
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the expression of chirality in a mechanically chiral rotaxane†
Charles E.
Gell
 ,
Timur A.
McArdle-Ismaguilov‡
and
Nicholas H.
Evans
*
,
Timur A.
McArdle-Ismaguilov‡
and
Nicholas H.
Evans
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 14th January 2019
Abstract
The expression of mechanical chirality by a hydrogen bond templated rotaxane, as detected by 1H NMR spectroscopy, may be modulated by affecting the co-conformational behaviour of the rotaxane through varying solvent or by addition of acid and base.
The synthesis and functional application of chiral rotaxanes and catenanes is a blossoming area of research within supramolecular chemistry.1–3 A mechanically interlocked molecule (MIM) may be made chiral by inclusion of a classical chiral element (e.g. a chiral centre, axis or plane).4–6 Alternatively, a MIM may be chiral by virtue of the mechanical bond, so-called mechanical chirality. Commonly encountered occurrences of mechanical chirality in MIMs are [2]catenanes consisting of two directional rings and [2]rotaxanes consisting of directional ring and axle components (Fig. 1a).7–9
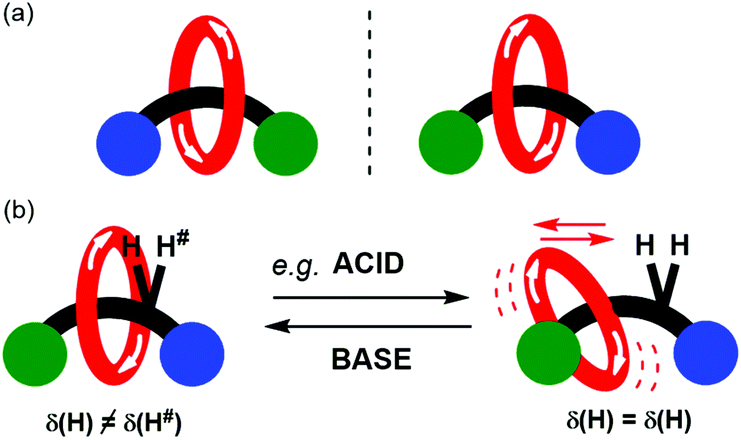 | ||
| Fig. 1 Schematic representations of: (a) the enantiomers of a mechanically chiral [2]rotaxane consisting of directional ring and axle; (b) stimuli controlled variation in co-conformational behaviour of a mechanically chiral [2]rotaxane, that leads to modulation in expression of chirality as detected by 1H NMR spectroscopy. | ||
Noteworthy recent achievements with chiral MIMs include the successful preparative scale preparation of enantiopure mechanically chiral rotaxanes without resorting to chiral HPLC for the separation of enantiomers;10,11 chiral MIMs being used as enantioselective receptors and asymmetric catalysts;12–19 and induction of single-handed helicity of a polymer by use of a mechanically chiral rotaxane as the source of chirality.20
MIMs are perhaps most well-known for the possibilities of controlled large amplitude motion of their interlocked components, through either change of environment, or application of a stimulus.21,22 Examples of such controlled motion in chiral MIMs are rare. Leigh and co-workers have reported upon rotaxanes where the expression of chirality (as detected by circular dichroism) was modulated by solvent and temperature23 and light.24 More recently the same group, disclosed a pH switchable rotaxane catalyst for controlled asymmetric Michael additions.25 Takata et al. have also demonstrated control of the handedness of a polyacetylene helix by use of a pendant chiral rotaxane switch.26 In all these rotaxanes, the chirality arose from inclusion of a classical chiral element.
Here we present the preparation of a novel mechanically chiral [2]rotaxane 1 whose detectable mechanical chirality may be modulated by solvent as monitored by 1H NMR spectroscopy. Further it also demonstrated that the expression of chirality may also be reversibly modulated by the addition of acid and then base (Fig. 1b). By dissolving the rotaxane in a hydrogen bond accepting solvent, or adding trifluoroacetic acid (TFA), the templating hydrogen bond interactions between the macrocycle and the amide of the axle are disrupted. The resulting changes in co-conformational behaviour are reported by variation in the appearance of a proton environment in the 1H NMR spectrum of the rotaxane.
Mechanically chiral [2]rotaxane 1 was prepared using our recently reported hydrogen bond templated strategy (Scheme 1).27 A novel rotationally asymmetric macrocycle 2 (for synthesis see page S3, ESI†) was dissolved in dichloromethane in the presence of 1.5 equivalents of azide 327 and 1.5 equivalents of alkyne 4.28 Catalytic Cu(CH3CN)4BF4 and TBTA and 1.6 equivalents of DIPEA were added and the reaction stirred overnight at room temperature under an inert atmosphere. After aqueous work-up and careful silica gel column chromatography, rotaxane 1 was isolated in 44% yield.
 | ||
| Scheme 1 Synthesis of mechanically chiral rotaxane 1 (only one enantiomer is shown for clarity). | ||
Rotaxane 1 was characterised by NMR and IR spectroscopy and mass spectrometry (see Fig. 2 and pages S18–S26, ESI†). Preliminary inspection of the 1H NMR spectrum (in CDCl3) reveals that the macrocyclic aromatic protons 10, 11, 19 and 20 are markedly downfield (6.10–6.75 ppm) of the typical aromatic region, consistent with interlocked structure formation (Fig. 2). A peak attributable to the molecular ion peak at m/z = 1037.3949 is also observed in the positive-ion electrospray mass spectra (see page S26, ESI†). An analytical sample of rotaxane 1 was submitted to chiral HPLC, with almost complete resolution of the two enantiomers achieved using a CHIRAL-PAK AD-H column (see Fig. 3).
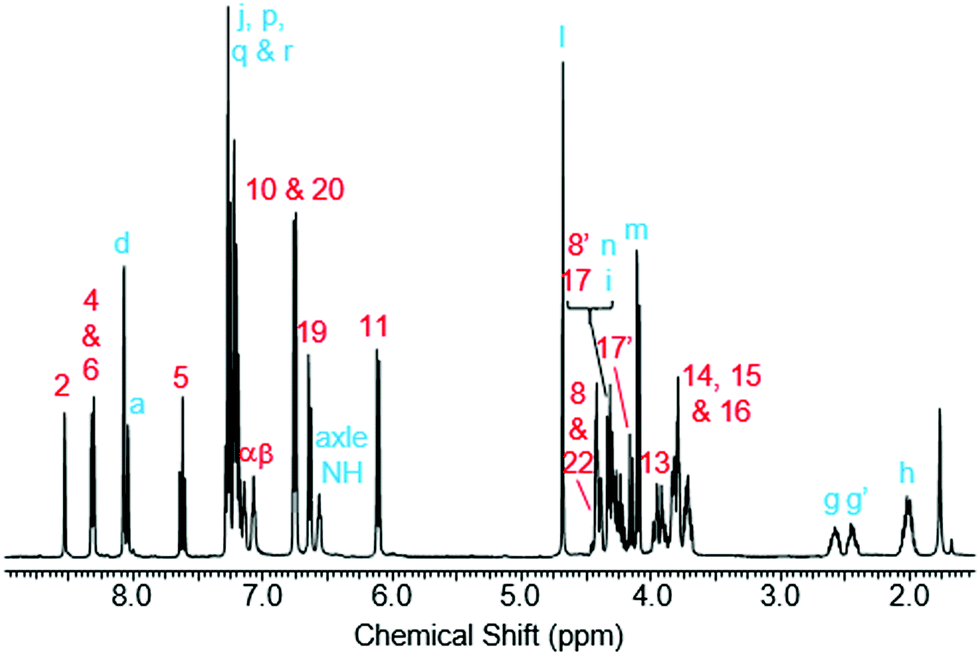 | ||
| Fig. 2 1H NMR spectrum of rotaxane 1 (CDCl3, 400 MHz, 298 K). Atom labels are defined in Scheme 1. | ||
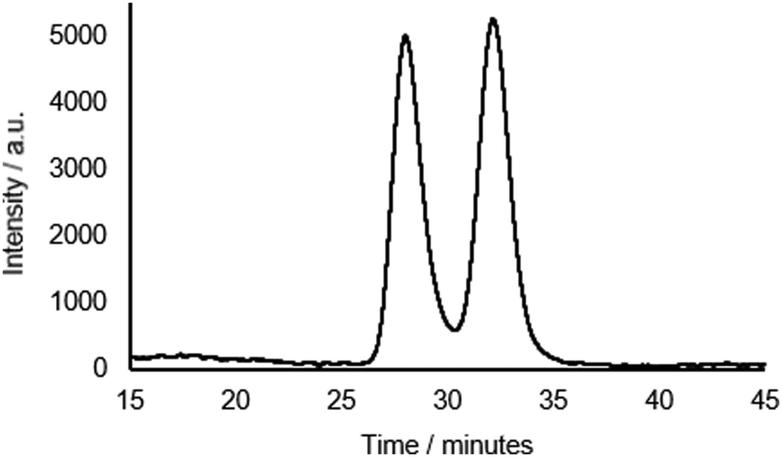 | ||
Fig. 3 Chiral HPLC trace of rotaxane 1 (CHIRAL-PAK AD-H 4.6 × 250 mm, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane:IPA. 1.5 mL min−1, λabs = 240 nm). :1 hexane:IPA. 1.5 mL min−1, λabs = 240 nm). | ||
Like for the analogous achiral rotaxane previously reported,27 the 1H NMR spectrum of rotaxane 1 has separate peaks for each of the two protons in environments 8 and 17 (but interestingly not 22), arising from the two faces of the macrocyclic ring being inequivalent due to the directionality of the threaded axle.
In addition, it can clearly be seen that the two axle protons g result in separate resonances, which was not observed in the previously reported achiral example.27 This is attributed to the two protons being diastereomeric, arising from the mechanical chirality of rotaxane 1. Considering that the neighbouring protons h are not split (to any significant extent), the close proximity of the macrocyclic ring, appears to be essential to observe diastereomeric behaviour. Evidence that the macrocycle is residing on the amide of the axle in chloroform (and hence is in closer proximity to protons g rather than h) is provided by the ROESY spectrum of rotaxane 1 in CDCl3 (see page S25, ESI†). This reveals through-space inter-component correlations between macrocycle protons and axle protons d, g and h, entirely consistent with the co-conformation depicted in Scheme 1. In addition, DFT calculations (see Fig. 4 and pages S44 and S45, ESI†) indicate that it is energetically favourable for the macrocycle to reside on the axle amide, rather than the triazole, which is the only other possible hydrogen bond donor/acceptor on the axle.29
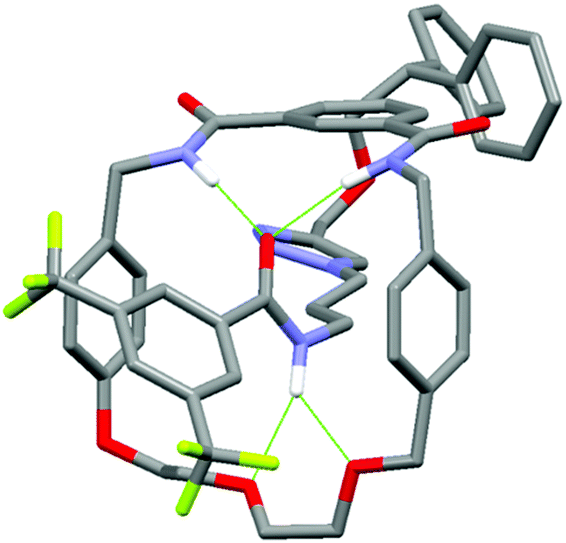 | ||
| Fig. 4 Minimum energy DFT calculated structure of rotaxane 1. Hydrogen bonds are represented by green dashed lines. For details of calculations, see ESI.† | ||
To test whether the co-conformational behaviour of 1 could be affected by varying the solvent (which may induce motion of the macrocycle by disruption of hydrogen bonding),30 a solvent screen was carried out (Fig. 5). It can be seen clearly that only in CDCl3 are the two resonances for g and g′ fully resolved, while in D6-DMSO they have (almost) completely coalesced. For the range of solvents tested here, there is at least a qualitative correlation between decreasing separation of resonances for g and g′ and ability of the solvent to compete as a hydrogen bond acceptor, as reflected by increasing Gutmann donor number of the solvent (chloroform (0) > acetonitrile (14) > acetone (17) > methanol (19) > DMSO (30)).31,32
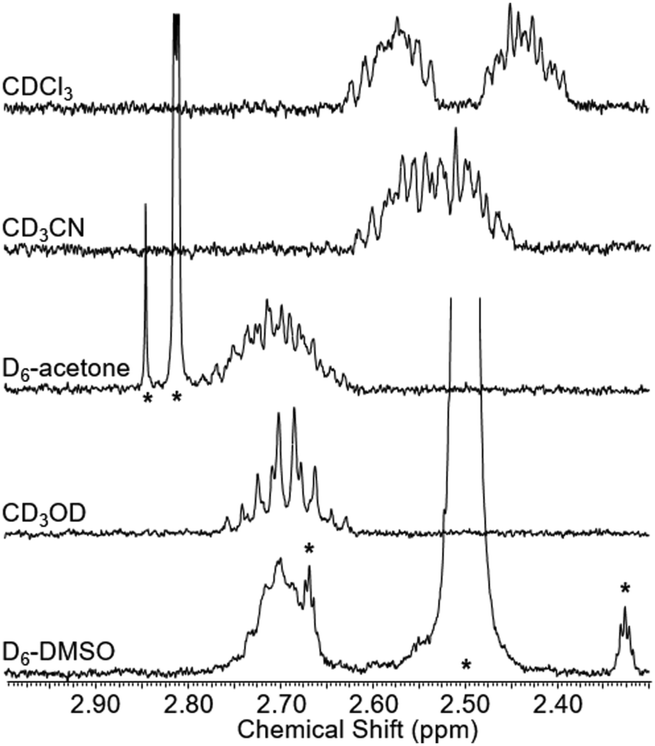 | ||
| Fig. 5 Partial 1H NMR spectra of rotaxane 1 in various solvents (2.5 mM, 400 MHz, 298 K). Solvent peaks marked with asterisks. | ||
Our attention then turned to whether the co-conformational behaviour of the rotaxane could be varied by the addition of acid and base.33 The addition of excess TFA to a sample of rotaxane 1 dissolved in CDCl3 leads to a downfield shift and merging of g and g′ in the 1H NMR spectrum (see Fig. 6 and page S36, ESI†). This is consistent with the macrocycle translating away from the axle amide: the alkyl protons g and g′ will be less shielded and no longer close enough to experience the rotational asymmetry of the macrocycle. Addition of D5-pyridine to neutralise the TFA reverses the shift and restores the separate peaks for g and g′.
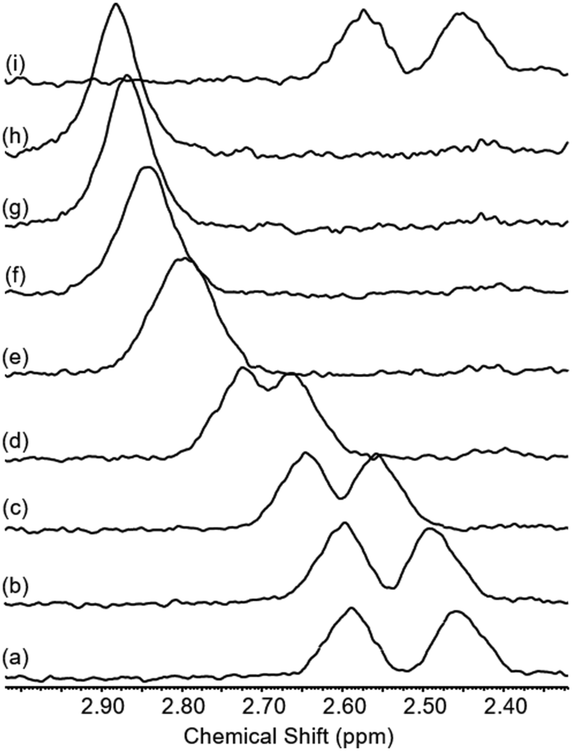 | ||
| Fig. 6 Partial 1H NMR spectra of rotaxane 1 with: (a) 0 eq. of TFA, (b) 2 eq. of TFA, (c) 5 eq. of TFA, (d) 10 eq. of TFA, (e) 20 eq. of TFA, (f) 30 eq. of TFA, (g) 40 eq. of TFA, (h) 50 eq. of TFA, (i) 50 eq. of TFA + 50 eq. of D5-pyridine (CDCl3, 400 MHz, 298 K. Spectra Fourier transformed with a line broadening of 3 Hz). | ||
Further insight into the co-conformational behaviour of rotaxane 1, upon the addition of TFA in CDCl3, was sought by studying peaks other than g and g′ in the 1H NMR spectrum (see Fig. 7 and page S36, ESI†). Protons h and l move upfield upon addition of TFA, i.e. these protons are experiencing greater shielding, implying greater residence within the macrocycle cavity. Meanwhile protons d move downfield suggesting they are further away from the macrocycle cavity. Macrocycle proton 2 moves upfield, consistent with a reduction in hydrogen bonding to the carbonyl O of the axle amide.
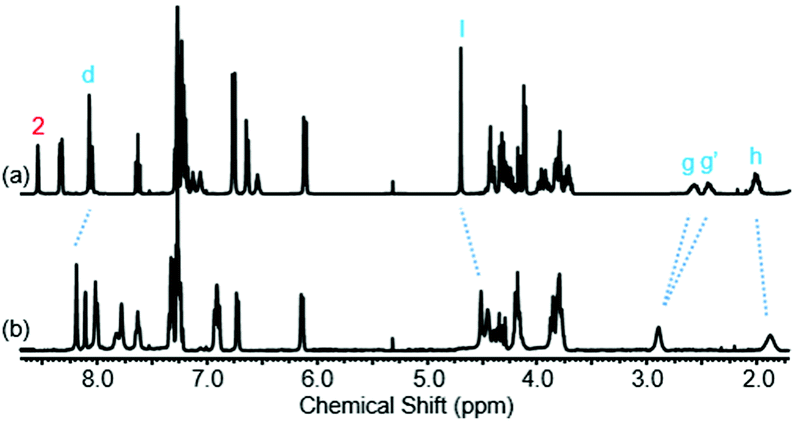 | ||
| Fig. 7 1H NMR spectra of rotaxane 1 with: (a) 0 eq. of TFA and (b) 40 eq. of TFA (CDCl3, 400 MHz, 298 K). Selected atom labels are defined in Scheme 1. | ||
Furthermore, a 1H–1H ROESY NMR spectrum was recorded (see page S43, ESI†). The addition of an excess of TFA resulted in a poor signal/noise ratio, necessitating a large number of scans to identify inter-component corrections, between macrocycle protons and those of axle protons d, g, h and l. Combining this result, with the study of the 1D 1H NMR spectra detailed above, and the apparent lack of splitting of any resonance arising from an axle proton environment, we suggest that in the presence of TFA, rotaxane 1 is dynamically occupying multiple co-conformations, rather than the single well-defined co-conformation depicted in Scheme 1. Multiple O atoms (plus the triazole N) may protonate possibly leading to a complicated set of hydrogen bond templated co-conformations.34 Undoubtedly, there is a significant change in the co-conformational behaviour of rotaxane 1 upon addition of TFA.35
In conclusion, it is possible to modulate the detectable chirality in the racemate of a mechanical chiral rotaxane by using two sets of achiral parameters – solvent and addition of acid and base – to affect the co-conformational behaviour of the rotaxane. Notably, to observe the influence of mechanical chirality upon the particular proton environment studied here, appears to require strict adherence of the rotaxane to a specific hydrogen bond supported co-conformation. In the future, we envisage that controlled variation between co-conformations of mechanically chiral rotaxanes could be used to create switchable molecular devices for various chiral applications. Research on chiral rotaxanes and their application is continuing in our laboratories and will be reported in due course.
We express our appreciation to the Faculty of Science and Technology at Lancaster University for a PhD studentship (C. E. G.) and the EPSRC (Institutional Sponsorship Grant: EP/P511183/1) for financial support of an Equality and Diversity internship (T. A. M.-I). N. H. E. also wishes to thank the Royal Society of Chemistry for a Research Grant that funded some preliminary experiments on chiral rotaxanes. At Lancaster University, we thank Dr Geoff Akien for donation of D5-pyridine and advice on NMR spectroscopy, Dr David Rochester for the recording of mass spectra and chiral HPLC, as well as Dr Michael Peach for advice on computational modelling. Underlying data for this paper are provided in the ESI, along with electronic copies of NMR spectra (including fid files) and computational output files being available from: DOI: 10.17635/lancaster/researchdata/159 & DOI: 10.17635/lancaster/researchdata/255.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. H. Evans, Chem. – Eur. J., 2018, 24, 3101–3112 CrossRef CAS PubMed.
- N. Pairault and J. Niemeyer, Synlett, 2018, 29, 689–698 CrossRef CAS.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- P. R. Ashton, I. Iriepa, M. V. Reddington, N. Spencer, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1994, 35, 4835–4838 CrossRef CAS.
- S. J. Cantrill, M. C. T. Fyle, A. M. Heiss, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem. Commun., 1999, 1251–1252 RSC.
- P. R. Ashton, S. E. Boyd, S. Menzer, D. Pasini, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White, D. J. Williams and P. G. Wyatt, Chem. – Eur. J., 1998, 4, 299–310 CrossRef CAS.
- D. K. Mitchell and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1988, 27, 930–931 CrossRef.
- C. Yamamoto, Y. Okamoto, T. Schmidt, R. Jäger and F. Vögtle, J. Am. Chem. Soc., 1997, 119, 10547–10548 CrossRef CAS.
- For further ways chirality may arise in rotaxanes and catenanes, please see reviews referenced above (ref. 1–3).
- R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed.
- M. A. Jinks, A. de Juan, M. Denis, C. J. Fletcher, M. Galli, E. M. G. Jamieson, F. Modicom, Z. Zhang and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 14806–14810 CrossRef CAS PubMed.
- R. Mitra, M. Thiele, F. Octa-Smolin, M. C. Letzel and J. Niemeyer, Chem. Commun., 2016, 52, 5977–5980 RSC.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584–588 CrossRef CAS PubMed.
- K. Hirose, M. Ukimi, S. Ueda, C. Onoda, R. Kano, K. Tsuda, Y. Hinohara and Y. Tobe, Symmetry, 2018, 10, 20 CrossRef.
- Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438–3439 CrossRef CAS PubMed.
- S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656–7659 CrossRef CAS PubMed.
- Y. Cakmuk, S. Erbas-Cakmak and D. A. Leigh, J. Am. Chem. Soc., 2016, 138, 1749–1751 CrossRef PubMed.
- R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 11456–11459 CrossRef CAS PubMed.
- F. Ishiwari, K. Nakazono, Y. Koyama and T. Takata, Angew. Chem., Int. Ed., 2017, 56, 14858–14862 CrossRef CAS PubMed.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- M. Asakawa, G. Brancato, M. Fanti, D. A. Leigh, T. Shimizu, A. M. Z. Slawin, J. K. Y. Wong, F. Zerbetto and S. Zhang, J. Am. Chem. Soc., 2002, 124, 2939–2950 CrossRef CAS PubMed.
- G. Bottari, D. A. Leigh and E. M. Pérez, J. Am. Chem. Soc., 2003, 125, 13360–13361 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, J. Am. Chem. Soc., 2014, 133, 4905–4908 CrossRef PubMed.
- F. Ishiwari, K. Nakazono, Y. Koyama and T. Takata, Chem. Commun., 2011, 47, 11739–11741 RSC.
- B. E. Fletcher, M. J. G. Peach and N. H. Evans, Org. Biomol. Chem., 2017, 15, 2797–2803 RSC.
- N. H. Evans, C. E. Gell and M. J. G. Peach, Org. Biomol. Chem., 2016, 14, 7972–7981 RSC.
- Multiple attempts to grow single crystals of rotaxane 1 suitable for X-ray diffraction structural determination have been undertaken, but alas none have been successful.
- For examples of well-defined hydrogen bonded co-conformational behaviour being disrupted in rotaxanes by varying solvent, see: (a) D. A. Leigh, M. Á. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 CrossRef CAS PubMed; (b) T. Da Ros, D. M. Guldi, A. F. Morales, D. A. Leigh, M. Prato and R. Turco, Org. Lett., 2003, 5, 689–691 CrossRef CAS PubMed; (c) J. M. Maloney, R. Shukla, R. A. Marshall, A. M. Beatty, J. Zajicek and B. D. Smith, J. Org. Chem., 2002, 67, 1436–1440 CrossRef.
- V. Gutmann, A. Steininger and E. Wychera, Monatsh. Chem., 1966, 97, 460–467 CrossRef CAS.
- To provide further insight into the stability of the macrocycle residing on the axle amide, NMR titrations experiments were undertaken where benzanilide was added to macrocycle 2 (see pages S28–S32, ESI†). While a pseudorotaxane forms in CDCl3, this does not occur in either D6-acetone or D6-DMSO.
- For examples of hydrogen bonded co-conformational behaviour of rotaxanes and catenanes varied by addition of acid and base, see: (a) M.-V. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1997, 36, 1904–1907 CrossRef; (b) C. M. Keaveney and D. A. Leigh, Angew. Chem., Int. Ed., 2004, 43, 1222–1224 CrossRef CAS PubMed; (c) K.-Y. Ng, V. Félix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC; (d) B. Hesseler, M. Zindler, R. Herges and U. Lüning, Eur. J. Org. Chem., 2014, 3885–3901 CrossRef CAS.
- Preliminary DFT calculations have been carried out to identify plausible protonated hydrogen bond templated co-conformations (see page S46, ESI)†.
- We also investigated whether it was possible to modulate the expression of chirality by the addition of the Li+ cation. Pirouetting of similar rotaxanes by addition of Li+ has been reported ( W. Zhou, J. Li, X. He, C. Li, J. Lv, Y. Li, S. Wang, H. Liu and D. Zhu, Chem. – Eur. J., 2008, 14, 754–763 CrossRef CAS PubMed ). It is also possible to imagine shuttling occurring by coordination of Li+ to the triazole of the axle and polyether of the macrocycle. To test this, LiPF6 was titrated into a sample of rotaxane 1 in CD3CN. A downfield shift and merging of peaks for g and g′ in the 1H NMR spectrum was observed (see page S38, ESI†). However, addition of LiClO4 (or Zn(ClO4)2) to rotaxane 1 in CD3CN resulted only in modest upfield shifts of g and g′ (see pages S40–S41, ESI†). We attribute the downfield shifts upon the addition of LiPF6 to the formation of acidic products from the hydrolysis of the hexafluorophosphate anion.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures; copies of spectral characterisation data; further data and protocols for titrations and details of computational modelling. See DOI: 10.1039/c8cc10044f |
| ‡ Current address: School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. |
| This journal is © The Royal Society of Chemistry 2019 |