
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking the catalytic potential of tris(3,4,5-trifluorophenyl)borane with microwave irradiation†
Jamie L.
Carden
a,
Lukas J.
Gierlichs
a,
Duncan F.
Wass
 ab,
Duncan L.
Browne
a and
Rebecca L.
Melen
*a
ab,
Duncan L.
Browne
a and
Rebecca L.
Melen
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, Cymru/Wales CF10 3AT, UK. E-mail: MelenR@cardiff.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, England BS8 1TS, UK
First published on 4th December 2018
Abstract
The catalytic activity of tris(3,4,5-trifluorophenyl)borane has been explored in the 1,2-hydroboration reactions of unsaturated substrates. Under conventional conditions, the borane was found to be active only in the hydroboration of aldehyde, ketone and imine substrates, with alkenes and alkynes not being reduced effectively. The use of microwave irradiation on the other hand has permitted alkenes and alkynes to be hydroborated in good yields.
Catalytic hydroboration is a well-known, efficient method of producing borylated substrates with wide synthetic applicability.1–3 Classically, there has been focus on the use of precious metal catalysts for this transformation,4–6 but recently there has been a surge of interest in using more abundant, non-toxic, and cheaper alternatives such as early transition metals and main group elements.1,7–25 Recent work has shown that Lewis acidic boranes and borocations can act as efficient catalysts for this process (Fig. 1).11–14 For example, Oestreich explored the use of tris[3,5-bis(trifluoromethyl)phenyl]borane (BArF3), whilst we have focused on the use of tris(2,4,6-trifluorophenyl)borane (B(2,4,6-ArF)3),11,12 with both catalysts proving to be more active than the archetypal tris(pentafluorophenyl)borane (B(C6F5)3). The aforementioned borane catalysts work efficiently but are sometimes hindered by lengthy reaction times and limited substrate scope, particularly in the case of ketones, alkenes and alkynes.
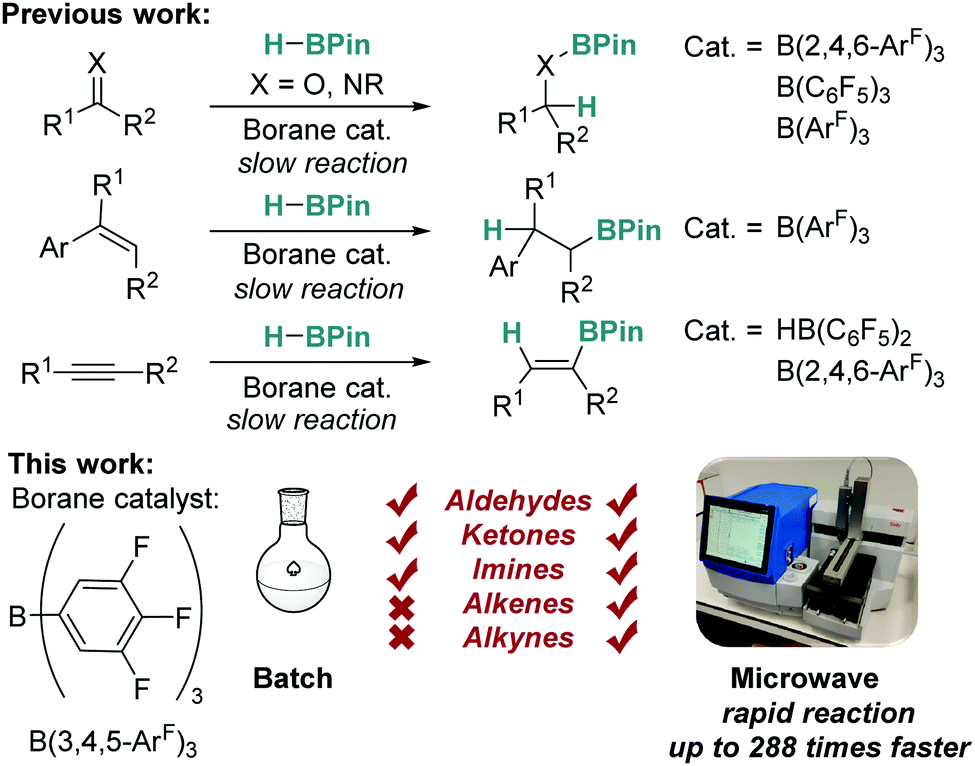 | ||
| Fig. 1 Previous work on borane catalysed hydroboration with conventional conditions (top) and this work, assisted with enabling technologies (bottom). | ||
One potential method to eliminate these problems is by employing microwave irradiation. The use of microwave assisted heating for synthesis and catalysis was first reported by Giguere and Gedye simultaneously in 1986.26,27 While early methods of microwave assisted synthesis involved the use of modified domestic microwave ovens, the field has expanded greatly to become an enabling technology for organic synthesis and catalysis.28,29 Microwave irradiation for heating offers many benefits compared to traditional heating methods, including the ability to heat reactions to higher temperatures and pressures in a safe manner, facilitating conventionally inaccessible chemistry, and allowing rapid reaction screening when combined with modern autosamplers.28,29 The use of microwave assisted synthesis is now commonplace in organic chemistry, for example in Suzuki cross-coupling reactions,30 but reports of microwave assisted catalytic hydroboration are rarer and are limited to transition metal catalysts.31–33 The application of microwaves in main group chemistry is confined to just a single example in which microwave irradiation was used for frustrated Lewis pair catalysed hydrogenation reactions.34 Microwave irradiation can enable chemists to access higher reaction temperatures, increased reaction yields, reduced reaction times and may provide access to reactions that cannot take place using traditional heating methods. Previous work in our research groups has focused on the use of enabling technologies such as flow chemistry to advance the field of main group catalysis.35
