
Rapid NMR assignments of intrinsically disordered proteins using two-dimensional 13C-detection based experiments†
Sujeesh
Sukumaran
ab,
Shahid A.
Malik
ab,
Shankararama Sharma
R.
bc,
Kousik
Chandra
*b and
Hanudatta S.
Atreya
 *b
*b
aSolid State and Structural Chemistry Unit (SSCU), Indian Institute Of Science, Bangalore – 560 012, India
bNMR Research Centre, Indian Institute of Science, Bangalore – 560 012, India. E-mail: hsatreya@iisc.ac.in
cIISc Mathematics Initiative, Indian Institute of Science, Bangalore – 560 012, India
First published on 10th June 2019
Abstract
An approach for rapid backbone resonance assignments in proteins using only two 2D NMR experiments is presented. The new method involves a combination of high-resolution 13Cα-detected NMR experiments and selective unlabeling of amino acid residues. The 13C detected 2D hNCA and 2D hNcoCA spectra of a uniformly labeled sample of the protein are analysed in concert with the 2D hNCA spectrum obtained for a selectively unlabeled sample. The combinatorial set of amino acid residues for selective unlabeling is chosen optimally to maximize the assignments. The method is useful for rapid assignment of proteins with low stability such as intrinsically disordered proteins and is applicable to deuterated proteins. This approach helped in assignments of 14.5 kDa human α-synuclein during the course of its aggregation.
Intrinsically disordered proteins (IDPs) are an important class of proteins comprising ∼30% of the total proteins in organisms, and characterized by the lack of a well-defined three-dimensional (3D) structure.1 IDPs are involved in various biological functions, such as regulation of transcription and translation, storage of small molecules, regulation of self-assembly of large multi-protein complexes, etc.2 Due to the absence of a well-defined 3D structure and high flexibility, structural studies of IDPs are difficult using most of the structural biology techniques.3 NMR spectroscopy plays a significant role in the study of IDPs, because it is the only method that allows a high resolution description of structural and dynamic features of IDPs in solution.4 The potential for NMR spectroscopy to study the dynamic properties of proteins under physiological conditions makes it a powerful technique to understand the functional properties of IDPs.5,6
The first step towards the structure determination of proteins using NMR spectroscopy is the assignment of resonances observed in the NMR spectra to individual nuclei of the protein.7 Although conventional approaches for connecting sequential spin systems using 3D and higher dimensional NMR experiments have been successful for structured proteins, they are time-consuming and become ineffective in the presence of spectral overlap or if the protein aggregates or degrades rapidly such as in the case of IDPs.8 The process of resonance assignments in IDPs is thus challenging due to their unique properties.9 Chemical shift dispersion is much less in the conventional 1H-detected experiments of IDPs. The signal overlap in the spectra of IDPs strongly limits the possibility of standard amide proton-detection based NMR experiments to provide sufficient resolution for their characterization.10 Heteronuclear NMR experiments based on 13C direct detection are useful in such cases, as they display a better chemical shift dispersion compared to 1H-detected experiments.11
In general, the conventional approach involves analysis of the NMR spectra obtained from multidimensional triple resonance experiments in concert, and linking the backbone 1H, 13C, and 15N chemical shifts of a continuous stretch of amino acids and mapping them onto the primary sequence of the protein for obtaining sequence-specific resonance assignments.7 When there is considerable spectral overlap like in the case of larger molecular weight proteins or IDPs, it is necessary to selectively identify spin systems corresponding to the different amino acid types to aid resonance assignments. Amino acid selective ‘‘unlabeling’’ is an efficient method for the selective identification of the type of spin systems.12 In this method, selective unlabeling of a set of amino acid type(s) in a polypeptide is performed by feeding the host microorganism with nutrients containing NMR active atoms. 15NH4Cl is used as a source of nitrogen and 13C (1H/2H)-D-glucose is used as a source of carbon. The desired set of amino acid(s) in the unlabeled form (i.e., containing 12C and 14N) are also added to the growth medium for protein expression.13 As a result, peaks due to these unlabeled residues will not be observed in the double and triple resonance spectra. The peaks corresponding to the unlabeled and labeled residues can be distinguished by comparing the spectrum recorded on the unlabeled sample with a control spectrum involving uniformly 13C or/and 15N labeled protein.12 Selective unlabeling is used in combination with other NMR methods to help in rapid assignment of resonances observed in the NMR spectra.12–15
By exploiting the advantages of 13C detection and optimal combinatorial selective unlabeling14 of amino acid residues, rapid resonance assignments can be obtained as shown here. In this method, we use two 2D NMR experiments: 13Cα detected 2D hNcoCa and 13Cα detected 2D hNCA. These experiments are recorded on a (13C, 15N) uniformly labeled and a selectively unlabeled protein sample. The 2D hNCA provides chemical shift correlation of Cα of residue i with that of 15N of residue i (Cαi, Ni) and 15N of residue i + 1 (Cαi, Ni+1) (Fig. 1a). Of these two correlations, the (Cαi, Ni+1) peak appears lower in intensity or may be absent. This can be recovered from the 2D hNcoCa spectrum, which gives a correlation between Cα of residue i with 15N of only residue i + 1 (Cαi, Ni+1) (Fig. 1b). The 2D hNcoCa thus helps distinguish between Ni and Ni+1 peaks in the 2D hNCA for a given Cαi. The 2D spectra are analysed in a combined manner yielding identification/assignment of a long peptide in a directional manner which can be mapped onto the primary sequence. The connected stretch of amino acids is categorized into specific patterns of X and U (X-labeled residue and U-unlabeled residue) with the help of selective unlabeling. If the pattern is unique in the sequence, then it is directly mapped onto the protein primary sequence to obtain backbone resonance assignment. The amino acid types corresponding to X and U need not be known.
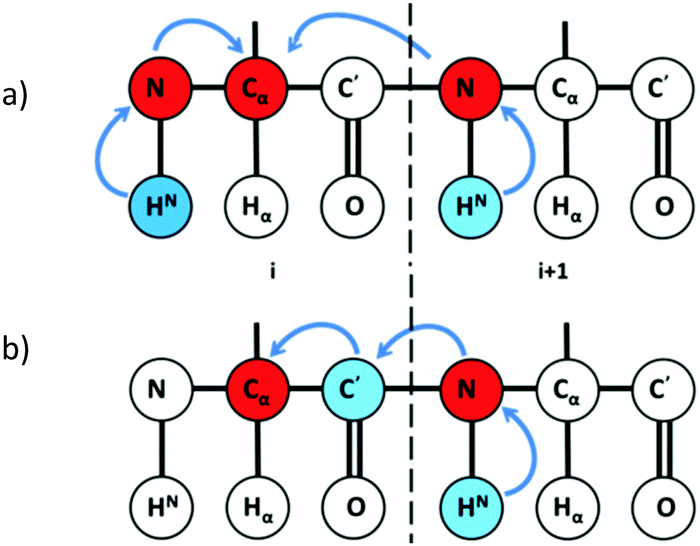 | ||
| Fig. 1 Magnetization transfer pathway in (a) 2D hNCA providing (Cαi, Ni) and (Cαi, Ni+1) chemical shift correlations and (b) 2D hNcoCA providing (Cαi, Ni+1) shift correlations. | ||
Two samples are used: a uniformly 13C, 15N labeled sample, and a (13C, 15N) labeled sample with simultaneous unlabeling of multiple amino acid types chosen optimally.
The assignment strategy is depicted in Fig. 2a and b. For any given residue Cαi as a starting point, 2D hNCA gives (Cαi, Ni) and (Cαi, Ni+1) correlations. The 2D hNcoCa helps identify the (Cαi, Ni+1) correlation. Next, using the Ni+1 shift thus identified, a search is carried out in 2D hNCA to identify the (Cαi+1, Ni+1) correlation (as indicated by the horizontal dotted line in Fig. 2a). Having identified Cαi+1 from 2D hNCA, the Ni+2 can be identified based on 2D hNcoCA at the Cαi+1 position as shown, and the assignment can be continued. In the same manner, starting from (Cαi, Ni) one can also proceed in the reverse direction.
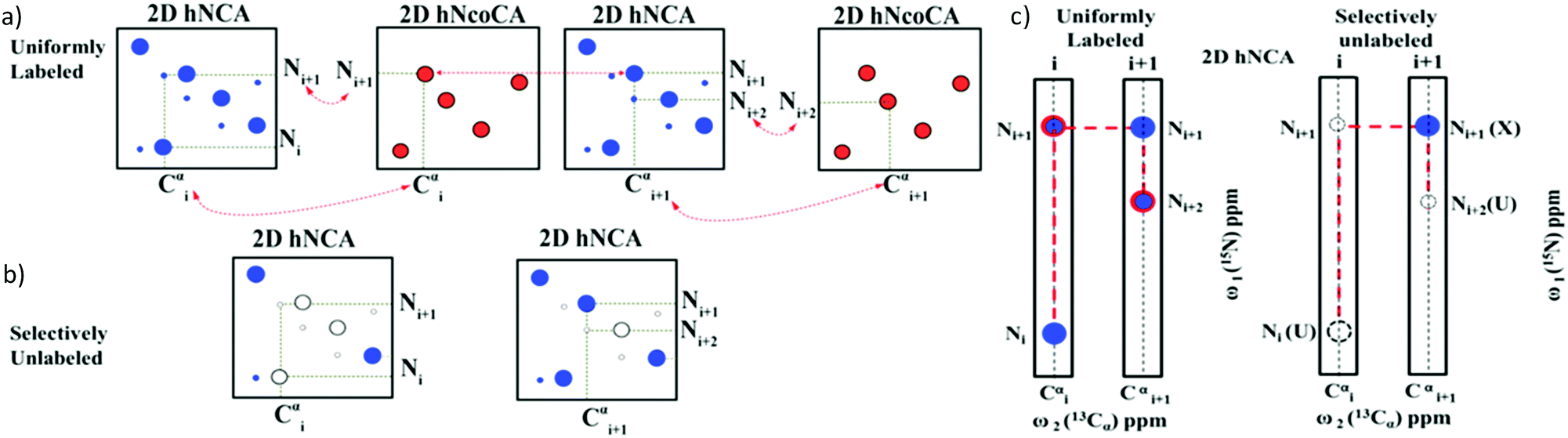 | ||
| Fig. 2 The assignment strategy using the 2D hNCA (blue) and 2D hNcoCA (red) spectra is illustrated for a tri-peptide stretch: U (i)–X (i + 1)–U (i + 2), where X corresponds to a labeled residue and U corresponds to any selectively unlabeled residue. (a) A sequential connectivity between the peaks can be established by linking them as shown. Peaks depicted by large and small circles correspond, respectively, to self (Cαi, Ni) and sequential (Cαi, Ni+1) correlations. (b) Based on the presence (shown by filled circles) or absence (empty circles) of (Cαi, Ni)/(Cαi, Ni+1) correlations in the 2D hNCA spectrum recorded on the selectively unlabeled sample, a particular residue can be identified as belonging to labeled (X) or unlabeled (U) amino acid category. (c) A strip plot shows the connectivity: U (i)–X (i + 1)–U (i + 2) established using (a and b). | ||
With the help of these two experiments we will be able to obtain a sequential connectivity between the peaks (Fig. 2c). These sequentially connected amino acids form a stretch which has to be directly mapped onto the primary sequence. We have to identify the type of amino acid residues for mapping onto the sequence. The 2D hNCA spectrum of the selectively unlabeled sample helps identify the amino acid type. A peak can be categorized into X (labeled) or U (unlabeled) based on whether it is present or absent in the 2D hNCA spectrum of the selectively unlabeled sample (Fig. 2b). Note that 2D hNcoCA of the selectively unlabeled sample is not necessary, because the residues that are selectively labeled or unlabeled can be identified from the hNCA spectrum itself. Thus, only three 2D spectra are recorded (2 on the uniformly labeled sample and one on the selectively unlabeled sample).
The connected stretch of resonances as shown in Fig. 2c can be written in terms of X and U. The primary sequence of amino acids can also be translated to the sequence of X and U based on the choice of amino acids for selective unlabeling. The sequentially connected stretch of amino acids forms a pattern of X and U (e.g., UXU). If this pattern is unique in the primary sequence (translated to the sequence of X and U), then we can directly map it onto the sequence to obtain backbone resonance assignments as shown below (also see Fig. S1, ESI†).
The choice of amino acids to be unlabeled is decided based on an in-house developed python-based computer program (available at http://nrc.iisc.ernet.in/hsa/software.htm) and based on the possibilities of isotope scrambling of 15N or 14N among amino acids during expression of the protein in E. coli.12 We have previously shown that based on the amino acid composition, a subset of amino acids from the following combinations: {Arg, Lys, Asn, Thr, Gly, Ser, Ala} can be chosen in any given protein for optimal selective unlabeling.13 These amino acids either do not cross-metabolize to other amino acid residues in the bacterial biosynthetic pathway or do so within the same set (e.g., Gly and Ser cross-metabolize to each other). This avoids the unlabeling of undesired residues. The program tries these combinations or their subsets taking the primary sequence of a protein as input. It then translates the amino acid sequence into a sequence of X, U and 1, where X is a labeled residue, U is a selectively unlabeled residue and 1 is assigned for proline. The program searches along the sequence for identifying unique amino acid stretches of different lengths: hexa, penta, tetra, tri, and di-peptide stretches and outputs the amino acid residues that can be assigned. This helps in selecting the best combination of amino acids to be unlabeled. The algorithm of the program is depicted in Fig. S2 of the ESI.† The set {Thr, Gly, Ser, Ala} was found to be optimal for selective unlabeling in α-synuclein; this constitutes 36.4% of residues in the protein.
Fig. 3 shows the 2D spectra obtained for α-synuclein (complete acquisition parameters are provided in Table S1, ESI†). Fig. 4a shows an example of sequential connection among a set of residues in α-synuclein. This was carried out using the strategy depicted in Fig. 2 (i.e., i − i + 1 − i + 2…). Based on the presence/absence of peaks in the 2D hNCA spectrum obtained for the selectively unlabeled sample (Fig. 4b), the stretch of amino acids is classified as XUXXUU. The XUXXUU pattern is unique in the protein sequence from residue number 63 to 68 (V63, T64, N65, V66, G67, G68). This stretch of amino acids can thus be directly assigned. The results of this method were validated by comparing the results with the assignment done on the protein using the traditional assignment strategy.16
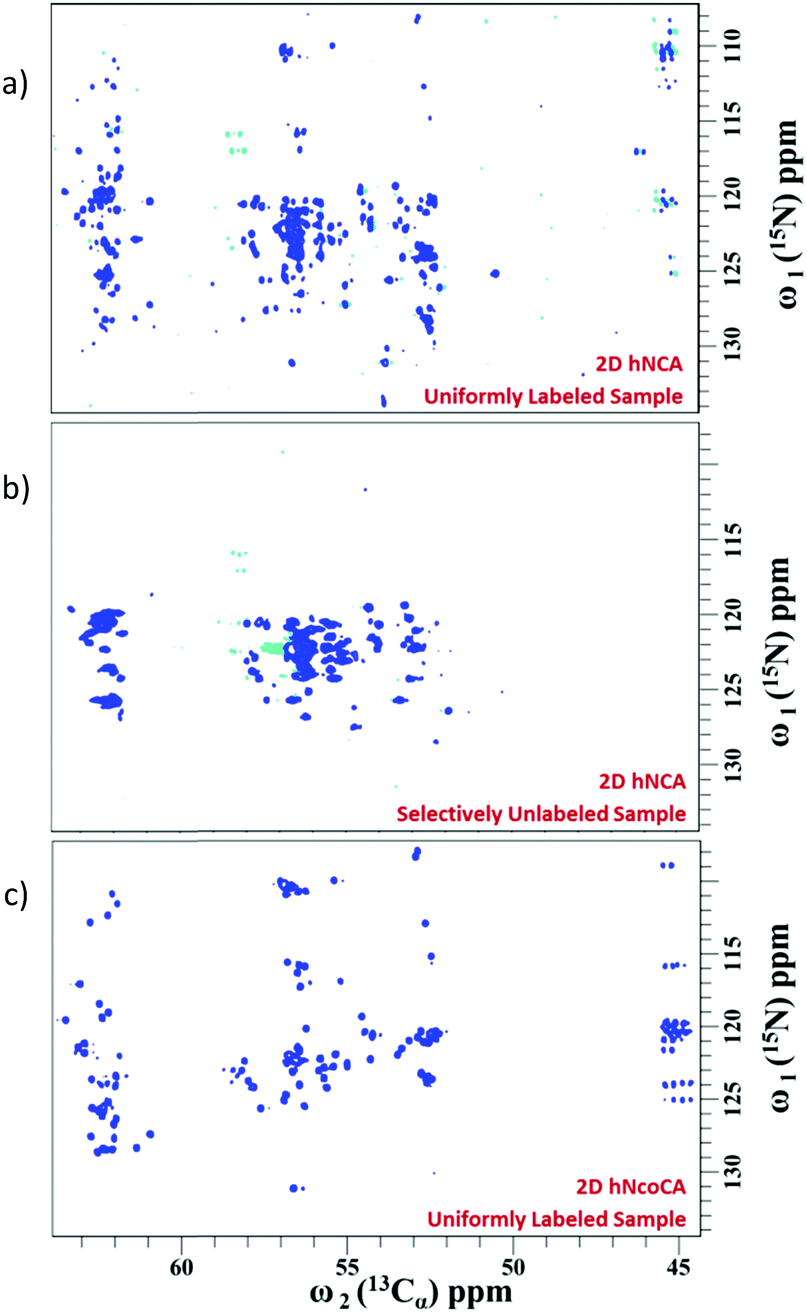 | ||
| Fig. 3 (a) The complete 2D hNCA spectrum of the uniformly labeled sample, (b) 2D hNCA of selectively unlabeled sample and (c) 2D hNcoCA spectrum of the uniformly labeled sample. Note that in this approach, the 2D hNcoCa spectrum of the selectively unlabeled sample is not needed (see text). | ||
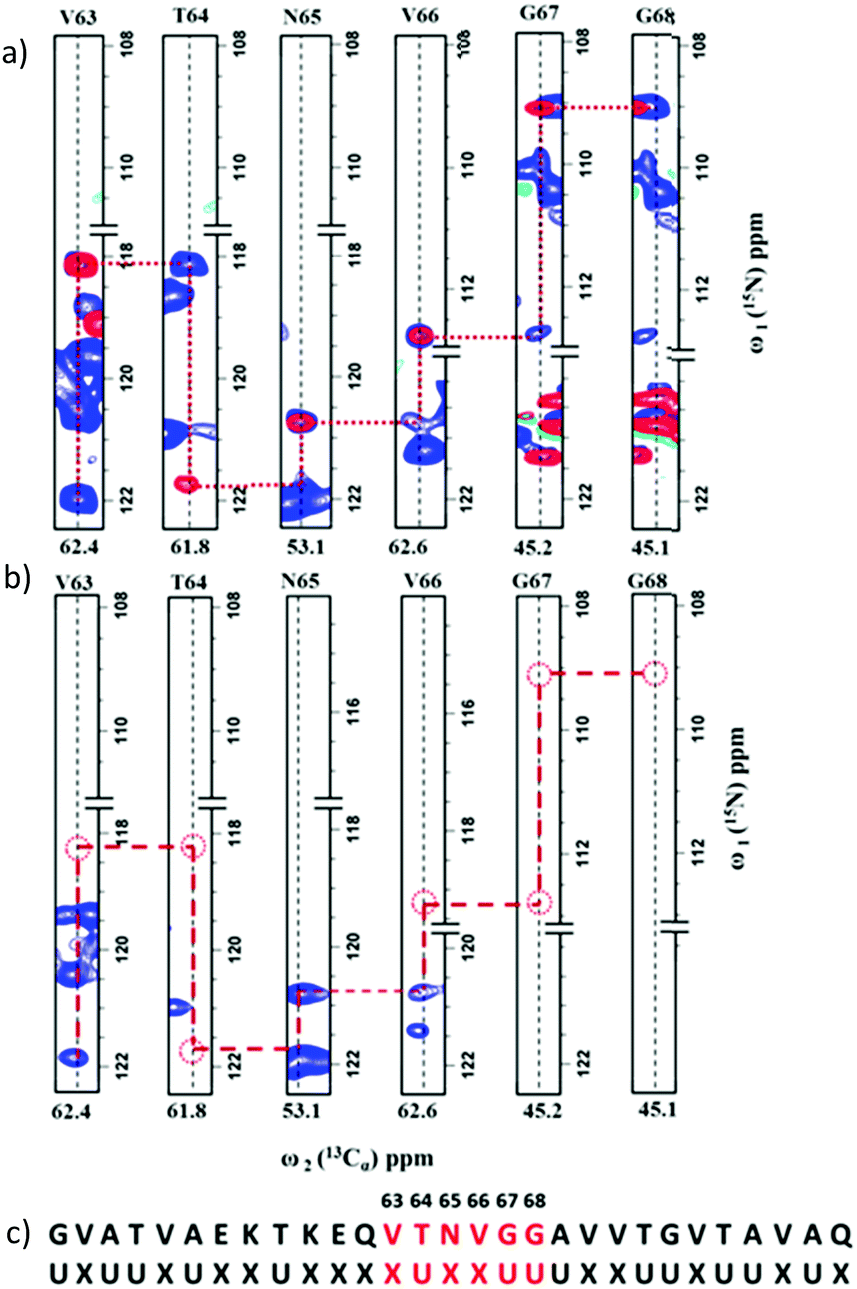 | ||
| Fig. 4 (a) An overlay of 2D strips from the 2D hNCA (blue) and 2D hNcoCA (red) spectra acquired on the uniformly labeled α-synuclein sample. The sequential connectivity between the stretch of residues shown is based on the assignment strategy depicted in Fig. 2. (b) For this stretch, the pattern of missing (Cαi, Ni) and/or (Cαi, Ni+1) correlations in the 2D hNCA spectrum of the selectively unlabeled sample indicate that the connected residues form a XUXXUU pattern. (c) This pattern is mapped on- to the protein sequence for sequence-specific assignments. | ||
Using this approach 121 spin systems were identified out of expected 134 spin systems at 283 K. The stretches of residues assigned along the polypeptide are depicted in Fig. S3 of the ESI.† Since α-synuclein is an intrinsically disordered protein and its aggregation rate (40–70 hours) is high under physiological conditions,17 the assignment of this protein using traditional methods is a difficult task. Our method is more effective in this case because it is fast and due to the high dispersion of peaks in 13C-detected experiments.18
Rapid protein assignments are of high utility where measurement time is crucial because of limited sample stability. Our method is rapid since it uses only two 2D NMR experiments which together can be completed in less than 20 hours of experiment time. The experimental time can be further reduced using non-uniform sampling.19 This method is useful not only in the case of IDPs, but also for structured proteins or for assignments of partially folded structures/intermediates formed during the course of protein folding or aggregation.20–22 Further the method is applicable to (per)deuterated proteins. The process of peak picking, sequential connection, pattern identification and sequence mapping can be accelerated with the help of automation.
A drawback of 13C detection with respect to 1H detection is its intrinsically lower sensitivity due to the lower gyromagnetic ratio of 13C with respect to that of 1H. Further loss of sensitivity can occur due to 13Cα transverse (T2) relaxation. In the two experiments, the 13Cα resides in the transverse plane during detection and for ∼25 ms within the pulse sequence.23 However, due to their high flexibility, the rotational correlation time of IDPs is lower compared to that of structured proteins of comparable size leading to higher sensitivity and sharper peaks. For instance, the rotational correlation time of 14.5 kDa α-synuclein is 4.3 ns at 283 K,24 which is equivalent to that of an ∼8 kDa globular structured protein. Thus, the 2D experiments are expected to exhibit sufficient sensitivity for larger size IDPs. Sensitivity can be further enhanced by deuteration and using cryogenic probes optimized for 13C-detection.
In summary, this represents the first approach based on a combination of selective unlabeling and 2D 13C-detection based NMR experiments for resonance assignments of proteins. These experiments are sensitive, can be performed rapidly, and analysed easily. The ability to carry out rapid assignments, the comparatively low experimental time, applicability to deuterated proteins and easy sample preparation are the highlights of this approach. These advantages make this method suitable for protein assignments in general and the assignment of samples with low stability such as intrinsically disordered proteins in particular.
This work was supported by the Department of Science and Technology (DST), Govt. of India (IR/S0/LU-007/2010). We thank the NMR Research Centre at IISc for providing facilities. We thank R. V. Hosur (the Centre for Excellence in Basic Science (CEBS), Mumbai) for providing the E. coli plasmid encoding the human α-synuclein gene.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. N. Uversky, Chem. Rev., 2014, 114, 6557–6560 CrossRef CAS PubMed.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS PubMed.
- V. N. Uversky, Protein Sci., 2013, 22, 693–724 CrossRef CAS PubMed.
- I. C. Felli and R. Pierattelli, IUBMB Life, 2012, 64, 473–481 CrossRef CAS PubMed.
- H. J. Dyson and P. E. Wright, Chem. Rev., 2004, 104, 3607–3622 CrossRef CAS PubMed.
- I. C. Felli and R. Pierattelli, J. Magn. Reson., 2014, 241, 115–125 CrossRef CAS PubMed.
- J. Cavanagh, W. Fairbrother, A. Palmer III, M. Rance and N. Skelton, Protein NMR Spectroscopy: Principles and Practice, Academic Press, Burlington, 2nd edn, 2007, pp. 114–270 Search PubMed.
- B. Brutscher, I. C. Felli, S. Gil-Caballero, T. Hošek, R. Kümmerle, A. Piai, R. Pierattelli and Z. Sólyom, Adv. Exp. Med. Biol., 2015, 870, 49–122 CrossRef CAS PubMed.
- R. Konrat, J. Magn. Reson., 2014, 241, 74–85 CrossRef CAS PubMed.
- W. Bermel, I. Bertini, I. C. Felli, M. Piccioli and R. Pierattelli, Prog. NMR Spectrosc., 2006, 48, 25–45 CrossRef CAS.
- W. Bermel, I. C. Felli, R. Kümmerle and R. Pierattelli, Concepts Magn. Reson., 2008, 32A, 183–200 CrossRef CAS.
- B. Krishnarjuna, G. Jaipuria, A. Thakur, P. D’Silva and H. S. Atreya, J. Biomol. NMR, 2011, 49, 39–51 CrossRef CAS PubMed.
- C. Prasanna, A. Dubey and H. S. Atreya, Methods Enzymol., 2015, 565, 167–189 CAS.
- G. Jaipuria, B. Krishnarjuna, S. Mondal, A. Dubey and H. S. Atreya, Adv. Exp. Med. Biol., 2012, 992, 95–116 CrossRef CAS PubMed.
- A. Dubey, R. V. Kadumuri, G. Jaipuria, R. Vadrevu and H. S. Atreya, ChemBioChem, 2016, 17, 334–340 CrossRef CAS PubMed.
- D. Eliezer, E. Kutluay, R. Bussell and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef CAS PubMed.
- M. M. Wördehoff and W. Hoyer, Bio-Protoc., 2018, 8, 2941 Search PubMed.
- W. Bermel, I. Bertini, I. C. Felli, Y.-M. Lee, C. Luchinat and R. Pierattelli, J. Am. Chem. Soc., 2006, 128, 3918–3919 CrossRef CAS PubMed.
- S. Robson, H. Arthanari, S. G. Hyberts and G. Wagner, Methods Enzymol., 2019, 614, 263–291 Search PubMed.
- R. P. R. Nanga, J. R. Brender, S. Vivekanandan and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2337–2342 CrossRef CAS PubMed.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312–316 CrossRef CAS PubMed.
- J. L. Neira, Arch. Biochem. Biophys., 2013, 531, 90–99 CrossRef CAS PubMed.
- K. Takeuchi, Z.-Y. J. Sun and G. Wagner, J. Am. Chem. Soc., 2008, 130, 17210–17211 CrossRef CAS PubMed.
- D. Bhattacharyya, R. Kumar, S. Mehra, A. Ghosh, S. K. Maji and A. Bhunia, Chem. Commun., 2018, 54, 3605–3608 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc03530c |
| This journal is © The Royal Society of Chemistry 2019 |