
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilylene induced cooperative B–H bond activation and unprecedented aldehyde C–H bond splitting with amidinate ring expansion†
V. S. V. S. N.
Swamy
ab,
K. Vipin
Raj
bc,
Kumar
Vanka
 *bc,
Sakya S.
Sen
*ab and
Herbert W.
Roesky
*d
*bc,
Sakya S.
Sen
*ab and
Herbert W.
Roesky
*d
aInorganic Chemistry and Catalysis Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: ss.sen@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cPhysical and Material Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: k.vanka@ncl.res.in
dInstitute of Inorganic Chemistry, Georg-August University, Tammannstrasse 4, D-37077, Göttingen, Germany. E-mail: hroesky@gwdg.de
First published on 25th February 2019
Abstract
The addition of HBpin to PhC(NtBu)2SiN(SiMe3)2 (1) results in the cleavage of the B–H bond in a cooperative fashion across the Si and amidinate-C sites. The reaction of 1 with benzaldehyde led to C–H bond activation with amidinate ring expansion leading to a five-membered heterocycle. In case of 4-fluorobenzaldehyde, a C–C bond coupling takes place leading to a dioxasilolane derivative as the major product.
The splitting of a B–H bond is well-known with transition metals both via oxidative addition1 and, of late, by metal–ligand cooperativity.2–6 The latter concept has only come to light in the past few years through the studies from the groups of Oestreich,2 Love,3 Iluc,4 Gessner,5,6 and others7 (Scheme 1). The cooperative bond activation in main group chemistry is the area of frustrated Lewis pairs and the B–H bond activation of HBcat using FLP, [tBu2RP (R = tBu, 2-C6H4(C6H5)) and B(C6F5)3] has been demonstrated.8 Apart from FLP, cAAC mediated B–H bond activation has been documented, but this has been seen to be the 1,1 oxidative addition,9 and not the cooperative activation. Nevertheless, Radosevich's group recently reported the first single-component system capable of a cooperative B–H bond activation of HBpin.10
 | ||
| Scheme 1 Selective examples of cooperative B–H bond activation of HBpin by single component systems. | ||
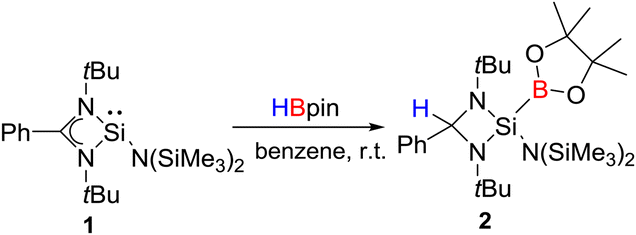
Ge(II), Al(III), Ga(III) compounds based on nacnac ligands with an exocyclic double bond are ambiphilic and have been found to undergo cooperative H–X bond activation.11–13 Berben and coworkers have demonstrated the activation of N–H and O–H bonds by a pincer-based aluminium complex via metal–ligand cooperation.14,15 However, no amidinate based main group compounds are known for cooperative bond activation. Due to our current interest in hydroboration chemistry,16 we were keen to examine the reaction of PhC(NtBu)2SiN(SiMe3)2 (1)17 with HBpin. Surprisingly, the addition of HBpin to PhC(NtBu)2SiN(SiMe3)2 (1) resulted in the cleavage of the B–H bond of HBpin through addition across the Si(II) and amidinate carbon center in a cooperative fashion (Scheme 2).
 | ||
| Scheme 2 The reaction of pinacolborane with silylene 1. | ||
Subsequent to the B–H bond activation of HBpin, we have approached the reaction of aldehydes with 1 to explore the possibility of applying such stoichiometric reactivity to catalysis. Aldehyde C–H bond activation has been promoted by late transition metals,18 but is not known with compounds with low valent main group elements. The reactions of 1 with benzaldehyde and 4-fluorobenzaldehyde led to the activation of the C–H bond (vide infra) accompanied by amidinate ring expansion with the formation of a five-membered heterocycle. While ring expansion chemistry of the N-heterocyclic carbenes is undergoing a great surge,19 related chemistry of the N-heterocyclic silylene is relatively less explored. Braunschweig and coworkers have reported the ring expansion of West's silylene (NHSi) upon reaction with PhBX2 (X = Cl, Br) to afford six-membered silaborinines.20 In a related work, So and coworkers have reported the insertion of the Si–H bond into the B–N bond of an amidinate borane in the presence of DMAP, leading to a ring expansion product.21
By monitoring the reaction of 1 with HBpin a new resonance appeared at δ 5.04 ppm in the 1H NMR spectrum, with the simultaneous disappearance of HBpin (q, BH, δ 2–3 ppm) resonances. This new resonance corresponds to the aliphatic CH proton, thereby indicating the formation of a cyclic four membered diamido Si(IV) compound. The 13C NMR of the CH carbon appears at δ 73.46 ppm. The four coordination of the silicon atom is reflected from the resonance at δ −52.41 ppm in the 29Si NMR spectrum. The 11B NMR spectrum shows a resonance at δ 37.84 ppm.
Colorless crystals of 2 suitable for single crystal X-ray structural analysis were grown from a saturated toluene solution at −32 °C in two days. 2 crystallizes in the orthorhombic space group Pbca and the molecular structure is shown in Fig. 1. The coordination around the silicon atom comprises of three nitrogen atoms (two from the amidinato ligand and one from the amide substituent moiety), and one boron atom, and features a distorted tetrahedral geometry. The formation of the diamido ligand is reflected from the shortening of the Si–N bond lengths (1.743(3) and 1.742(3) Å) in comparison to the bonds in the Si–Namidinate ligands (1.769(7) and 1.878(1) Å).17 The Si1–B1 bond length is 2.027(6) Å, which is in well agreement with the Si–B bond length in Aldridge's {B(NArCH)2}{N(SiMe3)Ar}SiH2 (2.016(2) Å) (Ar = 2,6-iPr2-C6H3)22 and Braunschweig's silaborinines (1.9899(15) and 2.019(3) Å).20
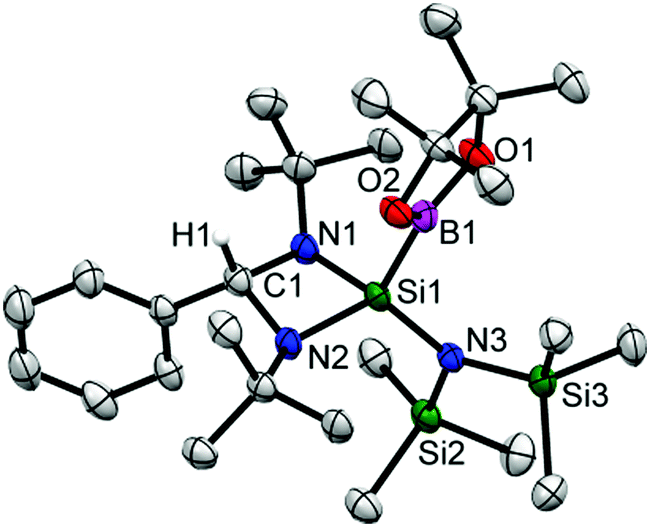 | ||
| Fig. 1 The molecular structure of 2. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except H1 bonded to C1) are omitted for clarity. Selected bond distances (Å) and bond angles (deg): C1–H1 1.03(6), Si1–B1 2.027(6), Si1–N1 1.743(4), Si1–N2 1.739(4), Si1–N3 1.742(4); N1–Si1–B1 109.8(2), N2–Si1–B1 114.4(2), N3–Si1–B1 113.3(2), N1–Si1–N2 77.4(2), N1–Si1–N3 120.0(2). | ||
We have also studied the mechanism of the B–H bond activation by 1. The reaction first proceeds through a transition state TS_1′′, with an energy barrier of 8.5 kcal mol−1, in which a Si–B bond is formed by electron donation from silicon to the vacant orbital of boron, which leads to an intermediate Int_1′′ (Fig. 2). Then the reaction surmounts a second transition state TS_2′′ that involves the transfer of a hydride ion from boron to carbon. This leads to a thermodynamically stable (−7.2 kcal mol−1) product. Therefore, the DFT calculations indicate that activation of a B–H bond by 1 is thermodynamically and kinetically feasible at room temperature. The feasibility of formation of a product arising from 1,1 oxidative addition at the silicon center was also calculated. Even though the oxidative addition is kinetically feasible (barrier: 19.1 kcal mol−1), the product is thermodynamically unfavourable by 11.6 kcal mol−1 with respect to 2.
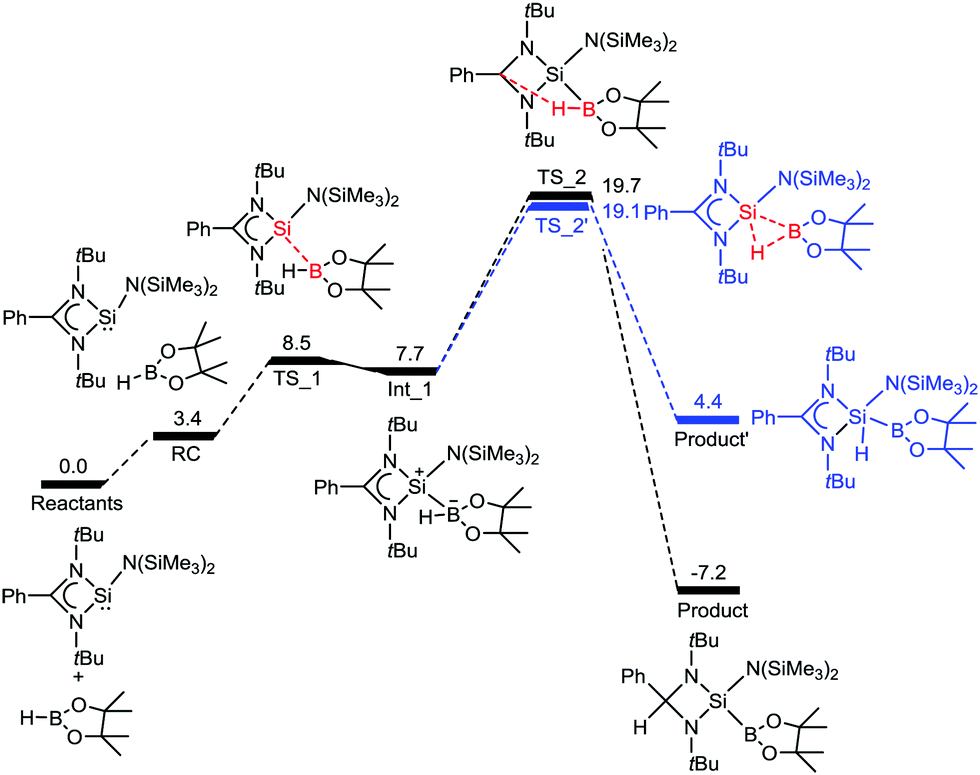 | ||
| Fig. 2 The reaction free energy profile diagram for the B–H bond activation by 1. The values (in kcal mol−1) have been calculated at the PBE/TZVP level of theory. | ||
The reactions of silylenes with ketones are well documented.23 In marked contrast, reactions of silylenes with aldehydes have remained rather cursory. To the best of our knowledge, the only reaction featuring a stable silylene and an aldehyde was reported by Jutzi et al. in 1996 and led to a product with a C–C bond formation, leaving the C–H moiety intact.24 The reaction of 1 with benzaldehyde led to the activation of the C–H bond along with expansion of amidinate ring (Scheme 3). This is quite unusual as the ring expansion usually requires a hydride source.19,21 The formation of 3 was substantiated by X-ray diffraction studies (Fig. 3) indicating the insertion of the benzoyl moiety in the C–N ring along with the formation of a C–C double bond. Additionally, one of the SiMe3 moieties migrates to another N atom bound to the tBu group and the hydride binds to the N atom bound to another SiMe3. The silicon atom is coordinated to three nitrogen atoms and one oxygen and exhibits a distorted tetrahedral geometry. The Si–N bond lengths are (∼1.718 Å), which are marginally longer than the Si–N single bonds of the compound 2 (∼1.727 Å). The C1–C2 bond length is of 1.345(4) Å, which confirms the formation of a double bond. This is further supported from the 13C NMR of C1 and C2, which resonate at δ 124.69 and 142.46 ppm. The N–H proton displays a broad resonance at δ 4.93 ppm. In the 29Si NMR, a signal δ –39.48 ppm appears for the four-coordinate central silicon atom. Surprisingly, we did not observe the formation of any dioxasilolane derivative (vide infra) even when changing the molar ratio of the reaction partners.
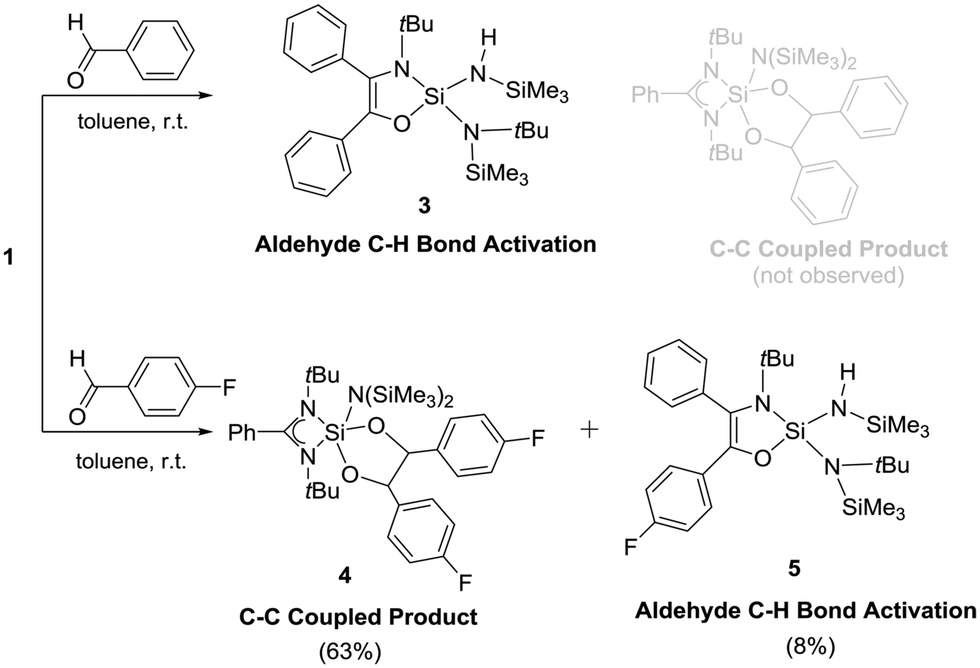 | ||
| Scheme 3 The reactions of aldehydes with silylene 1. | ||
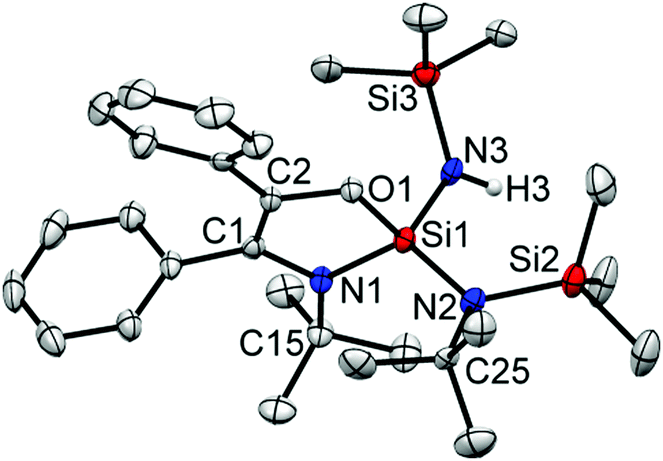 | ||
| Fig. 3 The molecular structure of 3. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except H3 is bonded to N3) are omitted for clarity. Selected bond distances (Å) and bond angles (deg): C1–C2 1.347(4), C2–O1 1.394(4), Si1–O1 1.650(2), Si1–N1 1.745(2), C1–N1 1.419(4), Si1–N2 1.714(3), Si1–N3 1.697(2), Si3–N3 1.725(2), Si2–N2 1.767(3); O1–Si1–N1 92.90(1), N1–Si1–N2 119.90(1), N1–Si1–N3 113.50(1), O1–Si1–N2 113.20(5), O1–Si1–N3 106.10(1), C1–N1–Si1 108.10(2), C2–O1–Si1 112.20(2), C1–C2–O1 113.40(3), N1–C1–C2 113.30(3), H3–N3–Si1 116.10(0), Si1–N2–Si2 118.80(1). | ||
Next, we have selected 4-fluorobenzaldehyde as a substrate to react with 1. The reaction has three possible outcomes: (i) C–F bond activation by silylene,25,26 (ii) aldehyde C–H bond activation and subsequent ring expansion (vide supra), and (iii) the C–C coupling reaction as reported by Jutzi et al.24 The reaction afforded the 1,3-dioxasilolane derivative (4) as the major product and C–H activation/ring expansion product, 5 as the minor product (Scheme 3). No aromatic C–F bond activation was observed. Mechanistically, the initial formation of a silaoxirane derivative is proposed, which undergoes C–C bond formation upon nucleophilic attack from the oxygen atom of another molecule of 4-fluorobenzaldehyde at the silicon center (see Fig. S2 in the ESI†). The formation of 4 is highly regio- and stereospecific; only the formation of the trans isomer was observed. A resonance at δ 6.63 ppm appeared in the 1H NMR spectrum of 4 indicates the C–H protons of the C–C bond. The 13C NMR spectrum of 4 exhibits two new resonances at δ 114.68 and 114.89 ppm corresponding to the C–C bond formation. In the 19F NMR spectrum of 4, resonances appear at δ −118.93 and −118.99 ppm, respectively. The 29Si NMR spectrum shows a resonance at δ −38.17 ppm. Despite repeated efforts, only relatively poor quality crystals of 4 could be obtained by crystallization. Although the X-ray diffraction study leaves no doubt about the constitution (Fig. 4), we refrain from a discussion of bonding parameters because of the low quality of the data.
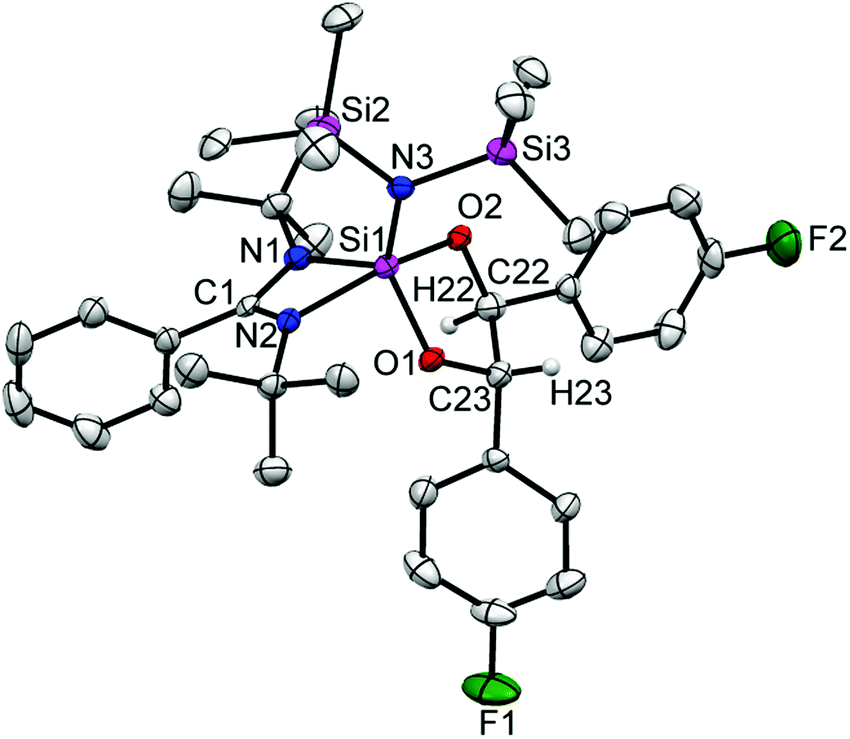 | ||
| Fig. 4 The molecular structure of 4. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except H22 and H23 are bonded to C22 and C23, respectively) are omitted for clarity. | ||
According to NMR spectroscopy, 5 is formed only in a small quantity. Despite several attempts, 5 could not be prepared in reasonable amounts allowing for a full spectroscopic characterization. Nevertheless, the appearance of a broad N–H resonance was observed at δ 4.90 ppm. The 19F NMR resonance of 5 was also identified at δ −118.96 ppm. Single-crystals of 5 were grown in the same flask of 4 in the same condition. The structural parameters of 5 (Fig. 5) are comparable to those in 3.
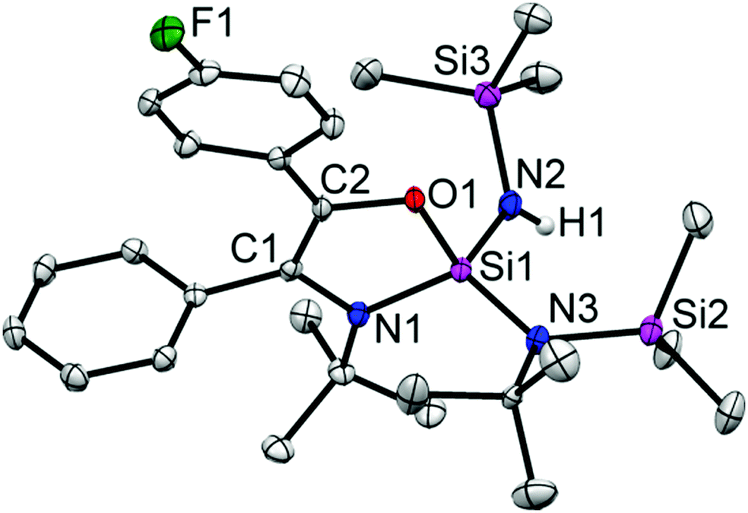 | ||
| Fig. 5 The molecular structure of 5. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except H1 is bonded to N2) are omitted for clarity. Selected bond distances (Å) and bond angles (deg): C1–C2 1.357(3), C2–O1 1.402(2), Si1–O1 1.664(1), Si1–N1 1.757(2), C1–N1 1.418(2), Si1–N2 1.700(2), Si1–N3 1.724(2), N3–Si2 1.778(2), N2–Si3 1.742(2), N3–C30 1.528(3), N2–H1 0.760(2); O1–Si1–N1 93.17(7), C1–N1–Si1 108.0(1), N1–Si1–N2 113.95(8), N1–Si1–N3 118.38(8), O1–Si1–N2 105.62(5), O1–Si1–N3 113.65(7), C2–O1–Si1 111.60(1), C1–C2–O1 113.50(2), N1–C1–C2 113.60(2), Si1–N2–H1 119.0(2), Si1–N3–Si2 119.08(9). | ||
After more than 150 years since the discovery of the pinacol coupling reaction by Wilhelm Rudolph Wittig,27 we have demonstrated that silylene (1) can also mediate a carbon–carbon covalent bond formation between two aldehydes leading to a 1,3-dioxasilolane derivative (4). The analogous reaction with benzaldehyde resulted in the cleavage of the aldehyde C–H bond and subsequent amidinate ring expansion via insertion of the benzoyl moiety into the C–N bond. This is also the first example of an aldehyde C–H bond activation by a silylene. The formation of the two distinctly different products can be attributed to the difference in the nature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in benzaldehyde and 4-fluorobenzaldehyde. The addition of HBpin to 1 resulted in a 1,3 B–H addition to 1 with the “Bpin” fragment translocating to the silylene center and the hydride migrating to the carbon center.
O bond in benzaldehyde and 4-fluorobenzaldehyde. The addition of HBpin to 1 resulted in a 1,3 B–H addition to 1 with the “Bpin” fragment translocating to the silylene center and the hydride migrating to the carbon center.
SSS, KV thank DST-SERB (SB/S2/RJN-073/2014) and (EMR/2014/000013) and HWR thanks DFG (RO 224/68-1) for providing financial assistance for this work. VSVSN thanks CSIR-India for a research fellowship. SSS is also grateful to Alexander von Humboldt Stiftung for a renewed research stay in Göttingen for two months.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected example, (a) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed; (b) D. Pun, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2007, 3297–3299 RSC; (c) M. V. Câmpian, E. Clot, O. Eisenstein, U. Helmstedt, N. Jasmin, R. N. Perutz, A. C. Whitwood and D. Williamson, J. Am. Chem. Soc., 2008, 130, 4375–4385 CrossRef PubMed; (d) A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2010, 49, 581–584 CrossRef CAS PubMed; (e) N. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Llyod-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef PubMed.
- (a) T. Stahl, K. Mether, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed; (b) L. Omann, C. D. F. Königs, H. F. T. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed.
- M. W. Drover, L. L. Schafer and J. A. Love, Angew. Chem., Int. Ed., 2016, 55, 3181–3186 CrossRef CAS PubMed.
- C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC.
- L. T. Scharf, J. Weismann, K.-S. Feichtner, F. Lindl and V. H. Gessner, Chem. – Eur. J., 2018, 24, 3439–3443 CrossRef CAS PubMed.
- K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC.
- (a) M. A. Rankin, K. D. Hesp, G. Schatte, R. McDonald and M. Stradiotto, Dalton Trans., 2009, 4756–4765 RSC; (b) A. Anaby, B. Butschke, Y. Ben-David, L. J. W. Shimon, G. Leitus, M. Feller and D. Milstein, Organometallics, 2014, 33, 3716–3726 CrossRef CAS.
