteration in the reaction conditions in the first alkenylation method, we established the optimal reaction conditions for the dual catalytic alkenylation of unactivated alkyl bromides with vinyl bromides.13 As highlighted in Table 2, the transformation under the dual catalysis system generally provided the corresponding alkene products in good yields (Table 2, 41–89% yields). In addition, it is general for alkyl bromides and shows good functional group compatibility. Furthermore, this protocol can also be extended to non-aromatic alkenes (3an, Table 2). Different from the formation of the mono-alkylated product 3ai using method A, the double alkylated compound 3am was obtained in good yield under the conditions of method B, demonstrating the different characteristics of the two presented methods.
| a Reaction conditions: alkyl bromides 1 (0.6 mmol), 6 (0.2 mmol), Na2CO3 (2.0 equiv.), (TMS)3SiH (0.24 mmol), [Ir] = Ir(dF(CF3)ppy)2(dtbbpy)PF6 (2 mol%), NiCl2·glyme (5 mol%), dtbbpy (5 mol%), THF (2.0 mL), Ar atmosphere, 3 W × 2 blue LEDs, 35 °C for 24 h; isolated yields. b 6.0 equiv. of 1a was used, DME as the solvent. |
|---|
|
To show the applicability of the dual photoredox/nickel catalysis systems, a gram-scale alkenylation reaction of alkyl bromide 1a and vinyl bromide 4a was conducted, for which the loading of the photocatalyst could be lowered to 0.5 mol% without affecting the reaction efficiency (Scheme 2a: 1.38 g, 87% yield).13 Moreover, the pharmacophore amitriptyline derivative 7 was prepared from tert-butyl(2-bromoethyl)carbamate 5 in 51% yield, which could be easily transferred to the antidepressant drug, amitriptyline.1d Considering the significance of utilizing renewable starting materials from nature,17 herein we realized the first photocatalytic alkenylation of glycosyl bromides with good efficiency and selectivity (Scheme 2c, 9aa–9ad, 65–71% yields). Furthermore, this strategy of visible light photocatalysis and silicon radical debromination was successfully extended to the alkynylation of alkynyl phenyl sulfone 10, albeit with a moderate yield (Scheme 2d: 44% yield).
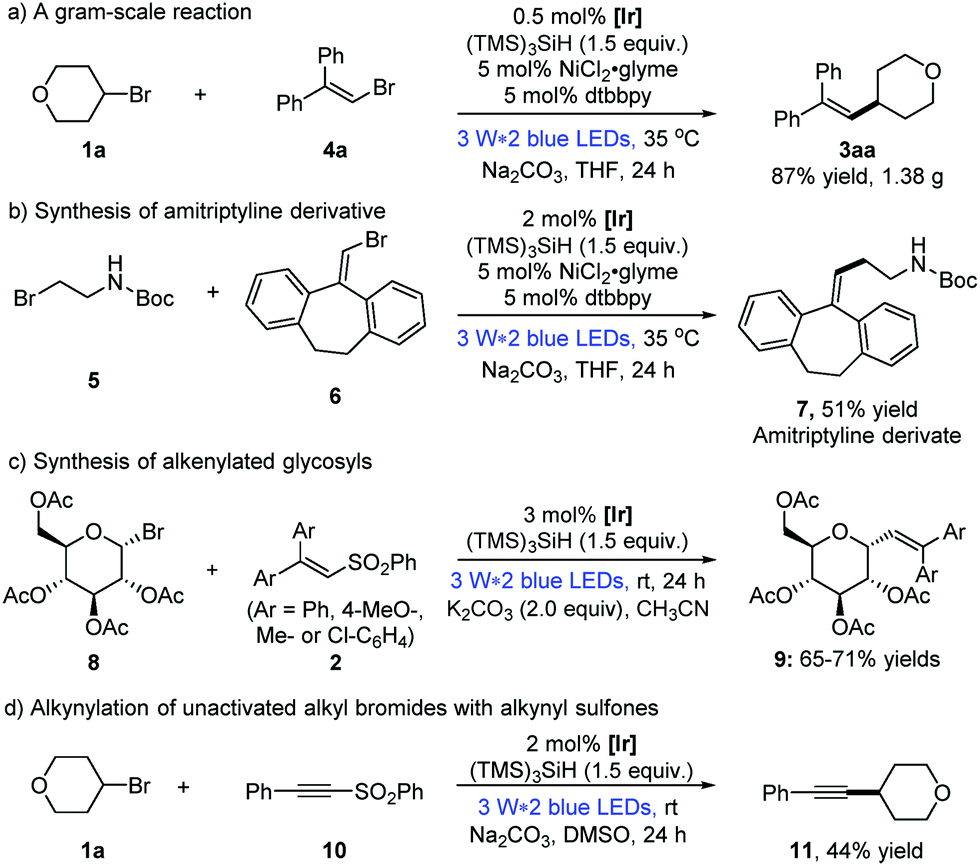 | ||
| Scheme 2 Demonstration of the synthetic utility of the methodology. | ||
