
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot, modular approach to functionalized ketones via nucleophilic addition/Buchwald–Hartwig amination strategy†
Jorn
de Jong‡
 ,
Dorus
Heijnen‡
,
Hugo
Helbert‡
and
Ben L.
Feringa
*
,
Dorus
Heijnen‡
,
Hugo
Helbert‡
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 18th February 2019
Abstract
A general one-pot procedure for the 1,2-addition of organolithium reagents to amides followed by the Buchwald–Hartwig amination with in situ released lithium amides is presented. In this work amides are used as masked ketones, revealed by the addition of organolithium reagents which generates a lithium amide, suitable for subsequent Buchwald–Hartwig coupling in the presence of a palladium catalyst. This methodology allows for rapid, efficient and atom economic synthesis of aminoarylketones in good yields.
The palladium catalysed amination of aromatic bromides was discovered in 1983 and has since proven to be one of the most important and versatile transformations in organic synthesis. Pioneered by Migita1 and co-workers, the groups of Buchwald and Hartwig among others developed the method towards its current status of privileged transformation in organic chemistry,2–6 with widespread applications in areas ranging from materials science to medicinal chemistry.6,7 Traditionally cross-coupling reactions to form a C–N bond require the use of additional base to achieve a full catalytic cycle. Stoichiometric amounts of tert-butanolate salts or cesium carbonate are often used for this purpose, though the use of lithium amide as base has also been reported to be a valuable substitute in Buchwald–Hartwig couplings.8 Alternatively, LiHMDS is chosen for its non-nucleophilic properties. Few reports describe aminations where lithium amides are directly playing the role of coupling partners.9,10 Our group recently disclosed a one pot procedure for the 1,2-addition of organolithium reagents to (Weinreb) amides, followed by alpha-arylation, which required no additional base11,12 (Scheme 