In this work, we seek to develop the scope of main group reactions that can be performed using microwave irradiation as an enabling technology (Fig. 1).
Initially we synthesised a range of Lewis acidic borane catalysts and screened these in the catalytic hydroboration of acetophenone using HBPin (1.1 equiv.) (Table 1). We were particularly interested in the tris(3,4,5-trifluorophenyl)borane (B(3,4,5-ArF)3) as a hydroboration catalyst. While a single stoichiometric reaction using this borane was reported, its catalytic properties remain underexplored.12 It was postulated that as this triaryl borane is devoid of ortho-fluorines, like BArF3, it would be an active catalyst for hydroboration reactions where B(C6F5)3 is not.12 This hypothesis was confirmed using 2 mol% catalyst loading of B(3,4,5-ArF)3, which gave quantitative conversion of acetophenone to the reduced boronate ester product after just one hour in CDCl3 at room temperature (entry 2, Table 1). Conversely, other boranes including BF3, BPh3, B(C6F5)3, and B(2,4,6-ArF)3 showed very poor conversions ranging from 3–27% after 24 h at room temperature (entries 3–6, Table 1). The reaction was also tolerant to both coordinating and non-coordinating solvents (entries 7 and 8, Table 1). However, for the convenience of in situ monitoring via1H NMR spectroscopy, we decided to continue with deuterated chloroform for the substrate screening. Using 1 equivalent of HBPin saw a reduced conversion as observed in other reports,11 and increasing the catalyst loading to 5 mol% did not significantly improve the rate of reaction (entries 9 and 10 respectively, Table 1). Other borylating agents such as 9-BBN showed no reactivity after 24 h (entry 11, Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Boron source (eq.) | Solvent | Time (h) | Conversiona (%) |
| Acetophenone (0.2 mmol, 24 mg).a Conversion determined by 1H NMR spectroscopy with mesitylene standard (0.1 mmol, 14 μL). | ||||||
| 1 | No catalyst | — | HBPin (1.1) | CDCl3 | 24 | 0 |
| 2 | B(3,4,5-ArF)3 | 2 | HBPin (1.1) | CDCl3 | 1 | >95 |
| 3 | BF3·Et2O | 2 | HBPin (1.1) | CDCl3 | 24 | 18 |
| 4 | BPh3 | 2 | HBPin (1.1) | CDCl3 | 24 | 27 |
| 5 | B(C6F5)3 | 2 | HBPin (1.1) | CDCl3 | 24 | 3 |
| 6 | B(2,4,6-ArF)3 | 2 | HBPin (1.1) | CDCl3 | 24 | 21 |
| 7 | B(3,4,5-ArF)3 | 2 | HBPin (1.1) | Et2O | 2 | >95 |
| 8 | B(3,4,5-ArF)3 | 2 | HBPin (1.1) | Toluene | 16 | >95 |
| 9 | B(3,4,5-ArF)3 | 2 | HBPin (1) | CDCl3 | 24 | 79 |
| 10 | B(3,4,5-ArF)3 | 5 | HBPin (1.1) | CDCl3 | 1 | >95 |
| 11 | B(3,4,5-ArF)3 | 2 | 9-BBN (1.1) | CDCl3 | 24 | 0 |
With the optimised conditions in hand, we moved our attention to the hydroboration of a range of aldehydes, ketones, and imines with HBPin to explore the aptitude of our borane catalyst at room temperature (Fig. 2). Hydrolysis of the boronate ester products (1) yielded the corresponding alcohols 2. Aldehydes with both electron donating and withdrawing groups worked well for this transformation giving the alcohols 2a–e in 87–98% isolated yield following hydrolysis in less than 24 h. Ketones also worked very well affording the secondary alcohols 2f–i in 89–96% isolated yield. Conversely, the more sterically demanding benzophenone substrate took 156 h to yield 2j in 84% yield. Aldimines were readily reduced to the amines (2k–s) typically giving quantitative conversions and 86–96% isolated yields in up to 60 h. The exception to this was in the synthesis of 2s which took 156 h to undergo complete hydroboration. B(3,4,5-ArF)3 could also catalyse the hydroboration of the ketimine N,1-diphenylethan-1-imine yielding the boronate ester 1t which was found to be stable to hydrolysis.
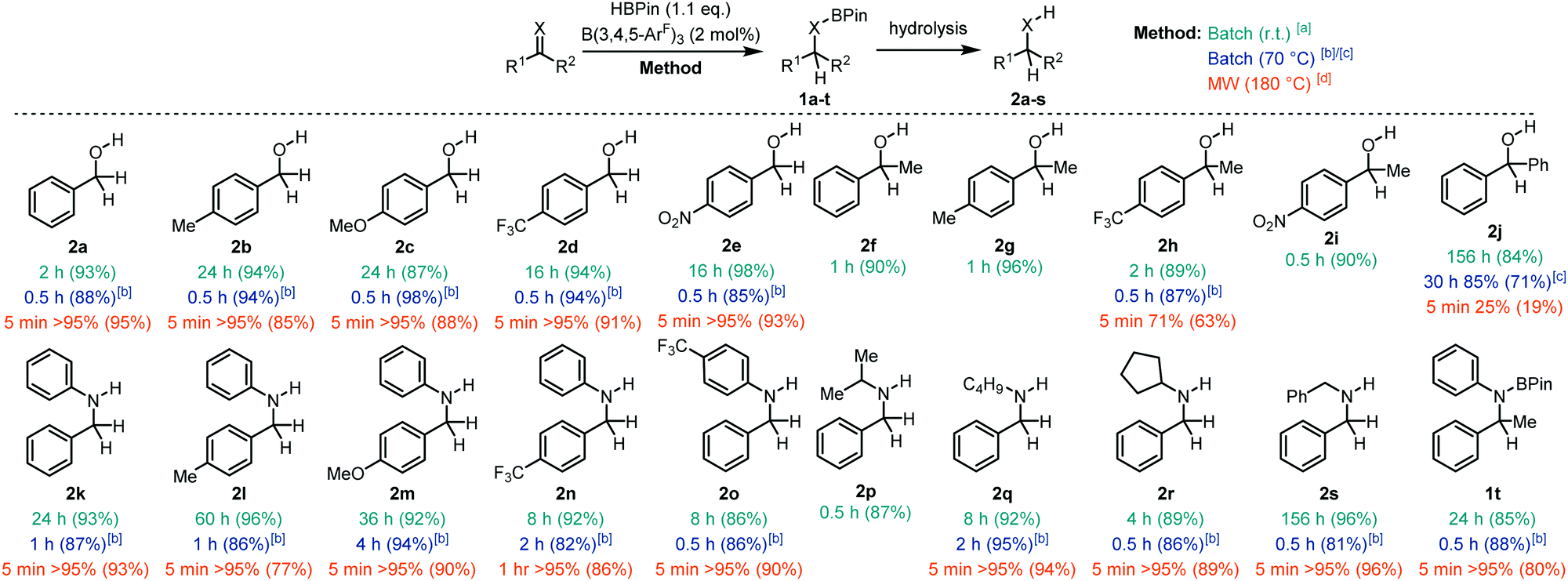 | ||
| Fig. 2 Hydroboration of aldehydes, ketones, and imines with HBPin using B(3,4,5-ArF)3. Conversions determined by 1H NMR spectroscopy. Isolated yields given in parentheses. a Time taken to reach quantitative conversion at room temperature. b Time taken to reach quantitative conversion at 70 °C. c Achieved maximum conversion of 85% at 70 °C and did not increase past this value. d Time taken and conversion in microwave at 180 °C. Isolated yields given in parentheses. | ||
While B(3,4,5-ArF)3 was found to be an efficient catalyst for a range of unsaturated substrates, sometimes long reaction times were required to reach quantitative conversion at room temperature.