As shown in eqn (1), the formation of 3aa was nearly completely inhibited by the addition of the radical scavenger TEMPO. Yet, we were able to detect the trapping product of the free alkyl radical.13 To further verify the presence of free alkyl radicals, a “radical-clock” experiment was performed with bromide 1y (eqn (2)); as a result, an 1,5-diene product 3ya was isolated in 52% yield via a radical-induced ring opening process.
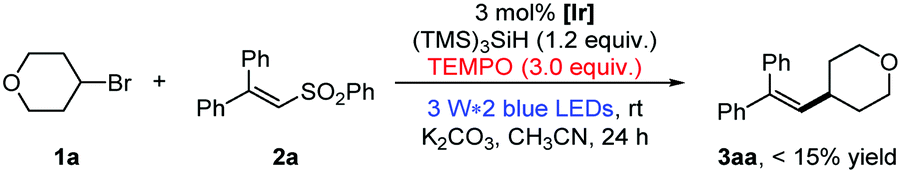 | (1) |
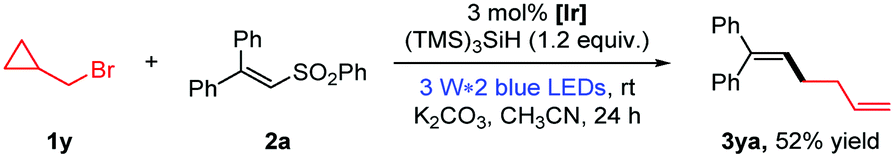 | (2) |
Based on these observation and related literature reports,8b,12 we proposed a possible mechanism for the coupling of unactivated alkyl bromides and vinyl phenyl sulfones (Scheme 3).18 Upon stirring a mixture of bromide 1a with a carbonate base for 6 h, small amounts of the unsaturated compound 12 were detected.18 The generated Br− can be oxidized by a photoredox catalyst, whereas the formed bromine radical can abstract a hydrogen atom from (TMS)3SiH.12e The generated silyl radical will abstract a bromine atom from 1a, and thereby the created alkyl radical is added to the double bond of 2a. A consecutive cleavage of the C–S bond will release the desired product and an open shell phenyl sulfonyl radical. Finally, the phenyl sulfonyl radical then undergoes a SET with the reduced photo-catalyst to close the catalytic cycle. It has to be noted that only trace amounts of the product were formed without the addition of a base. However, product formation was observed when LiBr was added as a bromide source instead of the carbonate base. Moreover, the yield could be further increased a little by supplemental addition of LiBr, demonstrating the necessity for the presence of bromide species for the photocatalytic cycle.18
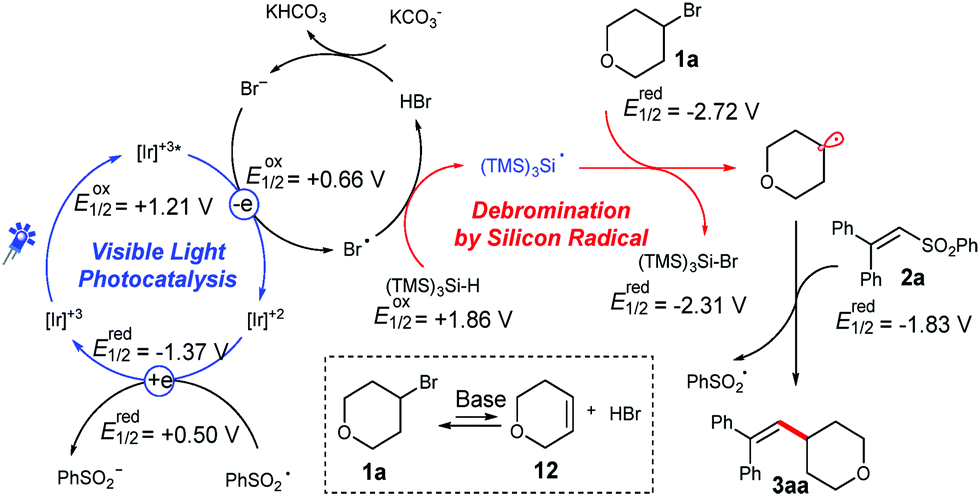 | ||
| Scheme 3 Proposed mechanism for the photocatalytic alkenylation of alkyl bromides with vinyl phenyl sulfones. | ||
In summary, we developed two photocatalytic alkenylation reactions of unactivated alkyl bromides with vinyl phenyl sulfones or vinyl bromides. The combination of visible light photocatalysis and silicon radical debromination was the key of this success. Moreover, this strategy was proven to be feasible for the alkenylation of bioactive molecules and glycosyl bromides, as well as the alkynylation of unactivated alkyl bromides.