1a). Optimizing the alpha-arylation step, the product arising from the competing Buchwald–Hartwig coupling was sometimes observed as a minor side product. We envisioned this amination process to be a very useful transformation. In the present study we take advantage of the nucleophilic properties of organolithium reagents by performing a 1,2-addition to an amide yielding a masked ketone.13–16 Collapse of the tetrahedral intermediate generates in situ a lithium amide17 allowing direct Buchwald–Hartwig amination without the need for additional base (Scheme 1b). This approach gives rapid access to a variety of amino substituted benzophenones which are widely used, among others, in cosmetics due to their UV protecting properties. Furthermore they are being employed as photoinitiator and have found use in medicinal chemistry, for instance as kinase inhibitors.18
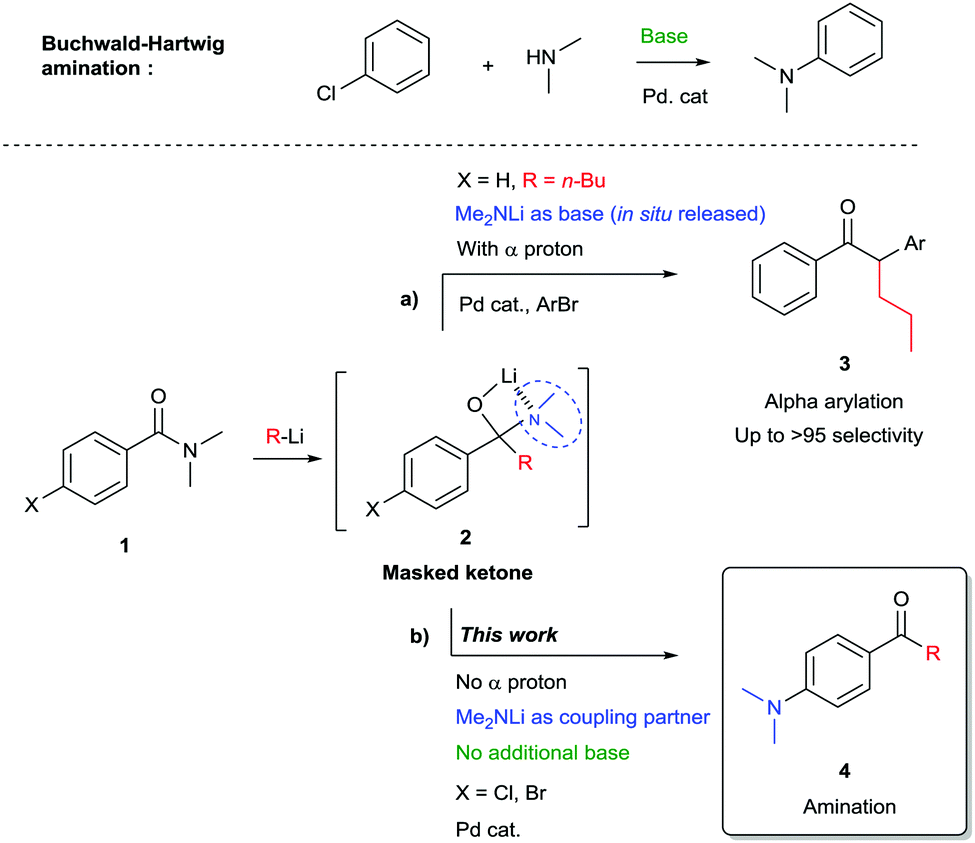 | ||
| Scheme 1 Lithium hemiaminal as a versatile building block. | ||
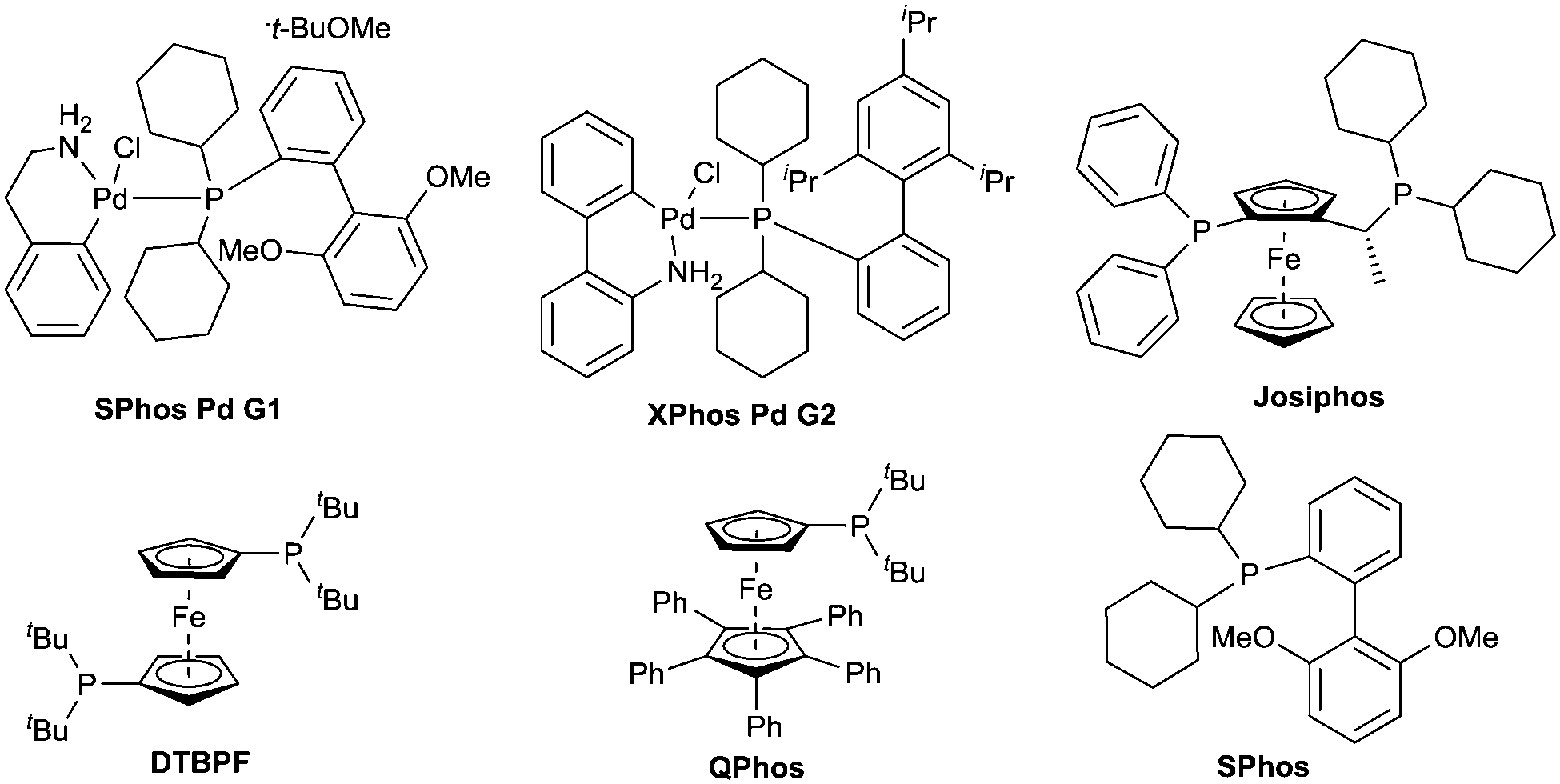
The competition between alpha-deprotonation or transmetallation to palladium could be avoided by the use of organolithium reagents lacking alpha hydrogen atoms. Therefore the lithium amide resulting from the 1,2-addition of an organolithium reagent to amide 1 – after dissociation of the tetrahedral intermediate 2 – is only available for Buchwald–Hartwig amination type reaction. In order to determine the optimal conditions for the reaction a ligand screening was performed and the results are shown in Table 1. XPhos palladacycle G2 (entry 2) proved to be the most efficient catalyst in toluene at 80 °C. Other well established catalysts for Buchwald–Hartwig amination based on mono and bidentate phosphine ligands19,20 (entries 3–6) or the use of palladacycle G1 (entry 1) were found to be less or not active in our case.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent (temp.) | Conversionb |