In response to this, we decided to test the catalytic properties of B(3,4,5-ArF)3 at reflux in CDCl3. We focused our attention on substrates that proceeded slower (>1 h) at room temperature. All of the aldehydes tested showed quantitative hydroboration to 1a–e within 0.5 h at 70 °C with isolated yields of 2a–e greater than 85% following hydrolysis. This demonstrates a significant reduction in reaction time from the 2–24 h reactions observed at room temperature. Ketone 1-(4-(trifluoromethyl) phenyl)ethan-1-one also showed quantitative conversion to 1h within 0.5 h giving the product 2h in 87% yield. Benzophenone also showed a reduction in reaction time from 156 h to 30 h, with 85% conversion to 2j. Secondary amines 2k–s could be synthesised quantitatively within 0.5–4 h, compared with reaction times up to 60 h at room temperature. Ketimines also worked well giving 1t in 80% isolated yields after 0.5 h compared to the 24 h reaction time needed at room temperature.
Having established that we have an active borane catalyst for the hydroboration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X (X = O, N) we then decided to determine if we could improve the reaction in terms of (i) conditions and (ii) substrate scope using microwave irradiation. With the capability of a Biotage® microwave reactor, we were able to heat the reaction samples safely to 180 °C to accelerate the hydroboration reactions. Although chloroform is an uncommon solvent for microwave assisted reactions due to its low loss factor and dielectric constant,29 it was chosen as the reaction medium to negate any possible solvent influence on catalysis and to allow a direct comparison to traditional heating methods. At this juncture, it was important to quench the reaction immediately after the heating process had ended to ensure that the conversion reflected the microwave reaction only, and not a combination of the conversion in the microwave and a continuing process in the reaction vessel after microwave irradiation had ceased. Therefore, we performed a basic workup immediately after microwave irradiation had ended to remove the catalyst and to hydrolyse the boronate ester to give the corresponding alcohol or amine. The majority of substrates showed much improved reactivity with microwave irradiation, giving full conversion (>95%) within 5 minutes. These slower conversions to 2h and 2n can be attributed to the more electron deficient ketone or imine substrates which gave a slightly lower conversion of 71% after 5 min (2h) and 95% after 1 h (2n). The increased steric encumberment of benzophenone resulted in 25% conversion after 5 min. Attempts to improve the yield by increasing the reaction time further to 0.5 h or 1 h showed a small increase in yield to 38% and 45% respectively.
X (X = O, N) we then decided to determine if we could improve the reaction in terms of (i) conditions and (ii) substrate scope using microwave irradiation. With the capability of a Biotage® microwave reactor, we were able to heat the reaction samples safely to 180 °C to accelerate the hydroboration reactions. Although chloroform is an uncommon solvent for microwave assisted reactions due to its low loss factor and dielectric constant,29 it was chosen as the reaction medium to negate any possible solvent influence on catalysis and to allow a direct comparison to traditional heating methods. At this juncture, it was important to quench the reaction immediately after the heating process had ended to ensure that the conversion reflected the microwave reaction only, and not a combination of the conversion in the microwave and a continuing process in the reaction vessel after microwave irradiation had ceased. Therefore, we performed a basic workup immediately after microwave irradiation had ended to remove the catalyst and to hydrolyse the boronate ester to give the corresponding alcohol or amine. The majority of substrates showed much improved reactivity with microwave irradiation, giving full conversion (>95%) within 5 minutes. These slower conversions to 2h and 2n can be attributed to the more electron deficient ketone or imine substrates which gave a slightly lower conversion of 71% after 5 min (2h) and 95% after 1 h (2n). The increased steric encumberment of benzophenone resulted in 25% conversion after 5 min. Attempts to improve the yield by increasing the reaction time further to 0.5 h or 1 h showed a small increase in yield to 38% and 45% respectively.