- M. A. Dureen, A. Lough, T. M. Gilbert and D. W. Stephan, Chem. Commun., 2008, 4303–4305 RSC.
- G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444–9447 CrossRef CAS PubMed.
- Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS PubMed.
- (a) Y. Xiong, S. Yao and M. Driess, Organometallics, 2009, 28, 1927–1933 CrossRef CAS; (b) A. Meltzer, S. Inoue, C. Präsang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS PubMed.
- (a) S. P. Sarish, S. S. Sen, H. W. Roesky, I. Objartel and D. Stalke, Chem. Commun., 2011, 47, 7206–7208 RSC; (b) A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2008, 48, 798–800 CrossRef PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed.
- T. W. Myers and L. A. Berben, J. Am. Chem. Soc., 2013, 135, 9988–9990 CrossRef CAS PubMed.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- (a) S. Yadav, S. Pahar and S. S. Sen, Chem. Commun., 2017, 53, 4562–4564 RSC; (b) M. K. Bisai, S. Pahar, T. Das, K. Vanka and S. S. Sen, Dalton Trans., 2017, 46, 2420–2424 RSC; (c) M. K. Bisai, T. Das, K. Vanka and S. S. Sen, Chem. Commun., 2018, 54, 6843–6846 RSC; (d) S. Yadav, R. Dixit, M. K. Bisai, K. Vanka and S. S. Sen, Organometallics, 2018, 37, 4576–4584 CrossRef CAS; (e) Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229 CrossRef CAS PubMed; (f) C.-C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190–194 CrossRef CAS PubMed.
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311–12316 CrossRef CAS PubMed.
- M. A. Garralda, Dalton Trans., 2009, 3635–3645 RSC.
- For selected examples on NHC ring expansion, please see: (a) M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall and M. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098–2100 CrossRef CAS PubMed; (b) D. Schmidt, J. H. J. Berthel, S. Pietsch and U. Radius, Angew. Chem., Int. Ed., 2012, 51, 8881–8885 CrossRef CAS PubMed; (c) S. K. Bose, A. Deißenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843–11847 CrossRef CAS PubMed; (d) S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson and E. Rivard, Chem. – Eur. J., 2012, 18, 13810–13820 CrossRef CAS PubMed.
- A. Gackstatter, H. Braunschweig, T. Kupfer, C. Voigt and N. Arnold, Chem. – Eur. J., 2016, 22, 16415–16419 CrossRef CAS PubMed.
- S. Khoo, Y.-L. Shan, M.-C. Yang, Y. Li, M.-D. Su and C.-W. So, Inorg. Chem., 2018, 57, 5879–5887 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- For selected references, please see: (a) R. S. Ghadwal, S. S. Sen, H. W. Roesky, M. Granitzka, D. Kratzert, S. Merkel and D. Stalke, Angew. Chem., Int. Ed., 2010, 49, 3952–3955 CrossRef CAS PubMed; (b) S. Ishida, T. Iwamoto and M. Kira, Organometallics, 2010, 29, 5526–5534 CrossRef CAS.
- P. Jutzi, D. Eikenberg, E.-A. Bunte, A. Möhrke, B. Neumann and H.-G. Stammler, Organometallics, 1996, 15, 1930–1934 CrossRef CAS.
- (a) V. S. V. S. N. Swamy, N. Parvin, K. V. Raj, K. Vanka and S. S. Sen, Chem. Commun., 2017, 53, 9850–9853 RSC; (b) M. Pait, G. Kundu, S. Tothadi, S. Karak, S. Jain, K. Vanka and S. S. Sen, Angew. Chem., Int. Ed., 2019, 58, 2804–2808 CrossRef CAS PubMed.
- (a) A. Jana, P. P. Samuel, G. Tavčar, H. W. Roesky and C. Schulzke, J. Am. Chem. Soc., 2010, 132, 10164–10170 CrossRef CAS PubMed; (b) R. Azhakar, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2013, 49, 1841–1843 RSC.
- R. Fittig, Justus Liebigs Ann. Chem., 1859, 110, 23–45 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, CIF files of 2, 3, 4 and 5, checkcifs, representative NMR and HRMS spectra and details of computational methodology are given. CCDC 1829919 (2), 1888645 (3), 1845412 (4) and 1888644 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00296k |
| This journal is © The Royal Society of Chemistry 2019 |