| a Reactions were carried on a 0.2 mmol scale, organolithium reagent was added dropwise and the mixture was stirred 15 min at 0 °C followed by the addition of Pd catalyst (5% loading) and stirred for additional 1 h at the indicated temperature. b Conversion determined by GC-MS. CPME = cyclopentyl methyl ether. | |||
| 1 | SPhos Pd G1 | Toluene (80 °C) | 42 |
| 2 | XPhos Pd G2 | Toluene (80 °C) | 83 |
| 3 | Pd2(dba)3/Josiphos | Toluene (80 °C) | 29 |
| 4 | Pd2(dba)3/DTBPF | Toluene (80 °C) | 0 |
| 5 | Pd2(dba)3/QPhos | Toluene (80 °C) | 49 |
| 6 | Pd2(dba)3/SPhos | Toluene (80 °C) | 69 |
| 7 | XPhos Pd G2 | THF (reflux) | 95 |
| 8 | SPhos Pd G1 | THF (reflux) | 94 |
| 9 | Pd2(dba)3/SPhos | THF (reflux) | 95 |
| 10 | PEPPSI-iPr | THF (reflux) | 87 |
| 11 | Pd2(dba)3/SPhos | 2-MeTHF (60 °C) | 62 |
| 12 | Pd2(dba)3/SPhos | 1,4-Dioxane (60 °C) | 93 |
| 13 | Pd2(dba)3/SPhos | MTBE (reflux) | 84 |
| 14 | Pd2(dba)3/SPhos | Hexane (60 °C) | 0 |
| 15 | Pd2(dba)3/SPhos | Et2O (reflux) | <5 |
| 16 | Pd2(dba)3/SPhos | CPME | 95 |
|
|||

Conversion towards the desired product was increased with the catalyst XPhos Pd G2 in THF at reflux (entry 7). Using Pd-PEPPSI21,22 (entry 10) in this solvent didn’t improve the conversion, but Pd2(dba)3/SPhos (entry 9) and SPhos palladacycle G1 (entry 8) gave comparable conversion. Pd2(dba)3/SPhos was preferred to palladacycles (G1, G2) as it is a cheaper catalyst. The influence of solvents was also tested, identifying THF at reflux (entry 9) as the solvent of choice for this reaction. Ethereal solvents show good conversion (entries 11–13) whilst the reaction doesn’t proceed in hexane (entry 14). Low conversion was observed when the reaction was performed in Et2O at reflux (entry 15) indicating that elevated temperature was required for the coupling step. 1,4-Dioxane (entry 12) also proved to be efficient and can be considered as an alternative if higher temperatures are required. Finally 2-MeTHF and cyclopentyl methyl ether (CPME)23 were tested. These solvents are reported as environmental friendly alternatives to THF. Whereas 2-MeTHF gave lower conversion (entry 11), CPME proved to be a valuable substitute (entry 16).