We are grateful to the National Science Foundation of China (No. 21822103, 21820102003, 21772052, 21772053, 21572074 and 21472057), the Foundation for the Author of National Excellent Doctoral Dissertation of PR China (No. 201422), the Natural Science Foundation of Hubei Province (2017AHB047) and the German Science Foundation (DFG, GRK 1626, Chemical Photocatalysis) for support of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, NJ, 2013 Search PubMed; (b) R. Álvarez, B. Vaz, H. Gronemeyer and A. R. de Lera, Chem. Rev., 2014, 114, 1 CrossRef PubMed; (c) M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. Van Otterlo, Chem. Rev., 2015, 115, 5462 CrossRef CAS; (d) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS.
- (a) B. E. Maryanoff and A. B. Reitz, Chem. Soc. Rev., 1989, 89, 863 CrossRef CAS; (b) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670 RSC.
- (a) H.-T. Chang, T. T. Jayanth, C.-C. Wang and C.-H. Cheng, J. Am. Chem. Soc., 2007, 129, 12032 CrossRef CAS; (b) C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932 CrossRef CAS.
- (a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS; (c) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514 CrossRef CAS; (d) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068 CrossRef CAS; (e) L. Firmansjah and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 11340 CrossRef CAS; (f) A. C. Bissember, A. Levina and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 14232 CrossRef CAS; (g) Y. Zou and J. Zhou, Chem. Commun., 2014, 50, 3725 RSC; (h) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS.
- (a) E. Negishi, X. Zeng, Z. Tan, M. Qian, Q. Hu and Z. Huang, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH Verlag GmbH, Weinheim, 2008; 815 Search PubMed; (b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 174 CrossRef; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (d) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC; (e) K. Zhu, J. Dunne, M. P. Shaver and S. P. Thomas, ACS Catal., 2017, 7, 2353 CrossRef CAS; (f) S. J. Meek, R. V. O’Brien, J. Liaveria and R. R. Schrock, Nature, 2011, 471, 461 CrossRef CAS; (g) T. Di Franco, A. Epenoy and X. Hu, Org. Lett., 2015, 17, 4910 CrossRef CAS; (h) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111 CrossRef CAS.
- (a) P. Knochel Handbook of Functionalized Organometallics: Applications in Synthesis, Wiley-VCH, Weinheim, 2005 CrossRef; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS; (c) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892 RSC.
- For selected reviews on visible light photocatalysis, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS; (d) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (e) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS; (f) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS; (g) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef; (h) L. Marzo, S. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed.
- For selected works on photocatalytic alkenylation reactions, see: (a) S. F. Wnuk, P. I. Garcia and Z. Wang, Org. Lett., 2004, 6, 2047 CrossRef CAS PubMed; (b) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef CAS PubMed; (c) V. Corce, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414 CrossRef CAS; (d) H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881 CrossRef CAS; (e) J. Li, J. Zhang, H. Tan and D. Z. Wang, Org. Lett., 2015, 17, 2522 CrossRef CAS; (f) J. Xie, J. Li, V. Weingand, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2016, 22, 12646 CrossRef CAS; (g) K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719 RSC; (h) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS; (i) S. Paul and J. Guin, Green Chem., 2017, 19, 2530 RSC; (j) G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888 CrossRef CAS.
- For selected examples on photocatalytic transformations of alkyl fluoroborate salts, see: (a) D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308 CrossRef CAS; (b) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS; (c) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (d) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS; (e) M. El Khatib, R. A. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS.
- For selected reviews on coupling reactions of alkyl halides, see: (a) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS; (b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS; (c) X. Hu, Chem. Sci., 2011, 2, 1867 RSC.
- (a) J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS; (b) D. Fernandez Reina, A. Ruffoni, Y. S. S. Al-Faiyz, J. J. Douglas, N. S. Sheikh and D. Leonori, ACS Catal., 2017, 7, 4126 CrossRef CAS; (c) Y. Shen, J. Cornella, F. Julia-Hernandez and R. Martin, ACS Catal., 2017, 7, 409 CrossRef CAS; (d) S. Sumino, M. Uno, H.-J. Huang, Y.-K. Wu and I. Ryu, Org. Lett., 2018, 20, 1078 CrossRef CAS PubMed; (e) D. Kurandina, M. Rivas, M. Radzhabov and V. Gevorgyan, Org. Lett., 2018, 20, 357 CrossRef CAS.
- (a) C. Chatgilialoglu, A. Alberti, M. Ballestri, D. Macciantelli and D. P. Curran, Tetrahedron Lett., 1996, 37, 6391 CrossRef CAS; (b) C. Chatgilialoglu, Chem. – Eur. J., 2008, 14, 2310 CrossRef CAS; (c) G. Rouquet, F. Robert, R. Mereau, F. Castet and Y. Landais, Chem. – Eur. J., 2011, 17, 13904 CrossRef CAS; (d) J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962 CrossRef CAS; (e) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (f) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010 CrossRef CAS PubMed.
- Please see the ESI† for details.
- Crystallographic data of 3xa CCDC 1879873†.
- For selected reviews and examples of photoredox/nickel dual catalytic transformations, see: (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS; (c) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS; (d) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed.
- (a) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS PubMed; (b) H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371 CrossRef CAS PubMed.
- (a) Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281 CrossRef CAS PubMed; (b) R. S. Andrews, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef CAS PubMed; (c) S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6610 CrossRef CAS PubMed.
- See the ESI† for more details: proposed mechanistic cycle of method B, control experiments, the measurements of the quantum yield and the measurements of the potential of the compounds are shown in Scheme 3.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data. CCDC 1879873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc08362b |
| ‡ Q.-Q. Zhou and S. J. S. Düsel contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |