
A molecular rotor-based turn-on sensor probe for amyloid fibrils in the extreme near-infrared region†
Niyati H.
Mudliar
a and
Prabhat K.
Singh
 *bc
*bc
aSVKM's Shri C. B. Patel Research Centre, Vile Parle, Mumbai, Maharashtra 400056, India
bHomi Bhabha National Institute, Training School Complex, Anushaktinagar, Mumbai 400094, India
cRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai 400085, India. E-mail: prabhatk@barc.gov.in; prabhatsingh988@gmail.com
First published on 5th March 2019
Abstract
A fluorescence turn-on probe for amyloid detection in the extreme near-infrared region (>750 nm) is a highly desirable technological evolution from the view point of potential in vivo applications. Herein, we report a molecular rotor-based amyloid sensor probe which, on binding to the insulin amyloid fibril, registers a large turn-on emission in the near-infrared region, and records an exceptionally large red-shifted emission wavelength of ∼770 nm along with a Stokes’ shift of ∼150 nm, the highest reported to date for any amyloid sensor probe, in the insulin fibril bound form, in the near-infrared region. Importantly, when bound to insulin fibrils, this probe also exhibits an exceptionally large red-shift of ∼120 nm in the absorption spectra, which enables the naked eye in vitro detection of amyloid fibrils.
Formation of highly organized fibrils of functional proteins and their subsequent deposition in the body are commonly associated with amyloid related disorders such as Parkinson's disease, Alzheimer's disease, type 2 diabetes and prion diseases.1–3 Presently, Alzheimer's disease is being clinically diagnosed by means of cognitive and mental scrutiny of the patients, however, the absolute confirmation of the disease can only be acquired post-mortem by examining the brain tissues histopathologically for deposition of neurofibrillary amyloid plaques and tangles. Thus, detection of this disease at an early asymptomatic stage is extremely desirable, but remains quite challenging. Deposition of amyloid fibrils is believed to occur several years before the appearance of clinical symptoms. Thus, efforts towards developing sensors that would probe the inception and progressive accumulation of amyloid plaques in the latent period itself hold a crucial importance for early detection, monitoring and evaluation of effective therapeutic measures.
Conventionally, MRI (magnetic resonance imaging) and PET (positron emission tomography) are the most widely used imaging techniques. However, apart from being very costly and complicated, the low sensitivity and selectivity of the MRI probes towards amyloid fibrils, the complicated PET probe synthesis due to scarcity of specific isotopes, the use of radioactivity and its obligatory usage within stipulated short half-lives of synthesized probes for PET scans, along with very limited accessibility of the technology, are some serious limitations associated with these techniques.4 Therefore, development of an alternative technique that can facilitate the sensitive and selective detection of amyloid fibrils, and that can evaluate the efficacy of therapeutic interventions, remains highly desirable. In this regard, fluorescence spectroscopy has gained immense interest for carrying out elaborative studies on amyloid fibrillation and protein aggregates in diverse systems. Owing to its various powerful attributes, such as, simplicity, extreme sensitivity, rapidity, versatility, etc., fluorescence spectroscopy has emerged as a superior non-invasive diagnostic tool that enables the real-time visualization of biomolecules, thus making it a more competent and economic tool for early detection of amyloidosis.
However, a large majority of the diagnostic fluorescent amyloid probes, that has been developed so far, possess their excitation and emission maxima below 600 nm, which suffers from various serious drawbacks, such as, auto-fluorescence from the biological components, photo-damage to the biological samples, large scattering from the tissue components, etc. In order to overcome these limitations, a desirable fluorescent probe is required to possess superior feature of emitting fluorescence in the near infrared (NIR) region (600–900 nm). The NIR probes offer various attractive advantages such as acceptable depth of penetration, non-invasive operation, and minimal interferences from auto-fluorescence of biological matter and minimal photo-damage to biological samples.4 Therefore, continuous efforts are made to shift the excitation and emission maxima of the fluorescent probes towards the NIR region. For this purpose, a number of probes have been developed, such as those based on the BODIPY architecture,5 curcumin architecture (CRANAD derivatives),6–8 donor–π-bridge–acceptor architecture utilizing a highly polarizable bisthiophene linker (NIAD derivatives),9,10 oxazine derivatives (AOI-987),11 DANIR probes,12etc., although with limited success. These probes suffer from one or more serious limitations such as emission wavelength below 650 nm in the amyloid bound form, narrow Stokes’ shift causing potential interference from Raman and Rayleigh scattering, poor contrast between the native and the amyloid form, high lipophilicity leading to non-specific binding, quenching of fluorescence upon binding of amyloid fibrils, etc. Hence, fluorescent sensor probes which can overcome these limitations and preferably emit in the NIR region remain a desirable goal for efficient detection of amyloid fibrils.