4-Bromo-N,N-dimethylbenzamide was also a suitable substrate for this reaction but aryl chlorides were preferred as they generate less waste, are generally cheaper and more widely available. Moreover by using aryl chlorides instead of aryl bromides we drastically decrease the risk of having undesired lithium–halogen exchange taking place.24
With the optimal conditions in hand (Table 1, entry 9), a range of amides was tested leading to a variety of aminobenzophenones represented in Scheme 2. The scope includes both meta (5a) and para chlorobenzamides (5b–5j). Cyclic (5c–5g) as well as acyclic (5a, 5b) amines were coupled in excellent yields. Interestingly, morpholine (5f), methylpiperazine (5g), indolines (5j–5l) and tetrahydroisoquinoline (5m) moieties were also successfully coupled in good yields providing a motif widely used in medicinal chemistry.25,26 Notably methylbenzylamine and dibenzylamine were also efficient coupling partners affording protected amines (5h, 5i) that can be further converted, after debenzylation, into free amines or mono-methylated amines. Methodologies to directly synthesize free anilines have been reported27–29 but they usually require large excess of ammonia, high pressure or long reaction time with the problem of selectivity for the primary amine versus secondary and tertiary amines. The methodology presented here can be considered as an attractive alternative to target primary and secondary arylamines.
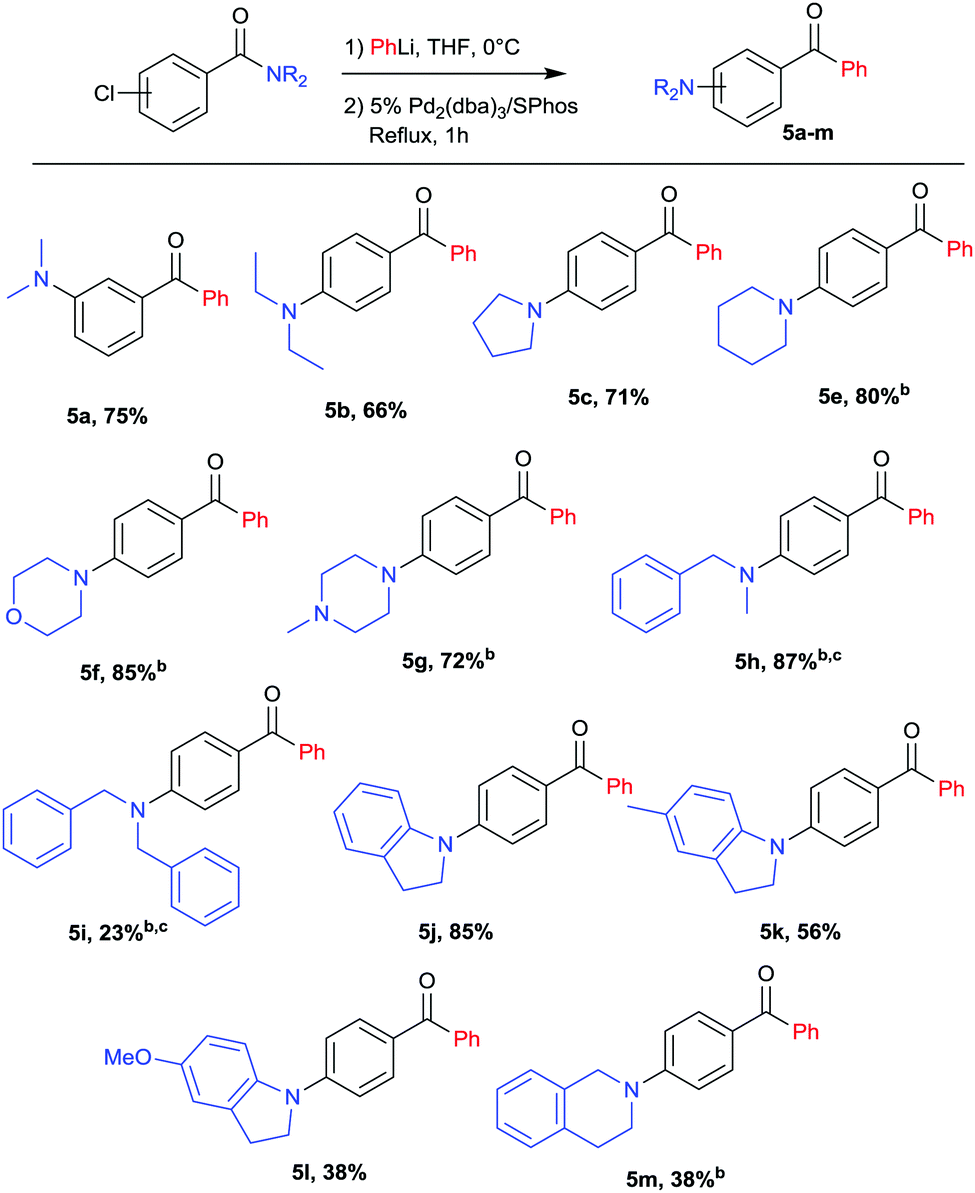 | ||
Scheme 2 Variation of amides.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yields refer to isolated yields after column chromatography, for an overview of unsuccessful attempts see ESI.†bPhLi added dropwise and reaction mixture stirred for 1 h before addition of the catalyst. cAfter addition of the catalyst the reaction mixture was heated at reflux overnight. Yields refer to isolated yields after column chromatography, for an overview of unsuccessful attempts see ESI.†bPhLi added dropwise and reaction mixture stirred for 1 h before addition of the catalyst. cAfter addition of the catalyst the reaction mixture was heated at reflux overnight. | ||
The scope of aryl lithium nucleophiles was investigated and the results are given in Scheme 3. Not only phenyllithium could be coupled (5n) but a diversity of aryllithium reagents, easily prepared via Li–halogen exchange or deprotonation, were found to be suitable reagents in this reaction. Aryllithium bearing ether, amine or electron withdrawing functionalities were successfully coupled (5o–5q). Heterocyclic lithium reagents such as 2-thienyllithium or 2-furyllithium were suitable coupling partners and provided the desired products 5r and 5s. Though lower yields were obtained when using more sterically hindered coupling partners the desired products were successfully isolated (5t, 5u).
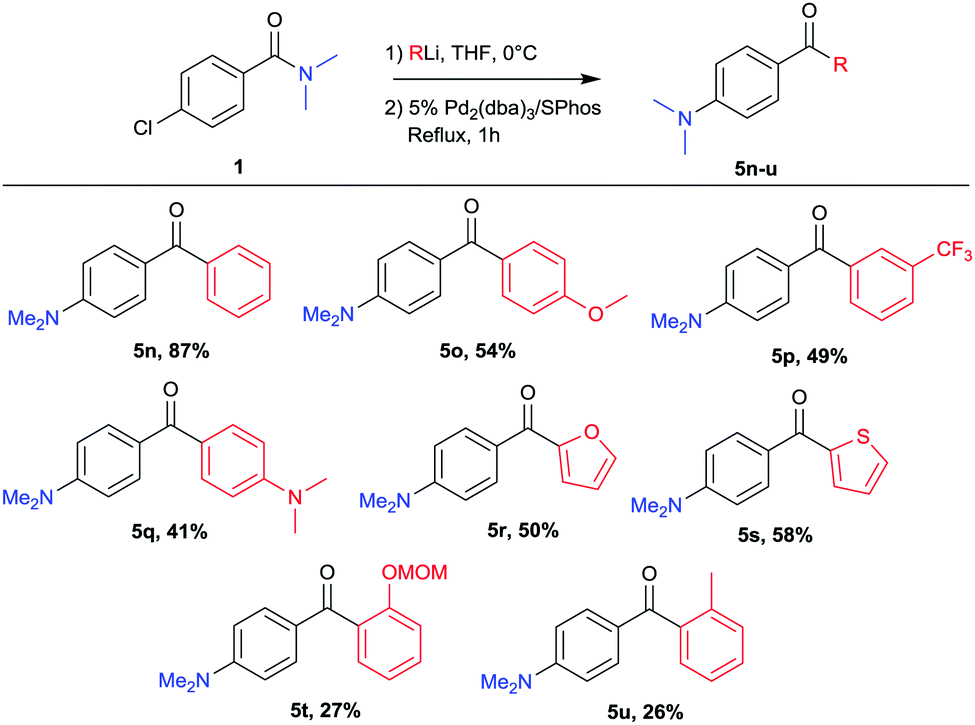 | ||
| Scheme 3 Variation of lithium reagents.a aYields refer to isolated yields after column chromatography. | ||
Small amounts of tertiary alcohol, arising from premature collapse of the tetrahedral intermediate were observed for most entries. It is the decreased reactivity in the second step however, that caused the lower yields for some of the substrates, providing (after hydrolysis) the chloro-ketone intermediate as a final product.
Previous reports already described the equilibrium between the intermediate 2 and products resulting from the 1,2-elimination of R2NLi being in favour of the tetrahedral intermediate 2.30,317Li and 1H-NMR analysis was performed to investigate the formation and stability of intermediate 2 under our reaction conditions (Fig. 1). As expected, when 1 eq. of PhLi (spectrum A-1) was added to (4-chlorophenyl)(morpholino)methanone 5f-StM, a single signal at 0.92 ppm was observed (spectrum A-2). This intermediate has a different chemical shifts compared to signals of lithium morpholin-4-ide at 0.87 ppm (spectrum A-5), indicating that the equilibrium is completely shifted towards the intermediate 2 (Scheme 1).
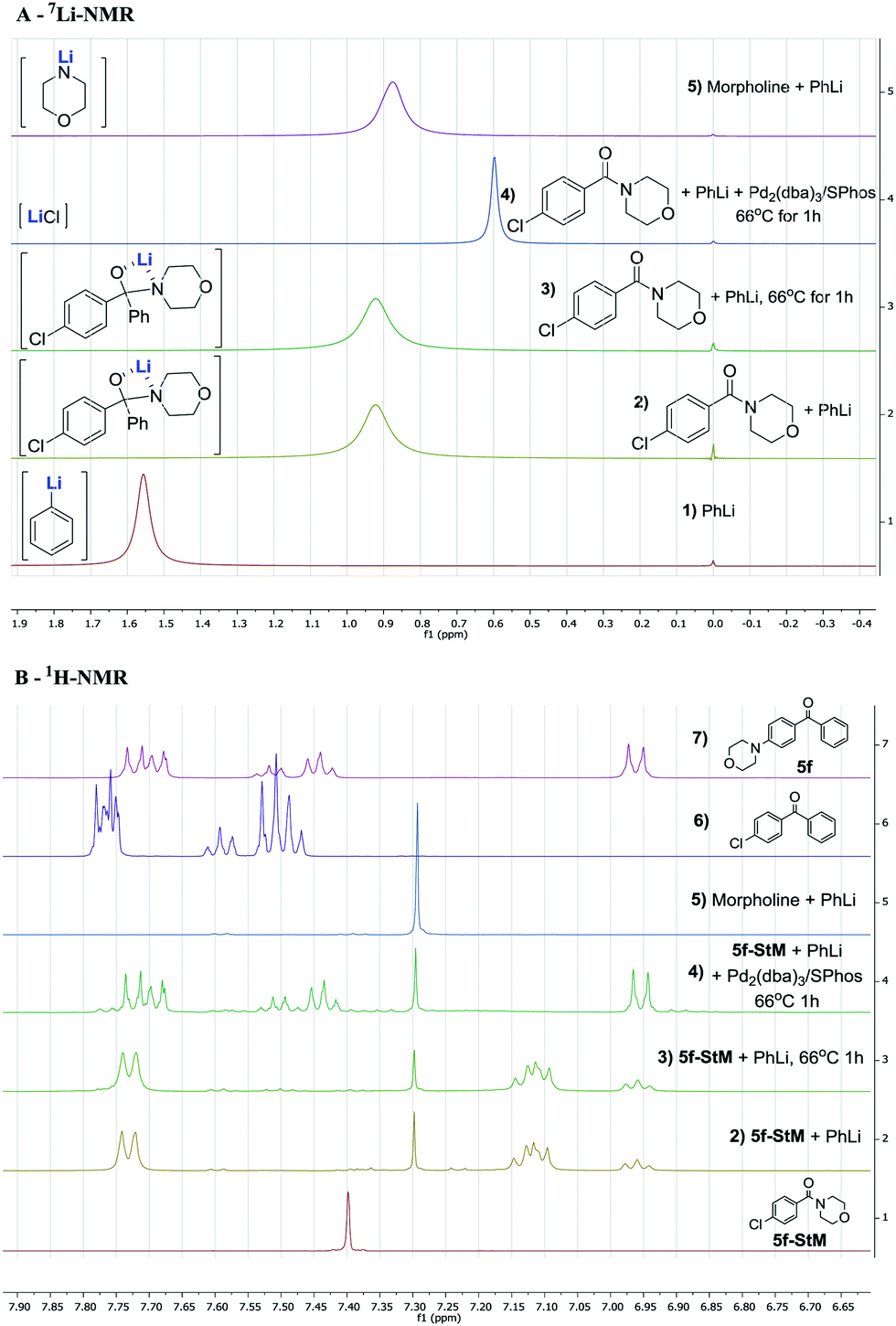 | ||
| Fig. 1 7Li (A) and 1H (B)-NMR analysis of the reaction with (4-chlorophenyl)(morpholino)methanone. Conditions: 0.1 mmol in 0.7 mL of THF-d8 at 25 °C with LiCl in D2O as internal standard. Hypothesized structure of lithium species between brackets. | ||
This hypothesis is also supported by 1H-NMR (full spectrum available in the ESI†) were no lithium morpholin-4-ide (B-5) nor 4-chlorobenzophenone (B-6) can be detected. The in situ formed intermediate (B-2) was then heated at reflux for 1 h but remain unaffected (B-3) and no product of 1,2-elimination was observed (comparison with B-6), showing the stability of this intermediate under our reaction conditions. When Pd2(dba)3/SPhos was added to the intermediate and the solution was heated at reflux for 1 h, the desired product 5f was selectively formed as confirmed by 1H-NMR (spectrum B-4 and B-7), resulting also in a sharp signal in 7Li-NMR at 0.60 ppm, corresponding to the LiCl (spectrum A-4) released. These experiments point toward the existence of a stable tetrahedral lithium hemiaminal intermediate 2 and a slow in situ release of lithium amide which is directly consumed in the amination reaction.
Finally, the versatility of this approach was demonstrated with the rapid and efficient synthesis of an isoform-specific phosphoinositide 3-kinase inhibitor.32 The herein described protocol drastically improves the yield compare to the previously reported synthesis and combines the formation of the C–N bond with a 1,2-addition in a one-pot procedure, leading to ketone 7 (a comparison of the two synthetic routes is available in the ESI†). The environmental friendly character of this transformation should also be highlighted as it exhibits a high reaction mass efficiency33 (RME) of 79% and produces only a light waste product LiCl with low toxicity (Scheme 4).
 | ||
| Scheme 4 Application in the synthesis of a kinase inhibitor (AMA37). | ||
In conclusion, a one-pot procedure toward the synthesis of amine aryl ketones has been developed, enabling Buchwald–Hartwig amination with in situ generated lithium amides. Starting from easily accessible amides and organolithium reagents, this strategy allows for fast and efficient modular functionalization with high atom economy generating only LiCl as stoichiometric waste. A diversity of aryllithium nucleophiles and amide coupling partners has been combined following this strategy and the applicability of the methodology was illustrated with the efficient synthesis of a selective kinase inhibitor.