After demonstrating that B(3,4,5-ArF)3 was not only an excellent catalyst for the hydroboration of heteronuclear CX (X = O, N) bonds both under conventional heating techniques and with microwave irradiation, we turned our attentions to more challenging substrates with homonuclear unsaturated bonds (CC and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C). Using phenylacetylene as our test substrate, we first explored the conventional hydroboration reaction catalysed by B(3,4,5-ArF)3. Using 2 mol% B(3,4,5-ArF)3, negligible conversion was observed after 24 h at 70 °C, and for 5 and 10 mol% B(3,4,5-ArF)3, it took 96 h for the reaction to reach 50% and quantitative conversion respectively (entries 1 and 2, Table 2). We then turned to microwave irradiation reactions to reduce the reaction time from days to something that would be considered more practical. The conditions for the microwave reaction were optimised for the hydroboration of phenylacetylene using 1.1 equivalents of HBPin (Table 2 and ESI†). The control reaction with no borane catalyst showed negligible conversion after 90 minutes (entry 3, Table 2). Adding 2 mol% of B(3,4,5-ArF)3 allowed the reaction to take place showing conversions of 47%, 63% and 71% after 20, 40 and 90 minutes respectively (entries 5–7, Table 2). Increasing the catalytic loading to 5 mol% saw much improved conversions of 77% after 20 minutes, 86% after 40 minutes and quantitative conversion was observed after 90 minutes (entries 8–10, Table 2). Importantly, identical reaction conditions using a Parr reactor vessel only yielded 40% conversion (entry 3, Table 2). For investigating the scope of the reaction, we chose to use a reaction time of 90 minutes and a 5 mol% catalyst loading. Using these conditions, hydroboration of both terminal and internal unsaturated homonuclear bonds was achievable (Fig. 3).
C). Using phenylacetylene as our test substrate, we first explored the conventional hydroboration reaction catalysed by B(3,4,5-ArF)3. Using 2 mol% B(3,4,5-ArF)3, negligible conversion was observed after 24 h at 70 °C, and for 5 and 10 mol% B(3,4,5-ArF)3, it took 96 h for the reaction to reach 50% and quantitative conversion respectively (entries 1 and 2, Table 2). We then turned to microwave irradiation reactions to reduce the reaction time from days to something that would be considered more practical. The conditions for the microwave reaction were optimised for the hydroboration of phenylacetylene using 1.1 equivalents of HBPin (Table 2 and ESI†). The control reaction with no borane catalyst showed negligible conversion after 90 minutes (entry 3, Table 2). Adding 2 mol% of B(3,4,5-ArF)3 allowed the reaction to take place showing conversions of 47%, 63% and 71% after 20, 40 and 90 minutes respectively (entries 5–7, Table 2). Increasing the catalytic loading to 5 mol% saw much improved conversions of 77% after 20 minutes, 86% after 40 minutes and quantitative conversion was observed after 90 minutes (entries 8–10, Table 2). Importantly, identical reaction conditions using a Parr reactor vessel only yielded 40% conversion (entry 3, Table 2). For investigating the scope of the reaction, we chose to use a reaction time of 90 minutes and a 5 mol% catalyst loading. Using these conditions, hydroboration of both terminal and internal unsaturated homonuclear bonds was achievable (Fig. 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Temperature (°C) | Time (min) | Conversiona (%) |
| Phenyl acetylene (0.4 mmol, 40.8 mg), HBPin (0.44 mmol, 63.8 μL), chloroform solvent (2 mL). Microwave reaction conditions −180 °C, 20 bar.a Conversion determined by 1H NMR spectroscopy.b Reaction completed in an oven heated Parr acid digestion vessel. | |||||
| 1 | B(3,4,5-ArF)3 | 5 | 70 (no MW) | 5760 (96 h) | 50 |
| 2 | B(3,4,5-ArF)3 | 10 | 70 (no MW) | 5760 (96 h) | >95 |
| 3 | B(3,4,5-ArF)3 | 5 | 180 (no MW)b | 90 | 40 |
| 4 | None | — | 180 | 90 | <5 |
| 5 | B(3,4,5-ArF)3 | 2 | 180 | 20 | 47 |
| 6 | B(3,4,5-ArF)3 | 2 | 180 | 40 | 63 |
| 7 | B(3,4,5-ArF)3 | 2 | 180 | 90 | 71 |
| 8 | B(3,4,5-ArF)3 | 5 | 180 | 20 | 77 |
| 9 | B(3,4,5-ArF)3 | 5 | 180 | 40 | 86 |
| 10 | B(3,4,5-ArF)3 | 5 | 180 | 90 | >95 |
| 11 | BH3·SMe2 | 5 | 180 | 90 | 11 |
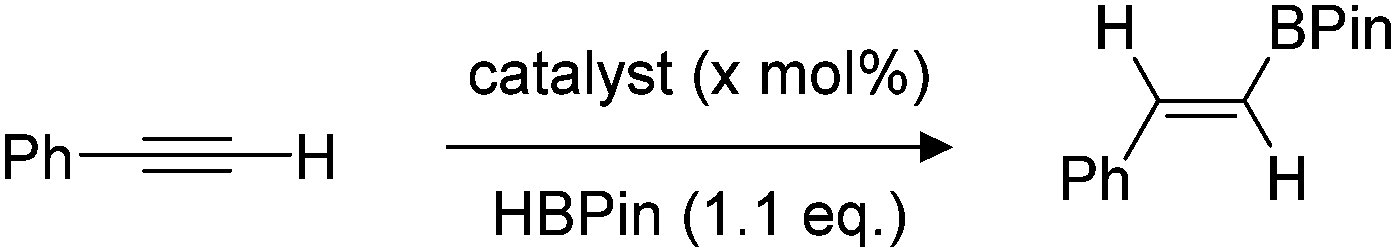
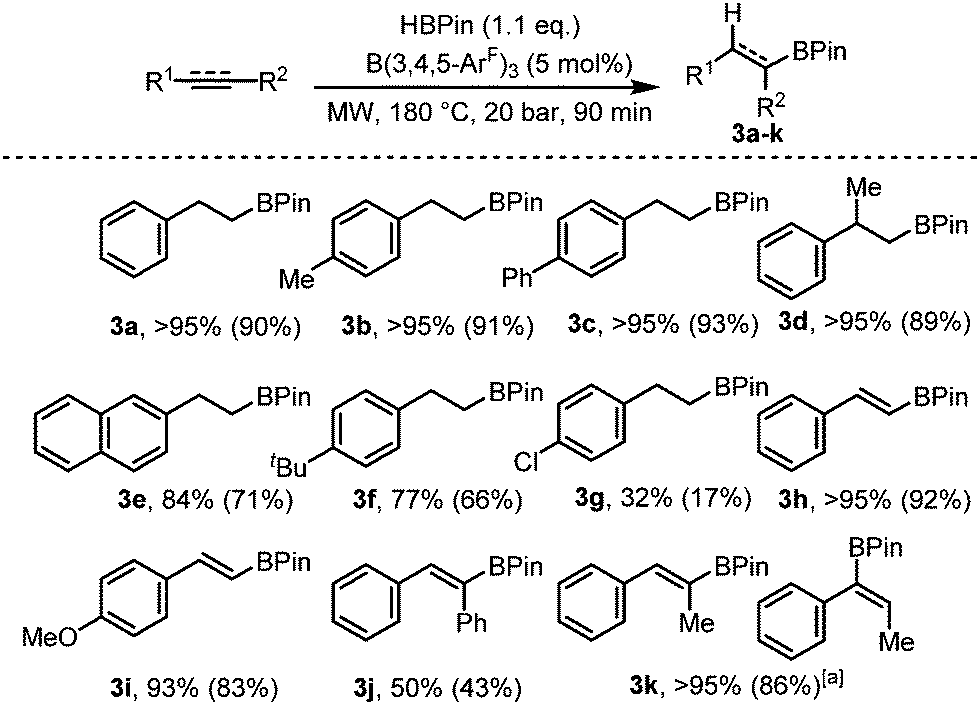 | ||
Fig. 3 Hydroboration of alkenes and alkynes. Conversions for 3a–k determined from 1H NMR spectroscopy, isolated yield in parentheses. a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 product ratio. :1 product ratio. | ||
Mono- and di-substituted terminal alkenes worked well in the microwave and gave the anti-Markovnikov alkane products 3a–d in quantitative yields. Other styrene derivatives were also isolated exclusively as the anti-Markovnikov product (3e–g), albeit with decreased yields. Terminal alkynes worked well yielding 3h and 3i in 92% and 83% respectively. Diphenylacetylene on the other hand resulted in just 50% conversion to 3j. The internal alkyne prop-1-yn-1-ylbenzene also worked well albeit producing an inseparable mixture of the Markovnikov and anti-Markovnikov products (3k). For the microwave reactions, it is likely that a new catalytic species HnB(3,4,5-ArF)3−n is generated in situ evidenced by the observation that BH3·SMe2 showed some conversion under the same conditions (Table 2, entry 11). Further studies to make and test the activity of HnB(3,4,5-ArF)3−n (n = 1, 2) are ongoing.
In conclusion we have found an efficient Lewis acidic borane catalyst for the hydroboration of a wide substrate scope, which is tolerant of a variety of functional groups. Notably we have shown that the hydroboration activity and the scope of this catalyst can be improved using microwave irradiation. Importantly, this approach permits ready access to higher temperatures and thus allows enhanced reactivity of substrates that were formerly recalcitrant under traditional approaches.
We would like to thank the EPSRC for funding (JLC, EP/L016443/1; RLM, EP/R026912/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS.
- C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS.
- P. Kaur, G. L. Khatik and S. K. Nayak, Curr. Org. Synth., 2017, 14, 665–682 CAS.
- J. Huang, W. Yan, C. Tan, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1770–1773 RSC.
- B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 14050–14054 CrossRef CAS PubMed.
- A. Edwards, M. Rubina and M. Rubin, Chem. – Eur. J., 2018, 24, 1394–1403 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS PubMed.
- M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed.
- N. Ang, C. Buettner, S. Docherty, A. Bismuto, J. Carney, J. Docherty, M. Cowley and S. Thomas, Synthesis, 2018, 803–808 CAS.
- A. Bismuto, M. J. Cowley and S. P. Thomas, ACS Catal., 2018, 2001–2005 CrossRef CAS.
- J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. – Eur. J., 2017, 23, 10997–11000 CrossRef CAS PubMed.
- Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381–2384 CrossRef CAS.
- Q. Yin, S. Kemper, H. F. T. Klare and M. Oestreich, Chem. – Eur. J., 2016, 22, 13840–13844 CrossRef CAS.
- M. Fleige, J. Möbus, T. vom Stein, F. Glorius and D. W. Stephan, Chem. Commun., 2016, 52, 10830–10833 RSC.
- D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2016, 22, 7158–7162 CrossRef CAS.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169–4175 RSC.
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841–844 CrossRef CAS.
- M. R. Adams, C.-H. Tien, R. McDonald and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660–16663 CrossRef CAS PubMed.
- D. M. C. Ould and R. L. Melen, Chem. – Eur. J., 2018, 24, 15201–15204 CrossRef CAS PubMed.
- A. Bismuto, S. P. Thomas and M. J. Cowley, Angew. Chem., Int. Ed., 2016, 55, 15356–15359 CrossRef CAS PubMed.
- J. S. McGough, S. M. Butler, I. A. Cade and M. J. Ingleson, Chem. Sci., 2016, 7, 3384–3389 RSC.
- M. A. Légaré, M. A. Courtemanche, É. Rochette and F. G. Fontaine, Science, 2015, 349, 513–516 CrossRef.
- R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Tetrahedron Lett., 1986, 27, 4945–4948 CrossRef CAS.
- R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett., 1986, 27, 279–282 CrossRef CAS.
- M. Larhed, C. Moberg and A. Hallberg, Acc. Chem. Res., 2002, 35, 717–727 CrossRef CAS.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2011, 40, 4925 RSC.
- S. W. Hadebe and R. S. Robinson, Tetrahedron Lett., 2006, 47, 1299–1302 CrossRef CAS.
- A. B. Dounay, L. E. Overman and A. D. Wrobleski, J. Am. Chem. Soc., 2005, 127, 10186–10187 CrossRef CAS PubMed.
- B. D. Stevens, C. J. Bungard and S. G. Nelson, J. Org. Chem., 2006, 71, 6397–6402 CrossRef CAS PubMed.
- S. Tussing and J. Paradies, Dalton Trans., 2016, 45, 6124–6128 RSC.
- L. C. Wilkins, J. L. Howard, S. Burger, L. Frentzel-Beyme, D. L. Browne and R. L. Melen, Adv. Synth. Catal., 2017, 359, 2580–2584 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc09459d |
| This journal is © The Royal Society of Chemistry 2019 |