In this communication, we report a styryl based probe (styryl-11, see Scheme S1, ESI†), which shows a large fluorescence turn-on response, specifically in response to insulin amyloid fibrils as compared to its native form, and registers a very large red-shifted emission maximum of ∼770 nm and an exceptional Stokes’ shift of ∼150 nm in the fibril bound form, which, to the best of our knowledge, is the highest reported to date for any probe, in the insulin fibril bound form, in the near IR region. Importantly, this probe also registers a mammoth red shift of ∼120 nm in its absorption maximum upon binding to insulin amyloid fibrils. Such a large change in the absorption maximum of the probe yields a visual colour change of the solution in the presence of amyloid fibrils.
Fig. 1A presents the steady-state fluorescence spectra of styryl-11, in response to the addition of insulin fibrils. The styryl-11 is very weakly emissive in bulk aqueous medium; however, upon addition of insulin fibrils, it achieves a massive increment in the fluorescence intensity of ∼500 fold in the presence of 30 μM of insulin fibrils with an emission maximum at 770 nm, which remains largely unchanged from its free form (Fig. S1, ESI†). The weakly emissive nature of styryl-11, in its free form such as, in aqueous solution, is attributed to its ability to form a twisted intramolecular charge transfer (TICT) state, in its excited state, which opens up a major non-radiative process for the molecule in the high polarity medium.13 In addition, this probe exhibits high conformational flexibility, where a donor and an acceptor moiety are joined by highly flexible methylene bridges, which further augments the non-radiative de-excitation process for the molecule in its excited state.13 The dominant contribution of formation of the TICT state, as well as large conformational flexibility, towards the non-radiative process of the molecule, has been recently supported by polarity and viscosity dependent measurements of photophysical properties of styryl-11.
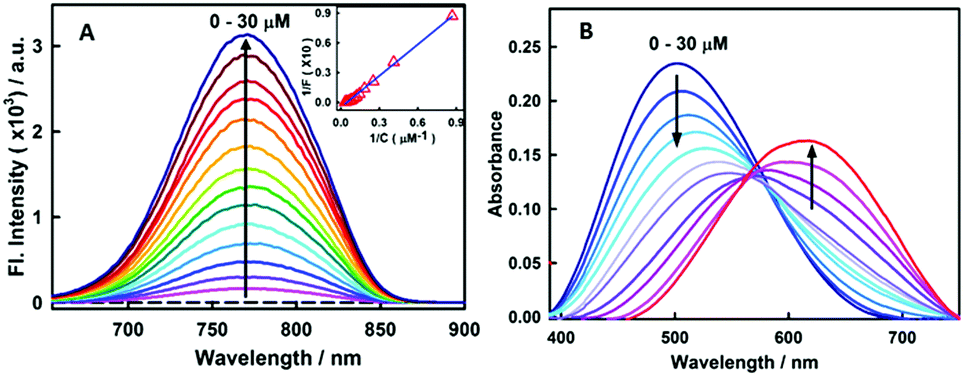 | ||
Fig. 1 (A) Fluorescence spectra of 15 μM stryl-11 with increasing concentration of insulin fibrils (0–30 μM) [λex = 630 nm]. Inset shows the double reciprocal plot (1/F vs. 1/C) and the solid line represents the fitting of the data points according to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. (B) Absorption spectra of 15 μM stryl-11 with the increase in concentration of insulin fibrils (0–30 μM). :1 binding model. (B) Absorption spectra of 15 μM stryl-11 with the increase in concentration of insulin fibrils (0–30 μM). | ||
These studies have convincingly demonstrated that the decrease in polarity and the increase in the viscosity of the medium significantly impact (increase) the emission yield of this molecule.13 Thus, the huge increase in the emission intensity of the styryl-11, in the presence of insulin fibrils, is a manifestation of a significant decrease in the non-radiative process of the molecule. The amyloid fibrils are known to provide non-polar hydrophobic binding channels to the probe molecules. Thus, it is very likely that the styryl-11 binds to hydrophobic channels present in the amyloid fibril medium (see molecular docking calculations later) which leads to a large reduction in the propensity of the molecule to form the energy wasting TICT state, leading to a large increase in the emission intensity. Furthermore, the conformational flexibility of the molecule, in the fibril bound state, is expected to be significantly impeded, where its aromatic rings are rigidly held, and these movements are restricted, which significantly contributes to the emission enhancement for the styryl-11, in the fibrillar medium. Thus, styryl-11 belongs to a well-known class of amyloid markers, termed as molecular rotors, and attains this remarkable fluorescence enhancement in the NIR region by a combination of reduced polarity, increased rigidity and reduced segmental flexibility in the fibril bound state. The other common type of amyloid marker is the one which is very sensitive to polarity and hydrophobicity of the surrounding environment in the structure of fibrils. Some uncommon but potentially important mechanisms which have been reported recently for a few amyloid markers are aggregation induced emission enhancement14 and disaggregation induced emission enhancement.15
Nonetheless, an important parameter for a fibril marker is that it should produce a significant contrast with the native form of the protein. Fig. 2(II) and Fig. S2 (ESI†) clearly suggest that the emission features of styryl-11 undergo only minor changes in the presence of native insulin, thus providing large specificity and contrast for fibrillar insulin over its native form, which is a pre-requisite for any potential amyloid marker probe to distinguish the fluorescence of the amyloid bound probe from its background.
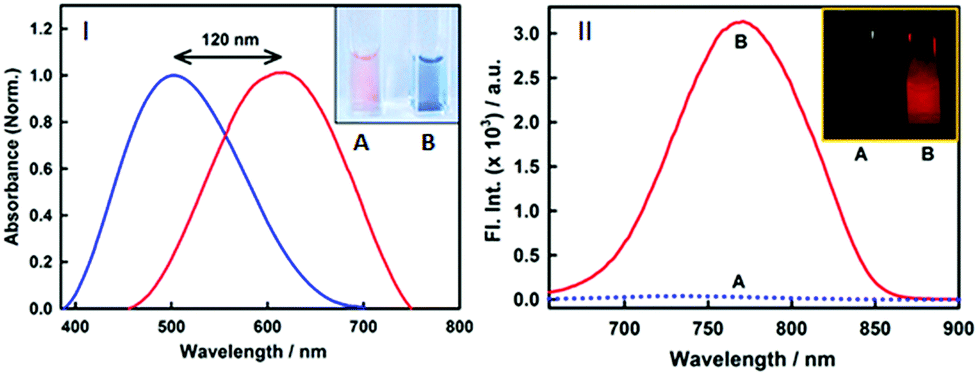 | ||
| Fig. 2 (I) Normalized absorption spectra of 15 μM styryl-11 at 0 (blue) and 30 μM fibril (red) showing a large red shift of the 120 nm shift in the peak maximum upon binding to fibrils. Inset shows the cuvettes with styryl-11 in native insulin protein (A) and insulin fibrils (B) under day light demonstrating visual colour change upon binding to fibrils. (II) Fluorescence spectra of 15 μM styryl-11 in insulin protein (dotted blue line) and insulin fibrils (red) showing large contrast for the fibrils. Inset of (II) shows the fluorescence images for styryl-11 in insulin protein (A) and insulin fibrils (B) under 630 nm excitation. | ||
To gain a quantitative idea about the binding strength of styryl-11 with insulin fibril medium, the fibril concentration-dependent fluorescence changes were analyzed with the 1:1 binding model, (Fig. 1A, inset) and the binding constant was found to be 5 × 104 M−1, suggesting a reasonably strong binding between styryl-11 and the insulin fibrils.
Styryl dyes are known to display rich solvatochromism in their absorption spectra with varying polarity.16 Thus, we have also performed ground-state absorption measurements to investigate the changes in the absorption spectrum and the binding interaction of styryl-11 with the addition of insulin fibrils. As is evident from the Fig. 1B, the addition of insulin fibrils to the aqueous solution of styryl-11 causes remarkable changes in the absorption spectra of the dye with a huge red shift of ∼120 nm observed in the presence of 30 μM of insulin fibrils (λmax,water = 500 nm and λmax,fibril = 620 nm). Such a remarkable red shift in the absorption spectra of the styryl-11 upon binding to the fibrillar medium suggests an extensively reduced polarity at the binding site for styryl-11. Importantly, such large shift in the absorption spectra of the dyes yields a visual colour change of the solution upon addition of insulin fibrils (Fig. 2A). Please note that the addition of the native insulin causes insignificant shift in the absorption spectra of styryl-11 (Fig. S3, ESI†). Note that the most extensively utilized amyloid marker probe, which works through the colorimetric approach, is Congo red, which shows a shift of only 50 nm in its absorption spectra upon binding to amyloid fibrils.17 Thus, our current probe, styryl-11, provides a superior route for in vitro colorimetric detection of the amyloid fibrils even by the naked eye.
Another desirable parameter for an amyloid marker is the presence of a large Stokes’ shift in the fibril bound form, which many amyloid markers in the NIR region, especially BODIPY based markers,5 fall short off. The presence of large Stokes’ shift helps in efficient reduction in the scattering of the excitation light. From this perspective, styryl-11 registers a mammoth Stokes’ shift of ∼150 nm in the fibril bound state. Note that, very few dyes in the near IR region have been reported to have such large Stokes’ shift; also, all these reports exist only for the dye in the unbound form.18
As revealed from steady-state emission measurements, the dye undergoes large modulations in its non-radiative processes, time-resolved emission measurements can provide important information on this aspect. As is evident from the Fig. 3B, styryl-11 undergoes very fast decay in bulk aqueous medium which has been attributed to the efficient non-radiative process in the molecule as discussed earlier.19 However, when styryl-11 is bound to insulin amyloid fibrils, a massive increase in the excited-state lifetime (0.024 ns to 0.9 ns) is recorded. This large increase in the excited-state lifetime of styryl-11 is a manifestation of the significant reduction in the conformational flexibility as well as the reduced tendency of the TICT state formation of the probe in the fibril bound state. The strong immobilization of the probe in the amyloid fibril medium is also supported by time-resolved anisotropy measurement (Fig. S4 and related discussion, ESI†).
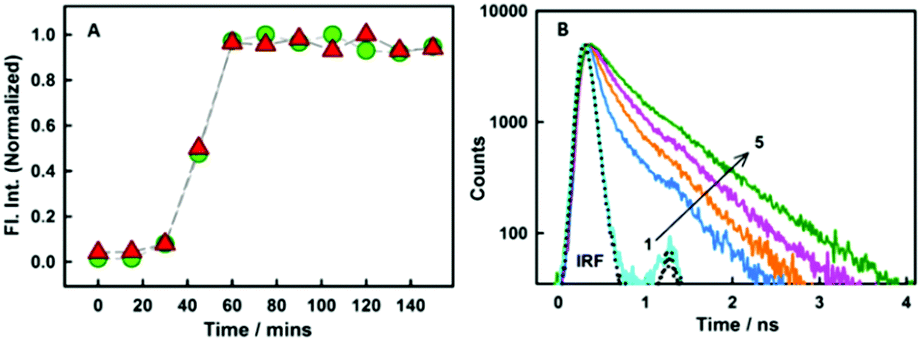 | ||
| Fig. 3 (A) Normalized plot for the variation in intensity for styryl-11 (triangles) and thioflavin-T (circles). (B) Fluorescence decay traces for 10 μM styryl-11 at various concentrations (in μM) of insulin fibrils (1) 0 (2) 1.45 (3) 4.2 (4) 7 and (5) 19.The dotted line represents the instrument response function (IRF). | ||
In addition to detecting amyloid fibrils, it is also desirable that the new amyloid probe should also be capable of monitoring the fibrillation kinetics which helps in both mechanistic understanding of protein aggregation as well as fast screening of amyloid inhibitors. To check this aspect for the styryl-11, we have monitored the protein fibrillation process for the insulin protein using both the most extensively utilized amyloid probe, thioflavin-T, and our current probe, styryl-11, and the results are presented in Fig. 3A. The fibrillation process of insulin protein is characterized by a lag phase followed by an elongation phase and finally a plateau phase leading to formation of mature fibrils, which is very nicely traced by both ThT and styryl-11. Thus, it is quite evident that, like ThT, styryl-11 can also be conveniently utilized for monitoring the kinetics of insulin fibrillation.
Amyloid binding probes are known to interact with amyloid fibrils via various types of interactions among which hydrophobic and electrostatic interactions are the main contributors. Since our probe consists of large hydrophobic groups with a cationic charge, it is expected to interact with the amyloid fibril via both hydrophobic and electrostatic interactions. However, the insignificant effect of ionic strength of the medium on the steady-state emission features (Fig. S5, ESI†) and transient decay traces (Fig. S6, ESI†) of the styryl–fibril complex clearly rules out any significant contribution of electrostatic interaction towards probe–fibril interaction. Thus, it can be inferred that styryl-11 interacts with the fibrillar matrix predominantly via hydrophobic interactions. This inference has also been supported by molecular docking calculations (see later). Thus, it can be concluded that, unlike the gold standard cationic amyloid probe, ThT, the fibril sensing by styryl-11 is immune to the changes in the ionic strength of the medium which is a highly desirable feature for an amyloid probe. Amyloid fibrils share a common structure which is rich in β-sheet and offers several binding sites, which include well-defined channels that are formed by the aromatic side chains of the residues of the β-strands, exposed grooves on the β-sheet surface and the ends of the extended β-sheet. Thus, molecular docking calculations were performed to identify the principal binding site of styryl-11 on the amyloid fibril matrix and to understand the nature of intermolecular interactions. Since structural information for insulin amyloid fibrils is not available, the solid state NMR structure of fibril from amyloid-β protein, Aβ1–42 (PDB ID = 2MXU)20 is used as the host fibril structure for docking purposes. The molecular docking studies suggest that styryl-11 prefers binding to the inner core of the fibril along its long axis (Fig. 4A). Molecular docking calculations also suggest that this kind of binding of styryl-11 into the inner core of the fibril is stabilized via various types of interactions which include van der Waals, pi-alkyl, amide-pi stacking interaction, C–H bond etc. (Fig. S7, ESI†). We have also performed docking calculations with two more recent structures of Aβ(1–42) i.e., PDB = 2NAO and 5OQV, and interestingly, in these two structures, as well, styryl-11 prefers to bind to the inner core of the fibril along its long axis (Fig. S8 and S9, ESI†). Thus, the docking calculations suggest that styryl-11 interacts with the inner core of the fibril with predominantly hydrophobic interactions which is the case observed for most of the amyloid markers.21,22
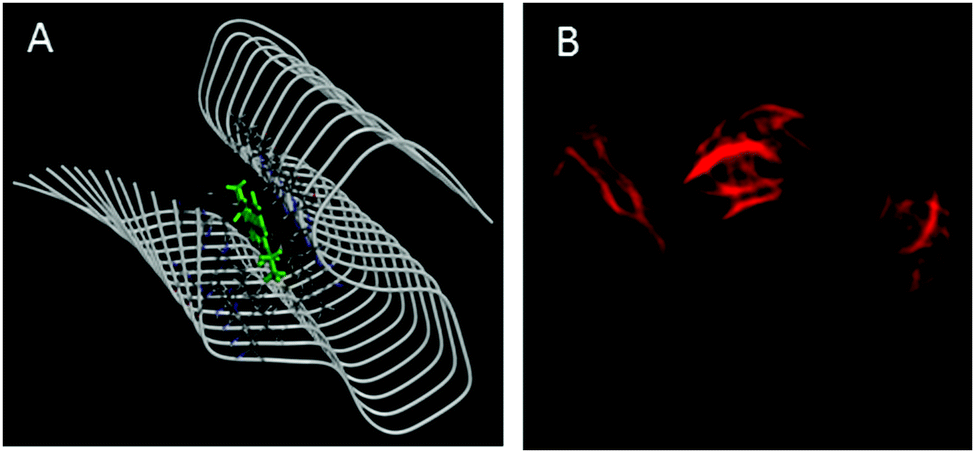 | ||
| Fig. 4 (A) Molecular docking diagram showing the location of the binding site of the styryl-11 in fibrils. (B) Fluorescence microscopic images of insulin fibrils using styryl-11. | ||
Fig. 4B shows fluorescence microscopic images of insulin fibrils using styryl-11. It is important to remark on the possibility of in vivo imaging using styryl-11. It has been reported in the literature that even cationic molecules can cross the Blood–Brain-Barrier (BBB), provided the molecule has good lipophilicity that is decided by its overall structure and molecular weight.11,23 In this regard, our current probe, styryl-11, has a clogP value (a parameter that indicates the ability of a molecule for its BBB permeability) of 0.8, which is close to the value of a few of the earlier reported cationic in vivo amyloid markers, and importantly, it also follows the desirable molecular weight criterion of <600 Da, i.e. 429 Da. Thus, we strongly believe that, despite bearing a cationic charge, styryl-11 can potentially cross the BBB, and combined with the remarkable modulations in its fluorescence properties, upon binding to amyloid fibrils, in the NIR region, it may prove to be a potential amyloid marker for in vivo imaging. To check the generality of styryl-11 as an amyloid marker, we have also tested the response of styryl-11 towards lysozyme fibrils (Fig. S10 and S11, ESI†) and as is evident from Fig. S10 (ESI†), styryl-11 also shows a large fluorescence enhancement in the presence of lysozyme fibrils, whereas it shows a very weak response towards the native form for the lysozyme protein. Note that styryl-11 does not share a common binding site with ThT in the fibril matrix (Fig. S12–S18, ESI†).
In summary, we have identified a sensitive and selective fluorescence turn-on probe for amyloid detection in the extreme near-IR region which registers a very large emission enhancement of around 500 fold, with emission maximum at ∼770 nm, in the presence of insulin amyloid fibrils. Importantly, the probe registers a magnificent Stokes’ shift of ∼150 nm and a very large red-shift of ∼120 nm in the absorption spectra, which to the best of our knowledge is the highest reported to date for any amyloid probe in the insulin fibril system. The extremely large shift in the absorption spectra for the fibril bound form of the probe leads to a visual colour change of the solution and enables naked eye detection of the fibrils. The probe shows a quite remarkable selectivity for the fibrillar form with respect to the native form of the protein, and is also able to monitor the protein fibrillation kinetics without interfering with the kinetic process of protein aggregation. Unlike the most extensively utilized cationic probe, thioflavin-T, the sensing performance of the current probe is unaffected by the ionic strength of the medium. Detailed time-resolved emission measurements suggest that the probe is conformationally immobilized in the amyloid fibril medium, which leads to a large reduction in the non-radiative process for the molecule, thus leading to a large fluorescence enhancement in the extremely advantageous and very desirable near-IR region. Another significant aspect of this probe is its commercial availability, which provides an advantage over the probes prepared through time-consuming and complex synthetic protocols. The attractive and powerful attributes of the near-IR amyloid probe reported in this work should be further explored for in vivo imaging.
The authors thank the Host Institutes, C. B. Patel Research Centre and Bhabha Atomic Research Centre for generous support during the course of this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. E. A. Meyer-Luehmann, Nat. Neurosci., 2003, 6, 1 CrossRef PubMed
.
- R. L. Nussbaum and C. E. Ellis, N. Engl. J. Med., 2003, 348, 1356 CrossRef CAS PubMed
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353 CrossRef CAS PubMed
- X. Zhang, Y. Tian, C. Zhang, X. Tian, A. W. Ross, R. D. Moir, H. Sun, R. E. Tanzi, A. Moore and C. Rana, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9734 CrossRef CAS PubMed
- M. Ono, M. Ishikawa, H. Kimura, S. Hayashi, K. Matsumura and H. E. A. Watanabe, Bioorg. Med. Chem. Lett., 2010, 20, 3885 CrossRef CAS PubMed
- C. Ran and A. Moore, Mol. Imaging Biol., 2012, 14, 293 CrossRef PubMed
- X. L. Zhang, Y. L. Tian, Z. Li, X. Y. Tian, H. B. Sun and H. E. A. Liu, J. Am. Chem. Soc., 2013, 135, 16397 CrossRef CAS PubMed
- C. Ran, X. Xu, S. B. Raymond, B. J. Ferrara, K. Neal, B. J. Bacskai, Z. Medarova and A. Moore, J. Am. Chem. Soc., 2009, 131, 15257 CrossRef CAS PubMed
- E. E. Nesterov, J. Skoch, B. T. Hyman, W. E. Klunk, B. J. Bacskai and T. M. Swager, Angew. Chem., Int. Ed., 2005, 44, 5452 CrossRef CAS PubMed
- S. B. Raymond, J. Skoch, I. D. Hills, E. E. Nesterov, T. M. Swager and B. J. Bacskai, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35(Suppl 1), S93 CrossRef PubMed
- M. Hintersteiner, A. Enz, P. Frey, A. L. Jaton, W. Kinzy and R. E. A. Kneuer, Nat. Biotechnol., 2005, 23, 577 CrossRef CAS PubMed
- M. Cui, M. Ono, H. Watanabe, H. Kimura, B. Liu and H. Saji, J. Am. Chem. Soc., 2014, 136, 3388–3394 CrossRef CAS PubMed
- H. Doan, M. Castillo, M. Bejjani, Z. Nurekeyev, S. V. Dzyuba, I. Gryczynski, Z. Gryczynski and S. Raut, Phys. Chem. Chem. Phys., 2017, 19, 29934 RSC
- Y. Hong, L. Meng, S. Chen, C. W. T. Leung, L.-T. Da, M. Faisal, D.-A. Silva, J. Liu, J. W. Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 1680–1689 CrossRef CAS PubMed
- F. Peccati, J. Hernando, L. Blancafort, X. Solans-Monforta and M. Sodupe, Phys. Chem. Chem. Phys., 2015, 17, 19718 RSC
- P. Sarkar, R. Luchowski, S. Raut, N. Sabnis, A. Remaley, A. G. Lacko, S. Thamake, Z. Gryczynski and I. Gryczynski, Biophys. Chem., 2010, 153, 61 CrossRef CAS PubMed
- W. E. Klunk, J. Histochem. Cytochem., 1989, 37, 1273 CrossRef CAS PubMed
- H. Tong, K. Lou and W. Wang, Acta Pharm. Sin. B, 2015, 5, 25 CrossRef
- R. Luchowski, Z. Gryczynski, P. Sarkar, J. Borejdo, M. Szabelski, P. Kapusta and I. Gryczynski, Rev. Sci. Instrum., 2009, 80, 033109(1) CrossRef PubMed
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499 CrossRef CAS PubMed
- F. Peccati, S. Pantaleone, V. Riffet, X. Solans-Monfort, J. Contreras-García, V. Guallar and M. Sodupe, J. Phys. Chem. B, 2017, 121, 8926–8934 CrossRef CAS PubMed
- N. A. Murugan, C. Halldin, A. Nordberg, B. Långström and H. Ågren, J. Phys. Chem. Lett., 2016, 7, 3313–3321 CrossRef CAS PubMed
- J.-S. Lee, Y. K. Kim, M. Vendrellc and Y.-T. Chang, Mol. BioSyst., 2009, 5, 411 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc01262a |
| This journal is © The Royal Society of Chemistry 2019 |