This work was supported financially by the European Research Council (Advanced Investigator Grant, No. 227897 to B. L. F.); The Netherlands Organization for Scientific Research (NOW-CW); funding from the Ministry of Education, Culture, and Science (Gravitation program 024.001.035); The Royal Netherlands Academy of Arts and Sciences (KNAW); and NRSC-Catalysis are gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Kosugi, M. Kameyama and T. Migita, Chem. Lett., 1983, 927–928 CrossRef CAS.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed., 1995, 34, 1348–1350 CrossRef CAS.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914–15917 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- S. Meiries, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3402–3409 CrossRef CAS.
- H. Xu and C. Wolf, Chem. Commun., 2009, 3035–3037 RSC.
- X. Huang and S. L. Buchwald, Org. Lett., 2001, 3, 3417–3419 CrossRef CAS PubMed.
- A. T. Wolters, V. Hornillos, D. Heijnen, M. Giannerini and B. L. Feringa, ACS Catal., 2016, 6, 2622–2625 CrossRef CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron, 1981, 22, 3815–3818 CrossRef CAS.
- R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CrossRef CAS.
- M. Adler, S. Adler and G. Boche, J. Phys. Org. Chem., 2005, 18, 193–209 CrossRef CAS.
- C. Liu, M. Achtenhagen and M. Szostak, Org. Lett., 2016, 18, 2375–2378 CrossRef CAS PubMed.
- D. A. Evans, G. Borg and K. A. Scheidt, Angew. Chem., Int. Ed., 2002, 41, 3188–3191 CrossRef CAS PubMed.
- A. Salomone, F. M. Perna, F. C. Sassone, A. Falcicchio, J. Bezenšek, J. Svete, B. Stanovnik, S. Florio and V. Capriati, J. Org. Chem., 2013, 78, 11059–11065 CrossRef CAS PubMed.
- M. Andrs, J. Korabecny, D. Jun, Z. Hodny, J. Bartek and K. Kuca, J. Med. Chem., 2015, 58, 41–71 CrossRef CAS PubMed.
- N. Kataoka, Q. Shelby, J. P. Stambuli and J. F. Hartwig, J. Org. Chem., 2002, 67, 5553–5566 CrossRef CAS PubMed.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS PubMed.
- M. G. Organ, M. Abdel-Hadi, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O’Brien, M. Sayah and C. Valente, Chem. – Eur. J., 2008, 14, 2443–2452 CrossRef CAS PubMed.
- Y. Zhang, V. César and G. Lavigne, Eur. J. Org. Chem., 2015, 2042–2050 CrossRef CAS.
- U. Azzena, M. Carraro, L. Pisano, S. Monticelli, R. Bartolotta and V. Pace, ChemSusChem, 2019, 12, 40–70 CrossRef CAS PubMed.
- R. Luisi and V. Capriati, Lithium Compounds in Organic Synthesis, From Fundamentals to Applications. Edited by Renzo Luisi and Vito Capriati., Wiley-VCH, Weinheim., 2014, vol. 53 Search PubMed.
- M. Al-Ghorbani, A. Bushra Begum, Z. Zabiulla, S. V. Mamatha and S. A. Khanum, Res. J. Pharm. Technol., 2015, 8, 611–628 CrossRef.
- N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS PubMed.
- G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 CrossRef CAS PubMed.
- Y. Aubin, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Chem. Soc. Rev., 2010, 39, 4130 RSC.
- J. Schranck and A. Tlili, ACS Catal., 2018, 8, 405–418 CrossRef CAS.
- M. Adler, M. Marsch, N. S. Nudelman and G. Boche, Angew. Chem., Int. Ed., 1999, 38, 1261–1263 CrossRef CAS PubMed.
- D. L. Comins, Synlett, 1992, 615–625 CrossRef CAS.
- Z. A. Knight, G. G. Chiang, P. J. Alaimo, D. M. Kenski, C. B. Ho, K. Coan, R. T. Abraham and K. M. Shokat, Bioorg. Med. Chem., 2004, 12, 4749–4759 CrossRef CAS PubMed.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General, experimental procedures and characterisation of compounds. See DOI: 10.1039/c8cc08444k |
| ‡ J. de Jong, D. Heijnen and H. Helbert contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |